
Enhancing the Biological Relevance of Machine Learning Classifiers for Reverse Vaccinology

Abstract
:1. Introduction
2. Results
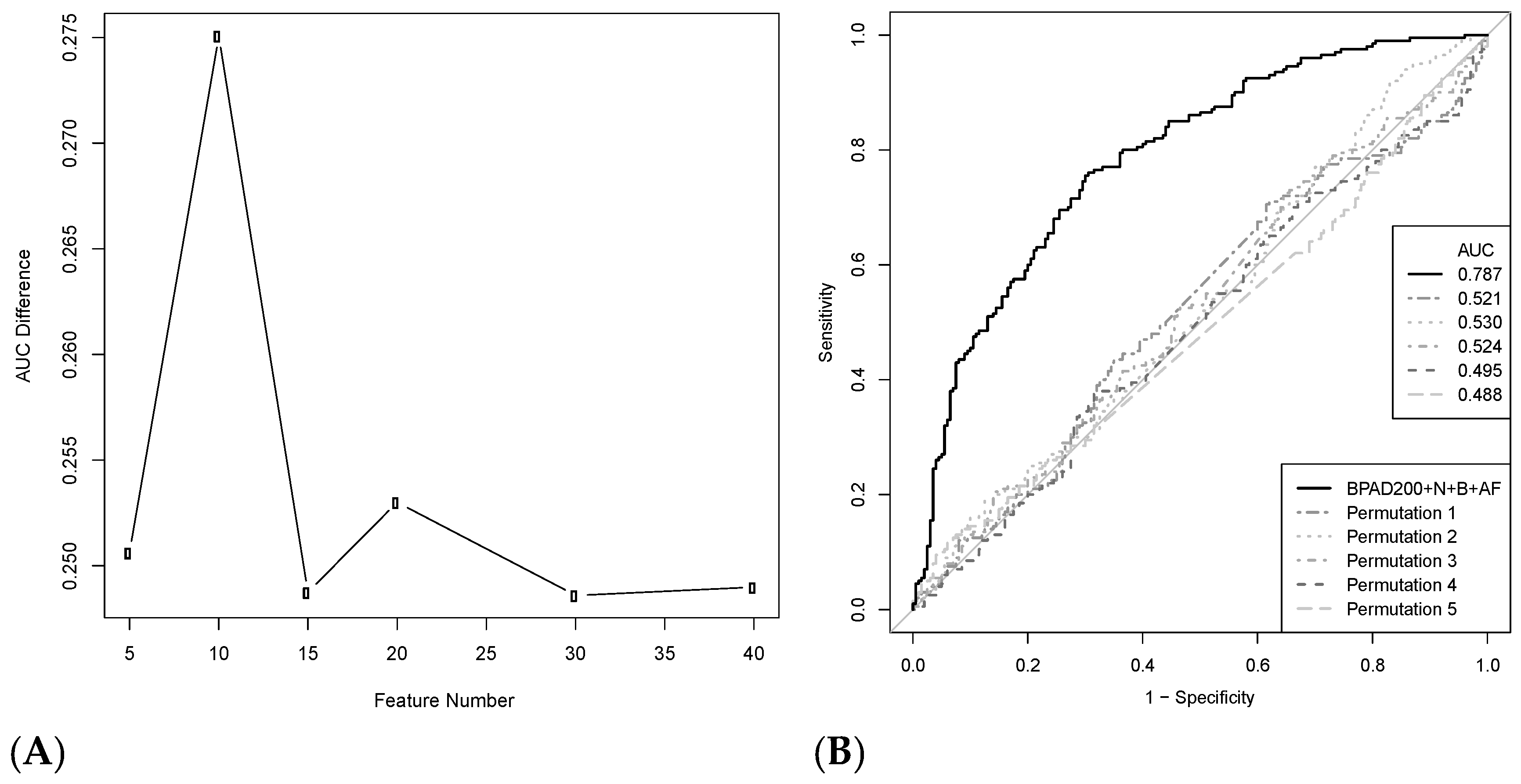
2.1. Permutation Analysis Reveals a Strong Protective Signal for BPAs Curated from the Literature
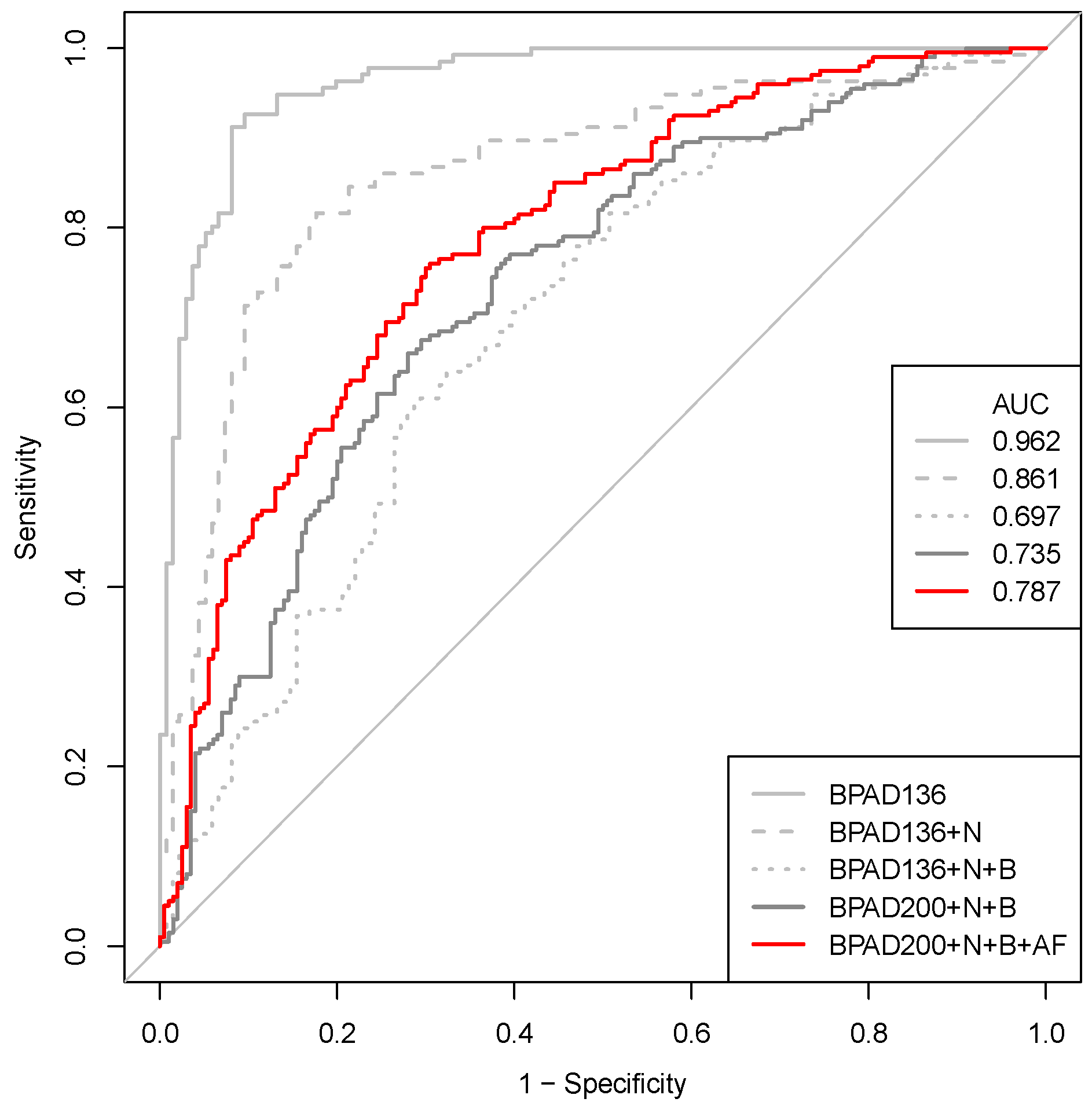
2.2. A Nested Approach Has a Significant Impact on the Ability of SVMs to Classify BPAs
2.3. Correcting a Bias in the Selection of Negative Training Data Impacts SVM Classification of BPAs
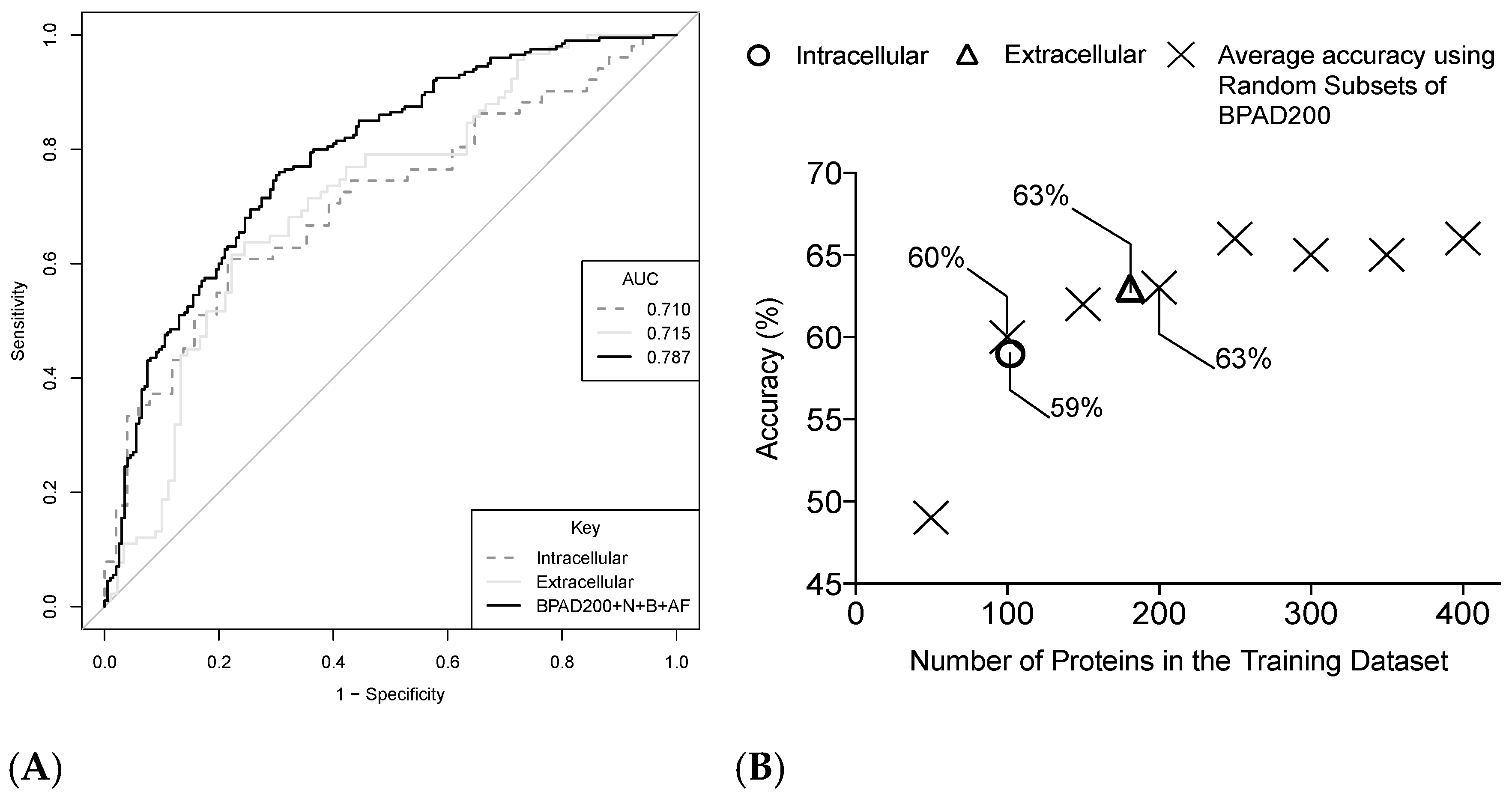
2.4. Increasing the Size of the Training Data Has a Positive Impact on the Ability of SVMs to Classify BPAs
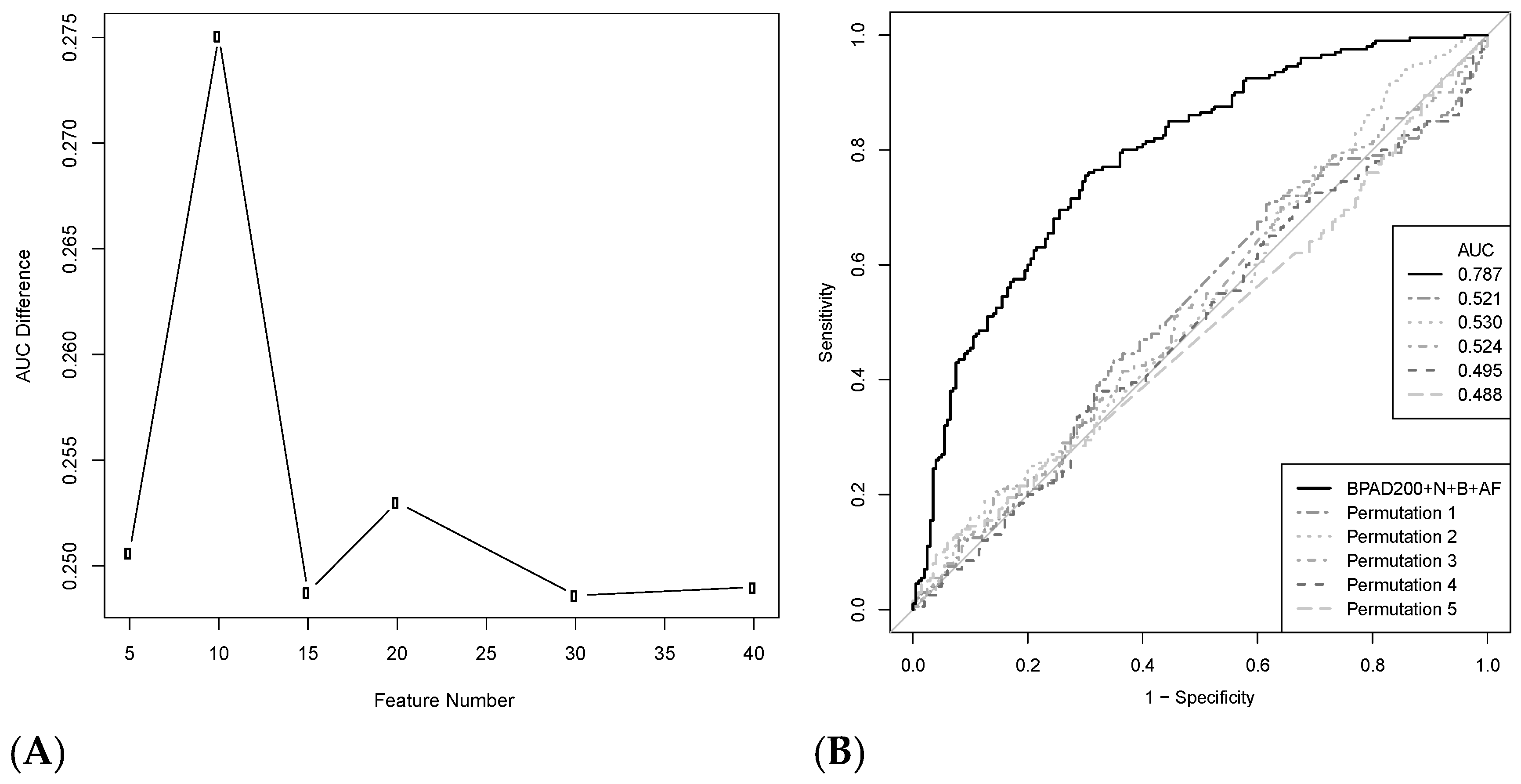
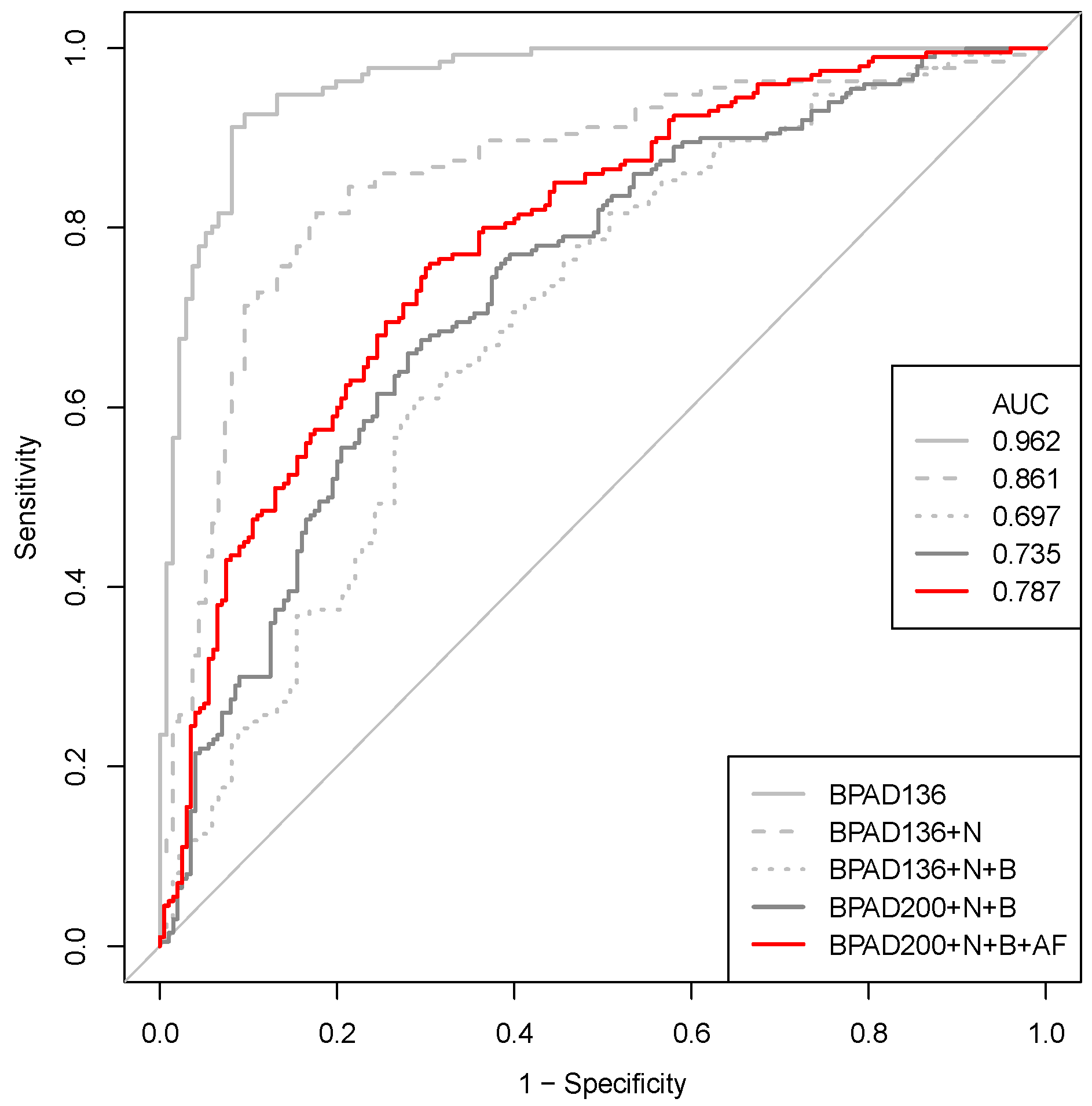
2.5. Increasing the Number of Protein Annotation Tools Enhances the Ability of SVMs to Classify BPAs
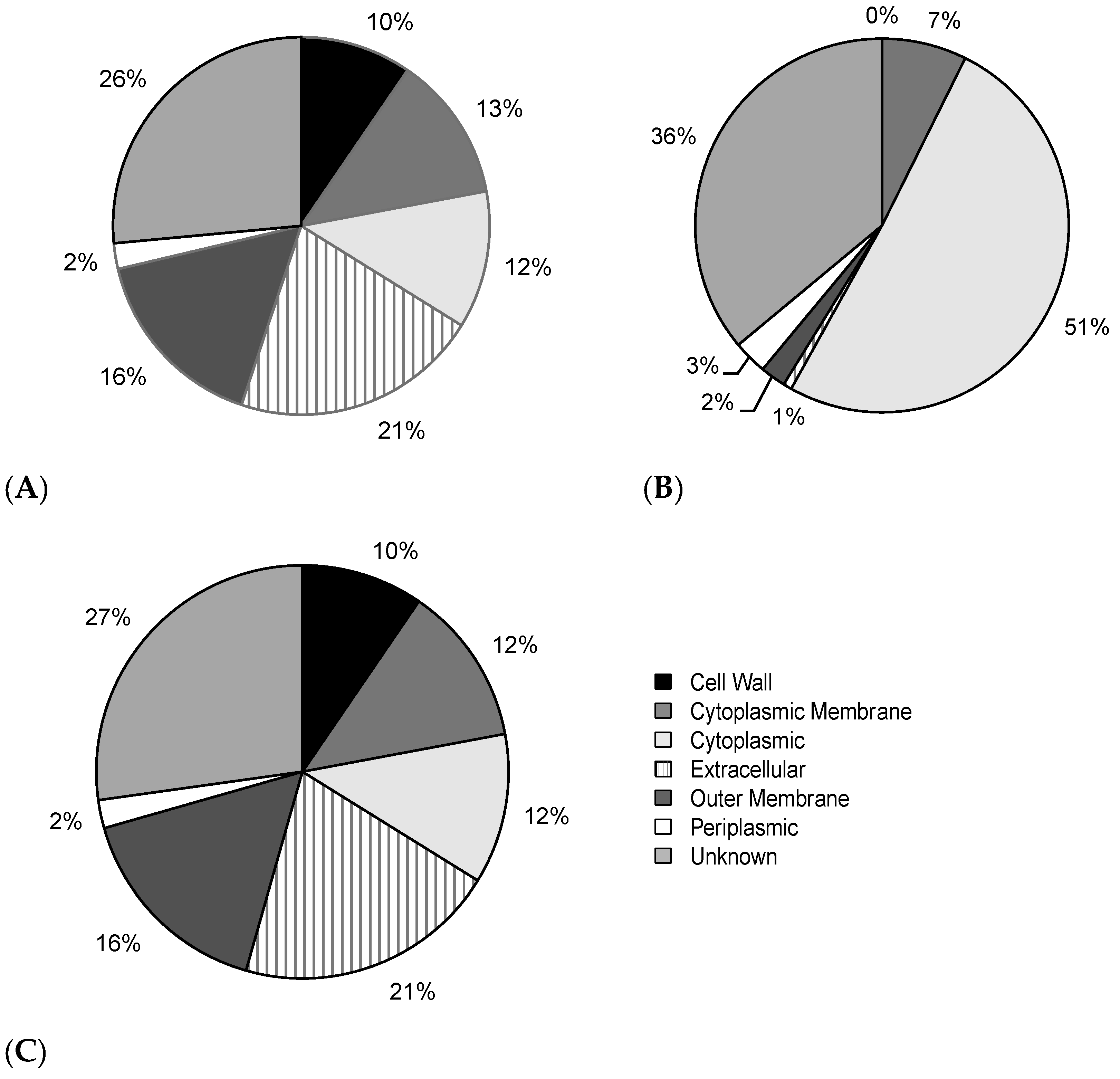
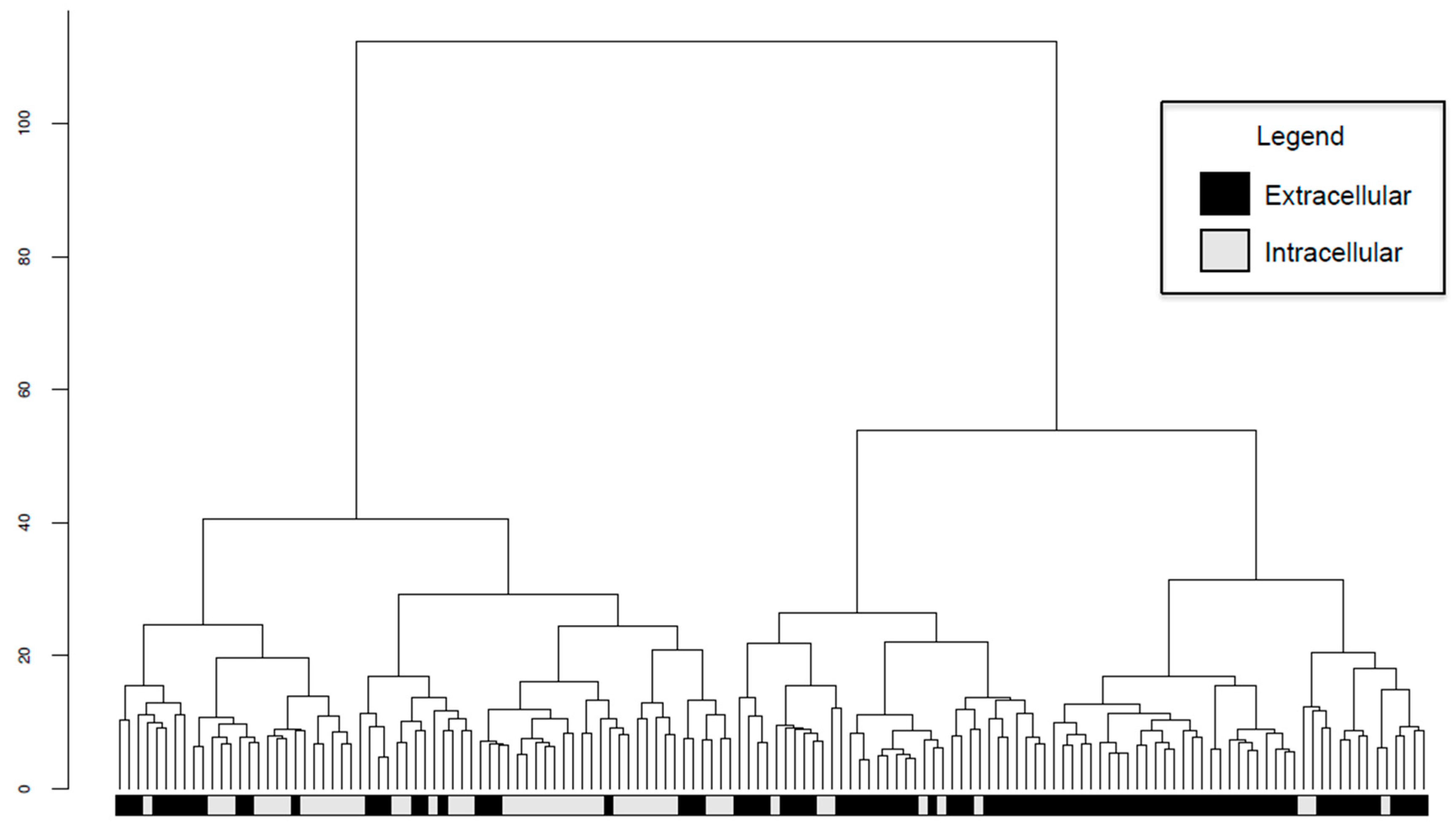
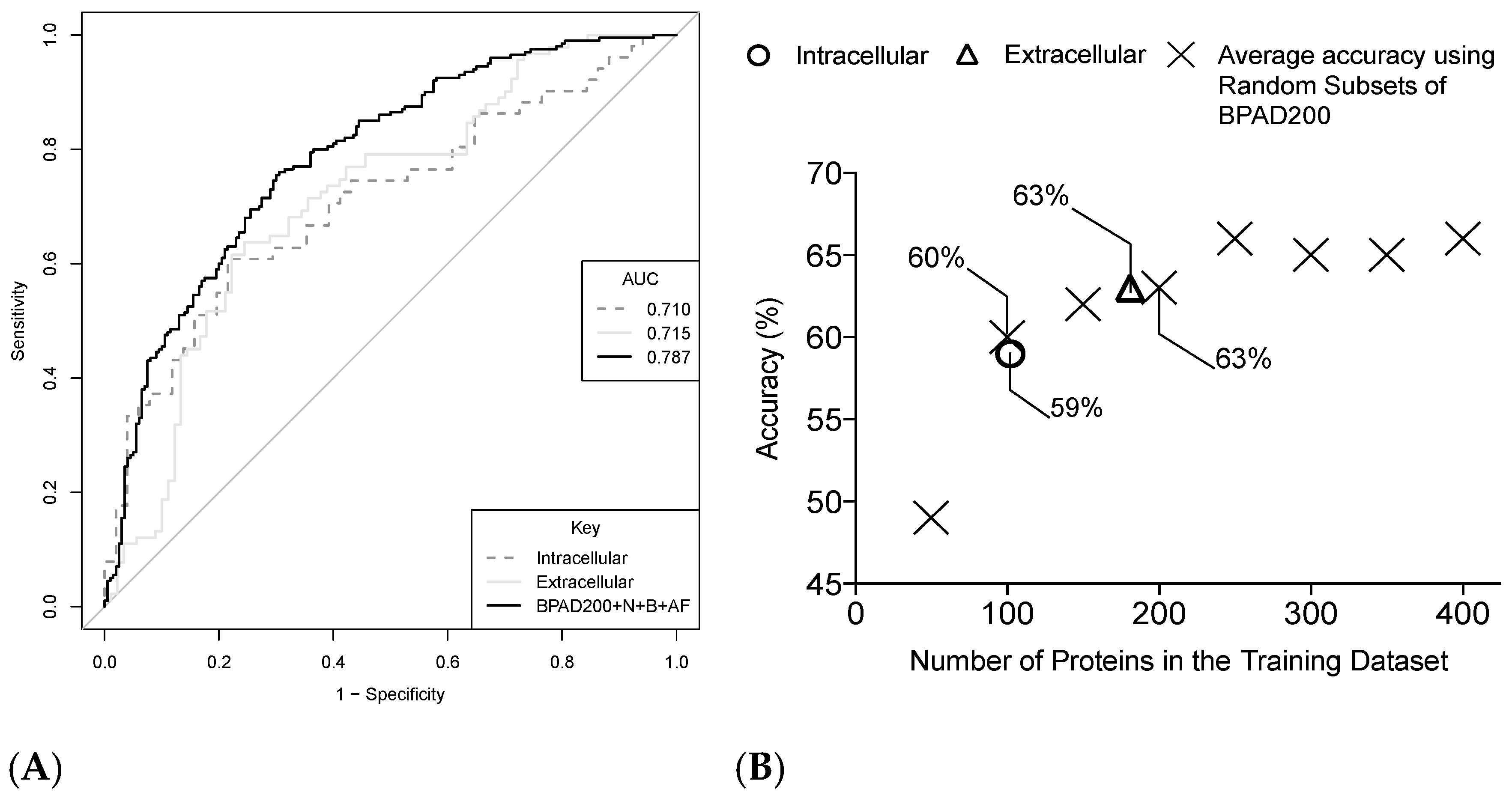
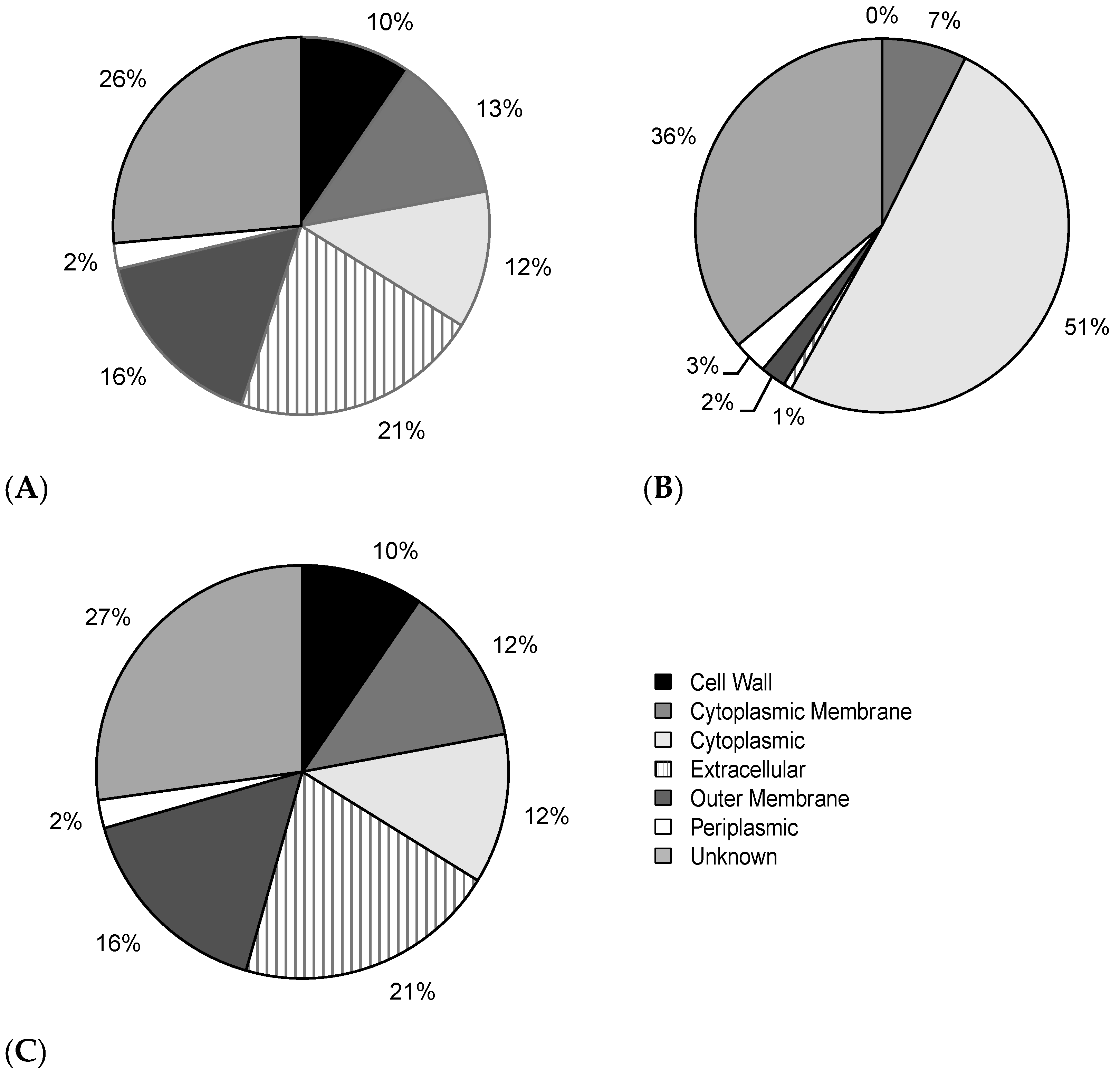
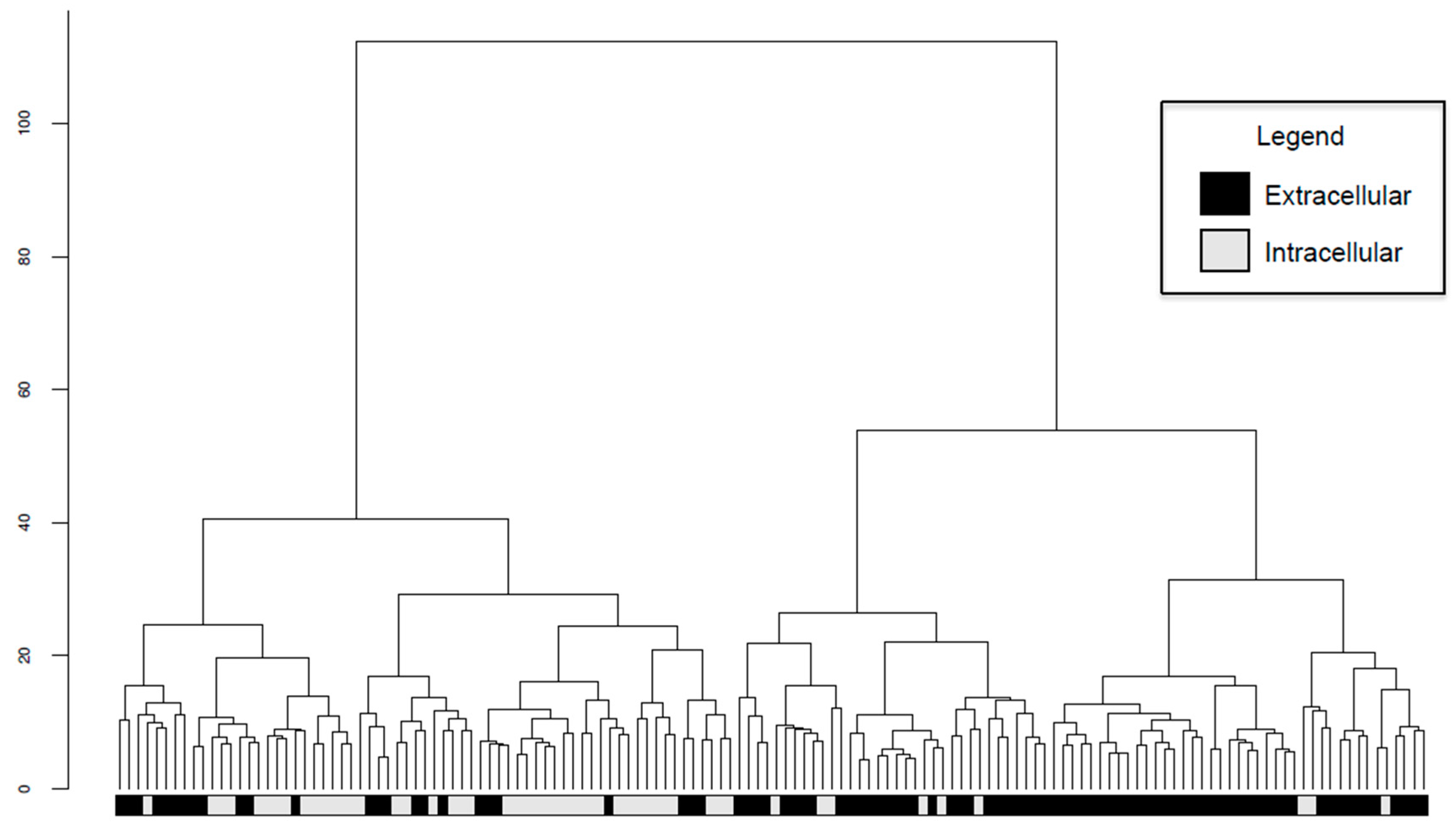
2.6. Intracellular and Extracellular BPAs Utilize Different Features for Classification
3. Discussion
4. Methods
4.1. Training Data
4.2. Permutation Analysis
4.3. Data Annotation
4.4. Machine Learning Classification
4.5. Statistics
4.6. Hierarchical Clustering
