Pharmacokinetics, Pharmacodynamics, Tolerability, and Food Effect of Cenerimod, a Selective S1P1 Receptor Modulator in Healthy Subjects

Abstract
:1. Introduction
2. Results
2.1. Pharmacokinetics
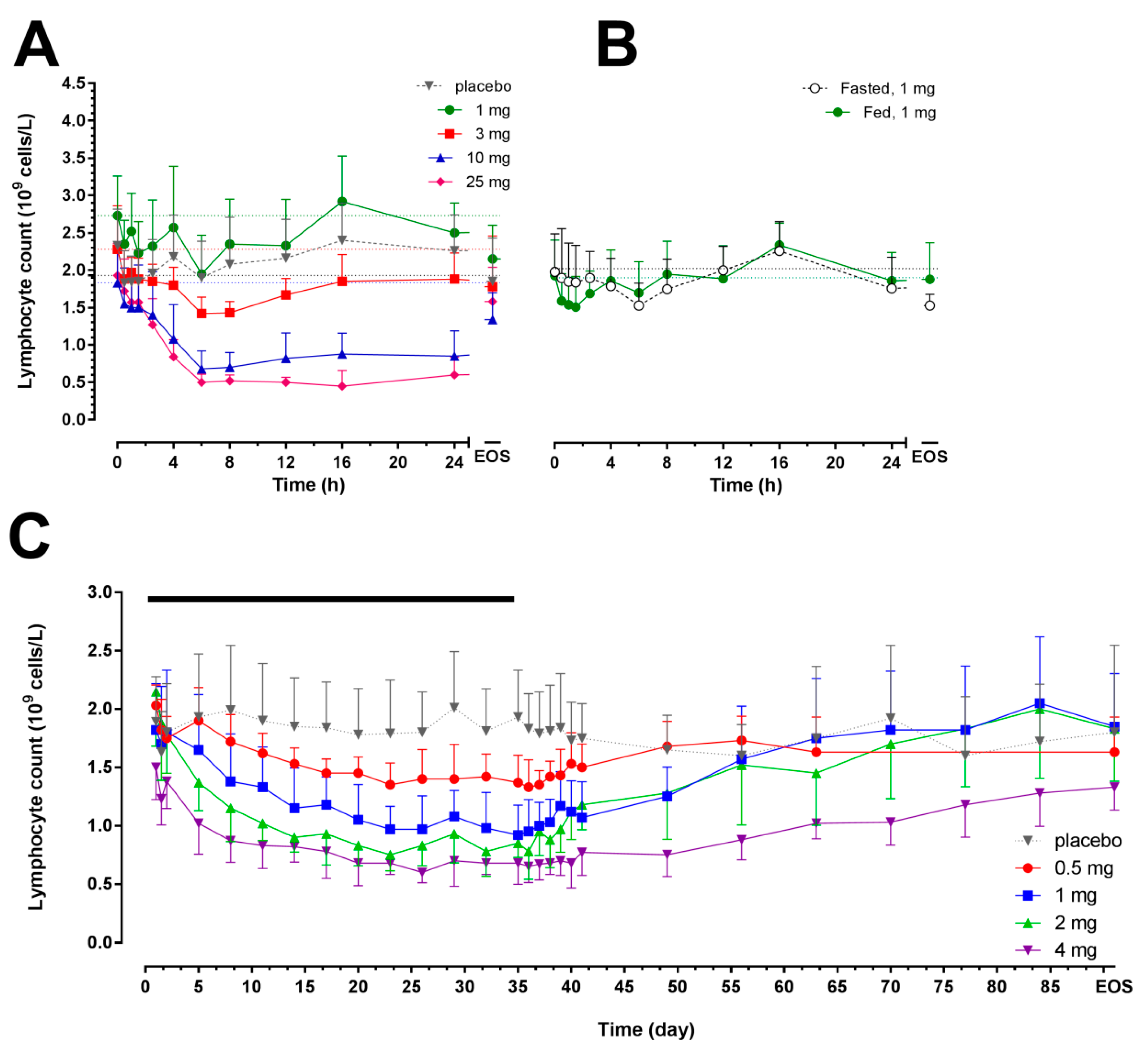
2.2. Pharmacodynamics
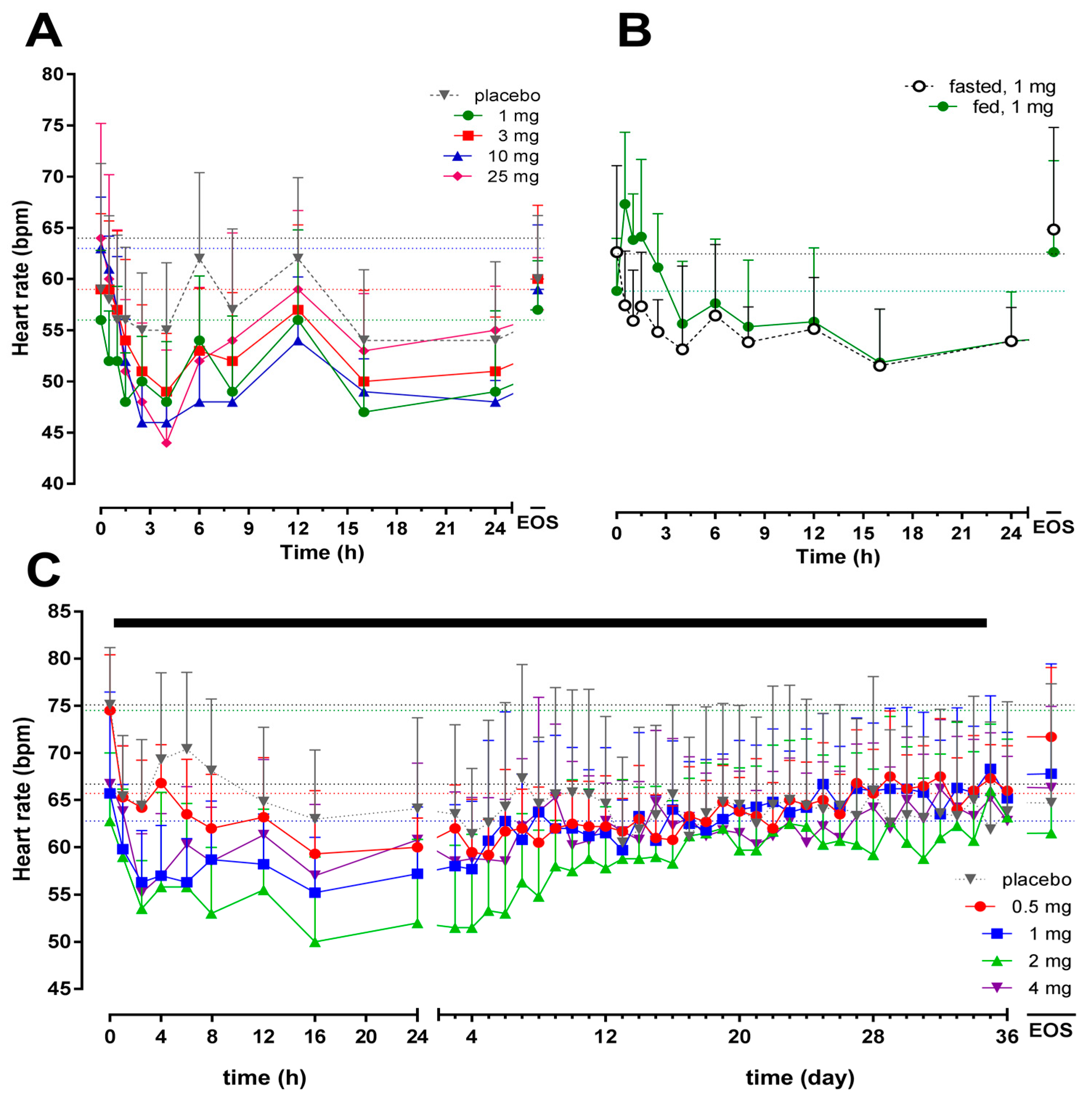
2.3. Pharmacokinetics/Pharmacodynamics
2.4. Safety and Tolerability
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Study Design
4.3. Pharmacokinetic Assessments
4.4. Pharmacodynamic Assessments
4.5. Safety and Tolerability Assessments
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AE | adverse event |
| AUC0–24 | area under the plasma drug concentration-time curve from 0 to 24 h |
| AUC0–∞ | area under the plasma drug concentration-time curve from 0 to infinity |
| AUC | area under the plasma drug concentration-time curve |
| AUEC | area under the effect-time curve |
| CI | confidence interval |
| CL/F | apparent oral clearance |
| Cmax | maximum plasma concentration |
| Emax | maximum effect |
| ECG | electrocardiogram |
| EDTA | ethylene di-amine tetra acetic acid |
| FEV1 | forced expiratory volume in 1 s |
| FVC | forced vital capacity |
| HR | heart rate |
| LS | least squares |
| o.d. | once daily |
| PD | pharmacodynamics |
| PK | pharmacokinetics |
| S1P | sphingosine-1-phosphate |
| S1P1R | S1P1 receptor |
| SD | standard deviation |
| t1/2 | terminal half-life |
| tmax | time to reach maximum concentration |
| Vss/F | apparent volume of distribution at steady-state |
References
- Rivera, J.; Proia, R.L.; Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 2008, 8, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, Y.; Okamoto, Y.; Yoshioka, K.; Takuwa, N. Sphingosine-1-phosphate signaling and biological activities in the cardiovascular system. Biochim. Biophys. Acta 2008, 1781, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Thangada, S.; Khanna, K.M.; Blaho, V.A.; Oo, M.L.; Im, D.S.; Guo, C.; Lefrancois, L.; Hla, T. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J. Exp. Med. 2010, 207, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Antel, J.; Comi, G.; Montalban, X.; O’Connor, P.; Polman, C.H.; Haas, T.; Korn, A.A.; Karlsson, G.; Radue, E.W.; et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. NEJM 2006, 355, 1124–1140. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Boster, A.; Fernandez, O.; Freedman, M.S.; Pozzilli, C.; Bach, D.; Berkani, O.; Mueller, M.S.; Sidorenko, T.; Radue, E.W.; et al. Oral ponesimod in relapsing-remitting multiple sclerosis: A randomised phase II trial. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Vaclavkova, A.; Chimenti, S.; Arenberger, P.; Hollo, P.; Sator, P.G.; Burcklen, M.; Stefani, M.; D’Ambrosio, D. Oral ponesimod in patients with chronic plaque psoriasis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 384, 2036–2045. [Google Scholar] [CrossRef]
- Forrest, M.; Sun, S.Y.; Hajdu, R.; Bergstrom, J.; Card, D.; Doherty, G.; Hale, J.; Keohane, C.; Meyers, C.; Milligan, J.; et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Gon, Y.; Wood, M.R.; Kiosses, W.B.; Jo, E.; Sanna, M.G.; Chun, J.; Rosen, H. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. PNAS 2005, 102, 9270–9275. [Google Scholar] [CrossRef] [PubMed]
- Sanna, M.G.; Liao, J.; Jo, E.; Alfonso, C.; Ahn, M.Y.; Peterson, M.S.; Webb, B.; Lefebvre, S.; Chun, J.; Gray, N.; et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004, 279, 13839–13848. [Google Scholar] [CrossRef] [PubMed]
- Brossard, P.; Derendorf, H.; Xu, J.; Maatouk, H.; Halabi, A.; Dingemanse, J. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br. J. Clin. Pharmacol. 2013, 76, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Brossard, P.; Scherz, M.; Halabi, A.; Maatouk, H.; Krause, A.; Dingemanse, J. Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: Favorable impact of dose up-titration. J. Clin. Pharmacol. 2014, 54, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Hoch, M.; D’Ambrosio, D.; Wilbraham, D.; Brossard, P.; Dingemanse, J. Clinical pharmacology of ponesimod, a selective S1P1 receptor modulator, after uptitration to supratherapeutic doses in healthy subjects. Eur. J. Pharm. Sci. 2014, 63C, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Brossard, P.; Chassard, D.; Hoch, M.; Dingemanse, J. Effects of ponesimod, a selective S1P1 receptor modulator, on the pharmacokinetics of a hormonal combination contraceptive. Eur. J. Clin. Pharmacol. 2014, 70, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Juif, P.E.; Kraehenbuehl, S.; Dingemanse, J. Clinical pharmacology, efficacy, and safety aspects of sphingosine-1-phosphate receptor modulators. Expert Opin. Drug Metab. Toxicol. 2016, 12, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Piali, L.; Birker-Robaczewska, M.; Lescop, C.; Froidevaux, S.; Schmitz, N.; Morrison, K.; Kohl, C.; Rey, M.; Studer, R.; Vezzali, E.; et al. Cenerimod, a novel selective S1P1 receptor modulator with unique signaling properties. Pharmacol. Res. Perspect. 2017. accepted. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Schmouder, R.; Barilla, D.; Riviere, G.J.; Wang, Y.; Hunt, T. Multiple-dose FTY720: Tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J. Clin. Pharmacol. 