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RV | Reverse Vaccinology |
| ML | Machine Learning |
| BPA | Bacterial Protective Antigen |
| BPAD | Bacterial Protective Antigen Dataset |
| SVM | Support Vector Machine |
| LTOCV | Leave Tenth Out Cross Validation |
| iBPAD | Intracellular Bacterial Protective Antigen Dataset |
| eBPAD | Extracellular Bacterial Protective Antigen Dataset |
| ELISA | Enzyme-linked Imunosorbent Assay |
| FACS | Fluorescence activated cell-sorting |
| DOMV | Detergent Extracted Outer Membrane Vesicle |
| ACC | Auto Cross Covariance |
| DA-PLS | Discriminant Analysis by Partial Least Squares |
| RBF | Radial Bias Function |
| TPR | True Positive Rate |
| FPR | False Positive Rate |
| ROC | Receiver Operating Characteristic |
| AUC | Area Under the Curve |
| N | Nested |
| B | Balanced |
| AF | Additional Features |
| HDL | High Density Lipoprotein |
| Th cells | T-helper cells |
References
- Pizza, M.; Scarlato, V.; Masignani, M.M.; Giuliani, B.; Arico, M.; Comanducci, G.T.; Jennings, L.; Baldi, E.; Bartolini, B.; Capecchi, B.; et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Crum-Cianflone, N.; Sullivan, E. Meningococcal Vaccinations. Infect. Dis. Ther. 2016, 5, 89–112. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [PubMed]