2004, 44, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, J.M.; Schmouder, R.; Barilla, D.; Wang, Y.; Kraus, G. Single-dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br. J. Clin. Pharmacol. 2004, 57, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Li, D.K.; Hartung, H.P.; Hemmer, B.; Kappos, L.; Freedman, M.S.; Stuve, O.; Rieckmann, P.; Montalban, X.; Ziemssen, T.; et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): An adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013, 12, 756–767. [Google Scholar] [CrossRef]
- David, O.J.; Kovarik, J.M.; Schmouder, R.L. Clinical pharmacokinetics of fingolimod. Clin. Pharmacokinet. 2012, 51, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Maeda, Y.; Shimano, K.; Mogami, A.; Kataoka, H.; Ogawa, K.; Hikida, K.; Kumagai, H.; Asayama, M.; Yamamoto, T.; et al. Amiselimod, a novel sphingosine 1-phosphate receptor-1 modulator, has potent therapeutic efficacy for autoimmune diseases, with low bradycardia risk. Br. J. Clin. Pharmacol. 2017, 174, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Krosser, S.; Wolna, P.; Fischer, T.Z.; Boschert, U.; Stoltz, R.; Zhou, M.; Darpo, B. Effect of ceralifimod (ONO-4641) on lymphocytes and cardiac function: Randomized, double-blind, placebo-controlled trial with an open-label fingolimod arm. J. Clin. Pharmacol. 2015, 55, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.; Hartung, J.; Timony, G.; Peach, R.; Boehm, M.; Rosen, H.; Smith, H.; Pan, C.; Brooks, J.; Gujrathi, S. Safety and tolerability of orally administered RPC1063, a novel S1P1 receptor modulator, in healthy adult volunteers, results of a phase 1 study. Neurology 2013, 80, P01.178. [Google Scholar]
- Marsolais, D.; Rosen, H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discov. 2009, 8, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Koyrakh, L.; Roman, M.I.; Brinkmann, V.; Wickman, K. The heart rate decrease caused by acute FTY720 administration is mediated by the G protein-gated potassium channel I. Am. J. Transplant. 2005, 5, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; MacDonell, K.L.; Giles, W.R. Effects of sphingosine 1-phosphate on pacemaker activity in rabbit sino-atrial node cells. Pflugers Arch. 1999, 438, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Bigaud, M.; Guerini, D.; Billich, A.; Bassilana, F.; Brinkmann, V. Second generation S1P pathway modulators: Research strategies and clinical developments. Biochim. Biophys. Acta 2014, 1841, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry Food-Effect Bioavailability and Fed Bioequivalence Studies; US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Washington, DC, USA, 2002.