- Corpet, F.; Servant, F.; Gouzy, J.; Kahn, D. ProDom and ProDom-CG: Tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res. 2000, 28, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G.; Pietrokovski, S. Blocks+: A non-redundant database of protein alignment blocks derived from multiple compilations. Bioinformatics 1999, 15, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, M.M.; Adu-Bobie, J.; Comanducci, M.; Arico, B.; Savino, S.; Santini, L.; Brunelli, B.; Bambini, S.; Biolchi, A.; Capecchi, B.; et al. A universal vaccine for serogroup B. meningococcus. Proc. Natl. Acad. Sci. USA 2006, 103, 10834–10839. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.S.; Turner, D.P. Clinical experience with the meningococcal B vaccine, Bexsero®: Prospects for reducing the burden of meningococcal serogroup B disease. Vaccine 2016, 34, 875–880. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Racz, R.; Sayers, S.; Lin, Y.; Todd, T.; Hur, J.; Li, X.; Patel, M.; Zhao, B.; Chung, M.; et al. Updates on the web-based VIOLIN vaccine database and analysis system. Nucleic Acids Res. 2013, 14, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Chanumolu, S.K.; Gupta, A.; Chauhan, R.S.; Rout, C. Jenner-predict server: Prediction of protein vaccine candidates (PVCs) in bacteria based on host-pathogen interactions. BMC Bioinform. 