- Gibaldi, M.; Perrier, D. Pharmacokinetics; Marcel Dekker: New York, NY, USA, 1982. [Google Scholar]
- Gough, K.; Hutchison, M.; Keene, O.; Byrom, B.; Ellis, S.; Lacey, L.; McKellar, J. Assessment of dose proportionality: Report from the statisticians in the pharmaceutical industry/pharmacokinetics UK joint working group. Drug Inf. J. 1995, 29, 1039–1048. [Google Scholar] [CrossRef]
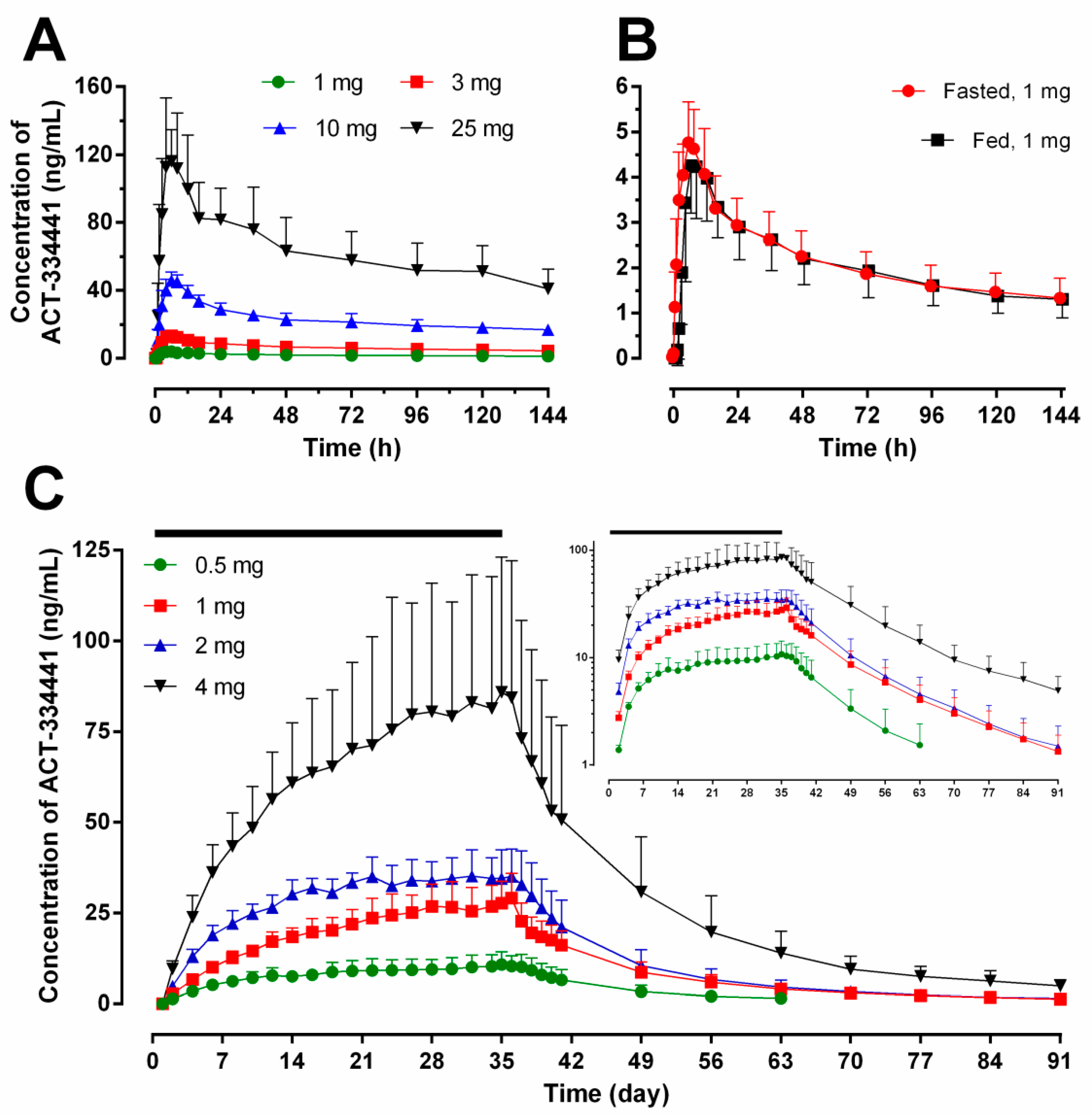
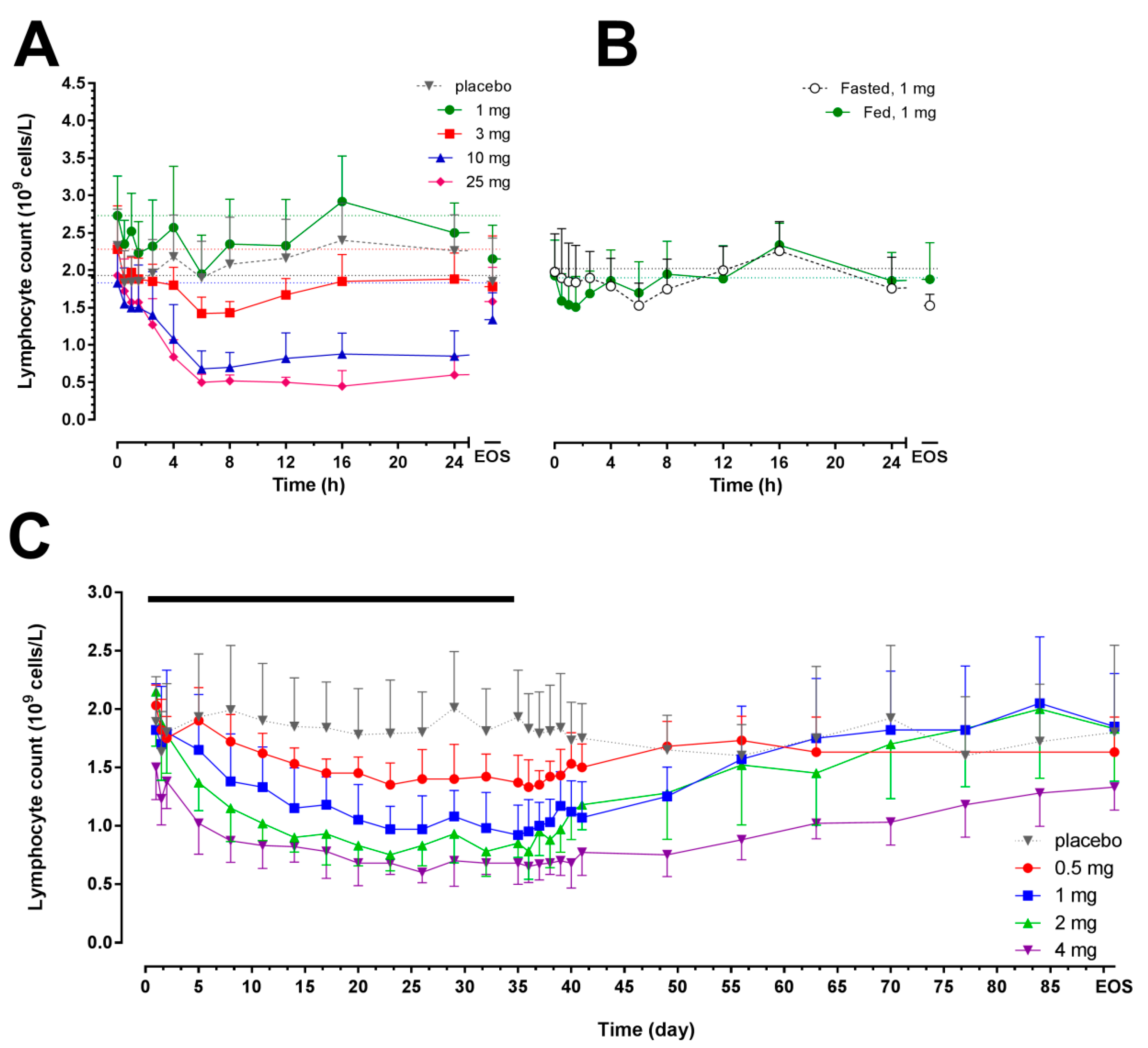
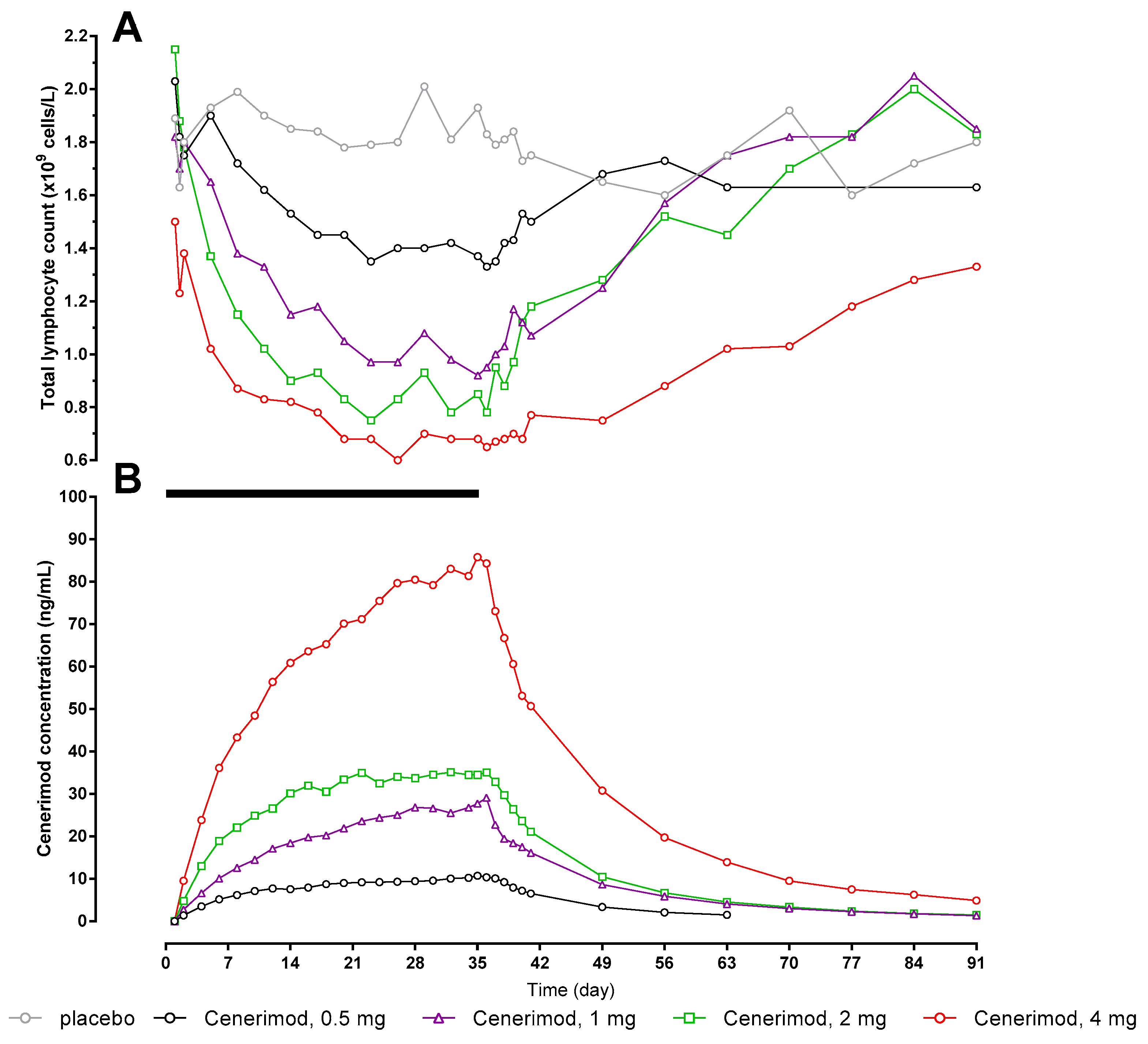


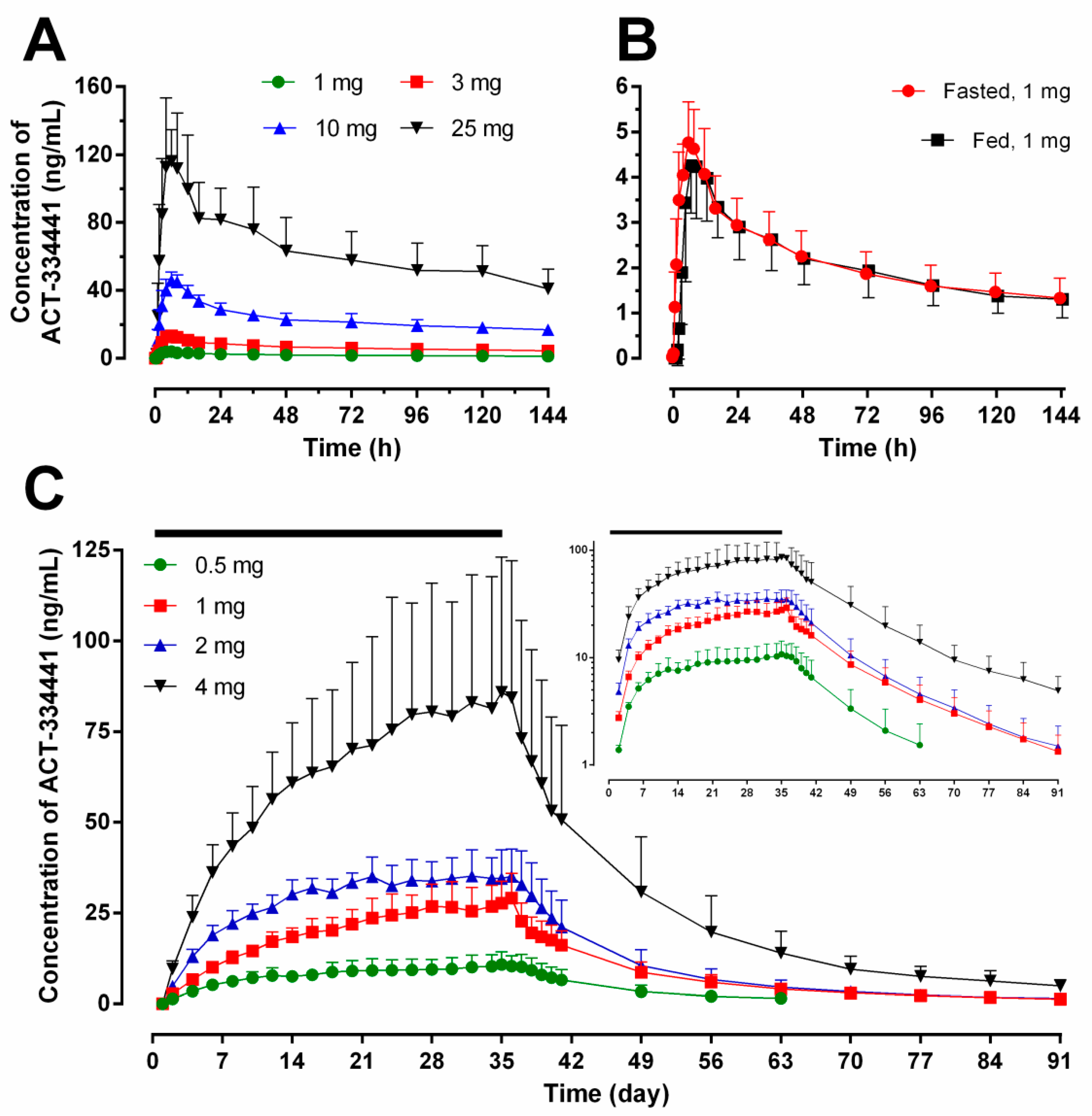{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Day 1 | Day 35 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose (mg) | Cmax (ng/mL) | tmax (h) | AUC0–24 (ng·h/mL) | AUC0–∞ (ng·h/mL) | t1/2 (h) | Cmax (ng/mL) | tmax (h) | AUC0–24 (ng·h/mL) | AUC0–∞ (ng·h/mL) | t1/2 (h) | R | ||
| Cmax | AUC0–24 | ||||||||||||
| Study 1 | 1 | 3.9 | 5.0 | 69.3 | 610 | 172 | NC | ||||||
| (2.9–5.2) | (4.0–8.0) | (52.4–90.3) | (504–735) | (125–232) | |||||||||
| 3 | 13.1 | 6.0 | 232 | 2070 | 184 | ||||||||
| (10.1–16.8) | (4.0–8.0) | (181–292) | (1470–2790) | (153–220) | |||||||||
| 10 | 46.7 | 6.0 | 823 | 8030 | 199 | ||||||||
| (42.4–51.3) | (6.0–8.0) | (732–923) | (6700–9550) | (188–211) | |||||||||
| 25 | 122 | 6.2 | 2030 | 18,700 | 170 | ||||||||
| (89.5–161.0) | (4.0–8.0) | (1480–2700) | (14,100–24,200) | (134–213) | |||||||||