2013, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Gutierrez, A.; Kibria, F.; Martin, R.; Tassone, R.; Liu, R.; Terry, F.; Martin, B.; de Groot, A.S. iVAX: An integrated toolkit for the selection and optimization of antigens and the design of epitope-driven vaccines. Hum. Vaccin. Immunother. 2015, 11, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Heinson, A.I.; Woelk, C.H.; Newell, M.L. The promise of reverse vaccinology. Int. Health 2015, 7, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Bhatnagar, R. GroEL provides protection against Bacillus anthracis infection in BALB/c mice. Mol. Immunol. 2010, 48, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Velikovsky, C.A.; Cassataro, J.; Giambartolomei, G.H.; Goldbaum, F.A.; Estein, S.; Bowden, R.A.; Bruno, L.; Fossati, C.A.; Spitz, M. A DNA vaccine encoding lumazine synthase from Brucella abortus induces protective immunity in BALB/c mice. Infect. Immun. 2002, 70, 2507–2511. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Xu, J.; Li, X.; Xie, Y.; Qiu, Y.; Du, X.; Yu, S.; Bai, Y.; Chen, Y.; Wang, T.; et al. Immunization of mice with recombinant protein CobB or AsnC confers protection against Brucella abortus infection. PLoS ONE 2012, 7, 29552. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kumar, S.; Dohre, S.; Afley, P.; Sengupta, N.; Alam, S.I. Identification of a protective protein from stationary-phase exoproteome of Brucella abortus. Pathog. Dis. 2013, 70, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Chen, C.S.; Palaniappan, R.U.; He, H.; McDonough, S.P.; Barr, S.C.; Yan, W.; Faisal, S.M.; Pan, M.J.; Chang, C.F. Immunogenicity of the recombinant leptospiral putative outer membrane proteins as vaccine candidates. Vaccine 2007, 25, 8190–8197. [Google Scholar] [CrossRef] [PubMed]
- Mizrachi Nebenzahl, Y.; Bernstein, A.; Portnoi, M.; Shagan, M.; Rom, S.; Porgador, A.; Dagan, R. Streptococcus pneumoniae surface-exposed glutamyl tRNA synthetase, a putative adhesin, is able to induce a partially protective immune response in mice. J. Infect. Dis. 2007, 196, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Fritzer, A.; Senn, B.M.; Minh, D.B.; Hanner, M.; Gelbmann, D.; Noiges, B.; Henics, T.; Schulze, K.; Guzman, C.A.; Goodacre, J.; et al. Novel conserved group A streptococcal proteins identified by the antigenome technology as vaccine candidates for a non-M protein-based vaccine. Infect. Immun. 2010, 78, 4051–4067. [Google Scholar] [CrossRef] [PubMed]