| Study 2 | 0.5 | 2.4 | 5.0 | 41.1 | NC | NC | 13.1 | 4.3 | 269 | 3390 | 283 | 5.5 | 6.6 |
| (2.0–2.8) | (4.0–6.0) | (37.0–45.5) | (9.8–17.6) | (2.5–8.0) | (198–366) | (2050–5590) | (210–381) | (4.6–6.5) | (5.3–8.1) | ||||
| 1 | 5.1 | 5.0 | 83.5 | 34.2 | 5.0 | 720 | 9750 | 436 | 6.7 | 8.6 | |||
| (4.4–5.9) | (4.0–6.0) | (72.9–95.6) | (26.8–43.7) | (2.5–8.0) | (560–926) | (7270–13,100) | (359–529) | (5.5, 8.3) | (7.3, 10.2) | ||||
| 2 | 7.7 | 6.0 | 134 | 43.9 | 6.0 | 925 | 11,900 | 415 | 5.7 | 6.9 | |||
| (6.1–9.7) | (4.0–8.0) | (109–164) | (35.9–53.8) | (6.0–8.0) | (767–1120) | (8340–16,900) | (358–481) | (4.3, 7.5) | (5.2, 9.3) | ||||
| 4 | 18.9 | 5.0 | 301 | 98.7 | 6.0 | 2100 | 31,900 | 539 | 5.2 | 7 | |||
| (14.4–24.9) | (4.0–6.0) | (238–380) | (68.0–143.0) | (2.5–12.0) | (1390–3170) | (21,200–48,000) | (492–591) | (4.4, 6.3) | (5.6, 8.7) | ||||
| Study 3 | 1 fasted | 4.73 | 6.0 | 83.7 | 649 | 200 | NC | ||||||
| (4.1, 5.5) | (6.0–8.0) | (71.9, 97.4) | (507, 832) | (166, 239) | |||||||||
| 1 fed | 4.35 | 7.0 | 75.5 | 630 | 191 | ||||||||
| (3.5, 5.4) | (6.0–12.0) | (62.0, 91.9) | (498, 798) | (157, 232) | |||||||||
| Dose (mg) | tmax (h) | Emax (×109/L) | AUEC (h·109/L) | tmax (day) | Emax (×109/L) | AUEC (day·109/L) | tmax (day) | Emax (×109/L) | AUEC (day·109/L) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 1 to Day 36 | Day 36 to EOS | ||||||||||
| Study 1 | 1 | 39.1 | 1.9 | 358 | Study 2 | 0.5 | 28.2 | 1.2 | 54 | 36.5 | 1.3 | 44 |
| (58.1) | (0.5) | (60) | (4.4) | (0.1) | (5) | (0.5) | (0.2) | (4) | ||||
| 3 | 13.7 | 1.3 | 1185 | 1 | 27.2 | 0.9 | 42 | 36.2 | 0.9 | 88 | ||
| (16.8) | (0.2) | (232) | (6.7) | (0.2) | (11) | (0.4) | (0.2) | (9) | ||||
| 10 | 7.7 | 0.6 | 801 | 2 | 25.2 | 0.6 | 35 | 36.7 | 0.8 | 85 | ||
| (2.3) | (0.2) | (188) | (7.3) | (0.2) | (5) | (1) | (0.1) | (22) | ||||
| 25 | 8 | 0.4 | 799 | 4 | 21 | 0.6 | 28.1 | 37.7 | 0.6 | 55 | ||
| (4) | (0.2) | (254) | (3.1) | (0.1) | (2) | (2) | (0.1) | (9) | ||||
| placebo | 66.5 | 1.5 | 1110 | placebo | 9.3 | 1.5 | 65.2 | 63.2 | 1.3 | 93 | ||
| (106.8) | (0.3) | (615) | (12.5) | (0.3) | (14.3) | (19) | (0.2) | (22) | ||||
| Study 3 | 1 fasted | 3.8 | 1.5 | 46 | ||||||||
| (2.5) | (0.3) | (8) | ||||||||||
| 1 fed | 3.1 | 1.4 | 47 | |||||||||
| (4) | (0.4) | (8) | ||||||||||
| Study 1 | Study 2 | Study 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | 1 mg | 3 mg | 10 mg | 25 mg | placebo | 0.5 mg | 1 mg | 2 mg | 4 mg | placebo | fasted | fed |
| Number of subjects dosed | 6 | 6 | 6 | 6 | 8 | 6 | 6 | 6 | 6 | 8 | 8 | 8 |
| Number of subjects with at least one AE (%) | 1 (17) | 6 (100) | 1 (17) | 4 (67) | 3 (38) | 4 (67) | 5 (83) | 6 (100) | 4 (66.7) | 8 (100) | 4 (50) | 3 (38) |
| Number of subjects reporting an event (%) | ||||||||||||