- Henningham, A.; Chiarot, E.; Gillen, C.M.; Cole, J.N.; Rohde, M.; Fulde, M.; Ramachandran, V.; Cork, A.J.; Hartas, J.; Magor, G.; et al. Conserved anchorless surface proteins as group A streptococcal vaccine candidates. J. Mol. Med. 2012, 90, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.N.; McAdam, P.R.; Vivona, S.; Zhang, J.X.; Luong, T.; Belew, R.K.; Sahota, H.; Guiney, D.; Valafar, F.; Fierer, J.; et al. Improving reverse vaccinology with a machine learning approach. Vaccine 2011, 29, 8156–8164. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinforma. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, K.A.; Fälker, S.; Dahlberg, S.; Normark, S.; Henriques-Normark, B. Bacterial adhesins in host-microbe interactions. Cell Host Microbe 2009, 5, 580–592. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.R.; DeLong, D.M.; Clarke-Pearson, D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988, 44, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, G.; Kumar, K.; Jain, P.; Ramachandran, S. SPAAN: A software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics 2005, 21, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, R.; Liu, Z.; Ren, J.; Ma, C.; Gao, T.; Zhou, Y.; Yang, Q.; Xue, Y. GPS-MBA: Computational analysis of MHC class II epitopes in type 1 diabetes. PLoS ONE 2012, 7, 33884. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Kesmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cao, J.; Gao, X.; Ma, Q.; Ren, J.; Xue, Y. GPS-CCD: A novel computational program for the prediction of calpain cleavage sites. PLoS ONE 2011, 6, 19001. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Pirillo, A.; Ammirati, E.; Catapano, A.L. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis 2012, 220, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Catapano, A.L. HDL and adaptive immunity: A tale of lipid rafts. Atherosclerosis 2012, 225, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, 6227. [Google Scholar] [CrossRef] [PubMed]
- Snapper, C.M.; Rosas, F.R.; Jin, L.; Wortham, C.; Kehry, M.R.; Mond, J.J. Bacterial lipoproteins may substitute for cytokines in the humoral immune response to T cell-independent type II antigens. J. Immunol. 1995, 155, 5582–5589. [Google Scholar] [PubMed]
- Haste Andersen, P.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Huang Jian, H.; Honda, W. CED: A conformational epitope database. BMC Immunol. 2006, 7, 7. [Google Scholar]
- Ansari, H.R.; Raghava, G.P. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res. 2010, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Fassler, J.C.P. BLAST Glossary, BLAST® Help; National Center for Biotechnology Information: Bethesda, MD, USA, 2011.