| Constipation | - | 4 | - | - | - | - | - | - | - | - | - | - |
| Headache | - | - | - | 3 | 1 | 2 | 2 | 2 | 3 | 1 | 1 | 1 |
| Dizziness | - | 1 | 1 | - | 1 | - | 1 | 1 | 2 | - | ||
| Presyncope | - | - | - | 2 | - | - | - | - | - | - | - | - |
| Bradycardia | - | - | - | 1 | - | - | - | - | - | - | - | - |
| Circulatory collapse | - | - | - | 1 * | - | - | - | - | - | - | - | - |
| Diarrhoea | - | - | - | 1 | - | - | 1 | - | 1 | - | ||
| Nausea | - | - | - | 1 | - | - | - | - | - | 3 | ||
| Pain in extremity | - | - | - | - | 1 | - | - | - | - | - | - | - |
| Paraesthesia | - | 1 | - | - | - | - | - | - | - | - | - | - |
| Rhinitis | - | 1 | - | - | - | - | - | - | - | - | - | - |
| Seborrheic dermatitis | - | - | - | - | 1 | - | - | - | - | - | - | - |
| Syncope | - | - | 1 | - | - | - | - | - | - | - | - | - |
| Upper respiratory tract infection | 1 | - | - | - | - | - | - | - | - | - | - | - |
| Chest pain | - | - | - | - | - | 1 | 2 | 2 | 3 | 2 | 2 | - |
| Nasopharyngitis | - | - | - | - | - | - | 1 | 1 | 2 | 3 | 2 | 1 |
| Abdominal pain | - | - | - | - | - | - | 1 | 1 | - | 1 | - | - |
| Back pain | - | - | - | - | - | - | - | 2 | 1 | - | - | - |
| Neck pain | - | - | - | - | - | - | 1 | 1 | - | 1 | - | - |
| Dyspepsia | - | - | - | - | - | - | 1 | 1 | - | - | - | - |
| Joint injury | - | - | - | - | - | - | - | 1 | - | 1 | - | - |