- Good, P. Permutation Tests: A Practical Guide To Resampling Methods For Testing Hypotheses; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM TIST 2011, 2, 27. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Lin, C.-J. Combining SVMs with Various Feature Selection Strategies. In Feature Extraction; Springer Science & Business Media: Berlin, Germany, 2006; pp. 315–324. [Google Scholar]
- Polat, K.; Güneş, S. A new feature selection method on classification of medical datasets: Kernel F-score feature selection. Expert Syst. Appl. 2009, 36, 10367–10373. [Google Scholar] [CrossRef]
- Vergara, J.R.; Estévez, P.A. A review of feature selection methods based on mutual information. Neural Comput. Appl. 2014, 24, 175–186. [Google Scholar] [CrossRef]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comp. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Simon, R.; Radmacher, M.D.; Dobbin, K.; McShane, L.M. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J. Natl. Cancer Inst. 2003, 95, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Coombes, K. ClassDiscovery: Classes and Methods for “Class Discovery“ with Microarrays or Proteomics, R Package Version 2.1. Available online: http://bioinformatics.mdanderson.org/Software/OOMPA (accessed on 1 December 2016).
- Galili, T. Dendextend: An R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Feature | Name of Bioinformatics Tool | Protein Annotation Tool Type | Correlated with BPA or Non-BPA |
|---|---|---|---|---|
| 1 | LipoP_Signal_Avr_Length | LipoP | Lipoprotein | BPA |
| 2 | YinOYang-T-Count | YinOYang | Glycosylation | BPA |
| 3 | NetPhosK-S-Count | NetPhosK | Phosphorylation | BPA |
| 4 | LipoP_SPI_Avr_Length | LipoP | Lipoprotein | BPA |
| 5 | NetMhcPan-B-AvgRank | NetMhc | T-Cell Epitope predictor (MHC Class II) | BPA |
| 6 | TargetP-SecretFlag | TargetP | Subcellular Compartmentalisation—In Eukaryotic Cells | BPA |
| 7 | YinOYang-Average-Difference1_Length | YinOYang | Glycosylation | Non-BPA |
| 8 | MBAAgl7_CorCount | GPS-MBA | T-Cell Epitope predictor | BPA |
| 9 | PickPocket-Average_score | PickPocket | MHC Peptide Binding | Non-BPA |
| 10 | PropFurin-Count_Score | ProP | Cleavage Sites—In Eukaryotic Cells | BPA |
| (A) | |||
| Rank | Feature | Protein Annotation Tool Type | Rank in Intracellular Classifier |
| 1 | Pad-value | Adhesin | 42 |
| 2 | DictOGlyc_Ser_Average_Threshold_Length | Glycosylation | 189 |
| 3 | LipoP_SPI_AvrScore | Lipoprotein | NF |
| 4 | Netsurfp_RSA_Exposed_AverageDiff | Surface accesibility and secondary structure | NF |
| 5 | PoloPhosphorylation_CorAvg | Phosphorylation | NF |
| 6 | Net_Chop_CorCount | Predicts cleavage sites | NF |
| 7 | DictOGlyc-No_Score_Sites_Length | Glycosylation | NF |
| 8 | GPS_SUMO_Sumoylation_Average_Score | Small ubiquitin like modifiers (SUMOs) binding site prediction | 9 |
| 9 | ProtParam-PercIsoleucine | General Annotation | 144 |
| 10 | ProtParam-PercGlutamicAcid | General Annotation | NF |
| (B) | |||
| Rank | Feature | Protein Annotation Tool Type | Rank in Extracellular Classifier |
| 1 | Bepipred-Count_Length | B-Cell Epitope | 149 |
| 2 | CCD_av_diff | Calpain Cleavage | NF |
| 3 | YinOYang-T-Average-Difference1_Length | Glycosylation | NF |
| 4 | ProtParam-GRAVY | General Annotation | 35 |
| 5 | NetOGlyc-T-Max-I | Glycosylation | 196 |
| 6 | YinOYang-T-Average_Length | Glycosylation | NF |
| 7 | ProtParam-PercAlanine | General Annotation | 97 |
| 8 | NetPhosK-Y-MaxScore | Phosphorylation | NF |
| 9 | GPS_SUMO_Sumoylation_Average_Score | Small Ubiquitin like modifiers (SUMOs) binding site predictor | 8 |
| 10 | MBAAgl7_CorAvg | T-Cell Epitope predictor | NF |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinson, A.I.; Gunawardana, Y.; Moesker, B.; Hume, C.C.D.; Vataga, E.; Hall, Y.; Stylianou, E.; McShane, H.; Williams, A.; Niranjan, M.; et al. Enhancing the Biological Relevance of Machine Learning Classifiers for Reverse Vaccinology. Int. J. Mol. Sci. 2017, 18, 312. https://doi.org/10.3390/ijms18020312
Heinson AI, Gunawardana Y, Moesker B, Hume CCD, Vataga E, Hall Y, Stylianou E, McShane H, Williams A, Niranjan M, et al. Enhancing the Biological Relevance of Machine Learning Classifiers for Reverse Vaccinology. International Journal of Molecular Sciences. 2017; 18(2):312. https://doi.org/10.3390/ijms18020312
Chicago/Turabian StyleHeinson, Ashley I., Yawwani Gunawardana, Bastiaan Moesker, Carmen C. Denman Hume, Elena Vataga, Yper Hall, Elena Stylianou, Helen McShane, Ann Williams, Mahesan Niranjan, and et al. 2017. "Enhancing the Biological Relevance of Machine Learning Classifiers for Reverse Vaccinology" International Journal of Molecular Sciences 18, no. 2: 312. https://doi.org/10.3390/ijms18020312