| Oropharyngeal pain | - | - | - | - | - | - | 1 | 1 | - | - | - | 1 |
| Excoriation | - | - | - | - | - | - | - | - | - | - | 1 | - |
| Nasal congestion | - | - | - | - | - | - | - | - | - | - | - | 1 |
| Rash | - | - | - | - | - | - | - | - | - | - | 1 | - |
| Dyspnoea | - | - | - | - | - | - | - | 1 | - | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juif, P.-E.; Baldoni, D.; Reyes, M.; Wilbraham, D.; Febbraro, S.; Vaclavkova, A.; Hoch, M.; Dingemanse, J. Pharmacokinetics, Pharmacodynamics, Tolerability, and Food Effect of Cenerimod, a Selective S1P1 Receptor Modulator in Healthy Subjects. Int. J. Mol. Sci. 2017, 18, 2636. https://doi.org/10.3390/ijms18122636
Juif P-E, Baldoni D, Reyes M, Wilbraham D, Febbraro S, Vaclavkova A, Hoch M, Dingemanse J. Pharmacokinetics, Pharmacodynamics, Tolerability, and Food Effect of Cenerimod, a Selective S1P1 Receptor Modulator in Healthy Subjects. International Journal of Molecular Sciences. 2017; 18(12):2636. https://doi.org/10.3390/ijms18122636
Chicago/Turabian StyleJuif, Pierre-Eric, Daniela Baldoni, Maribel Reyes, Darren Wilbraham, Salvatore Febbraro, Andrea Vaclavkova, Matthias Hoch, and Jasper Dingemanse. 2017. "Pharmacokinetics, Pharmacodynamics, Tolerability, and Food Effect of Cenerimod, a Selective S1P1 Receptor Modulator in Healthy Subjects" International Journal of Molecular Sciences 18, no. 12: 2636. https://doi.org/10.3390/ijms18122636