
Extending Immunological Profiling in the Gilthead Sea Bream, Sparus aurata, by Enriched cDNA Library Analysis, Microarray Design and Initial Studies upon the Inflammatory Response to PAMPs

 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. Identification of Immune-Related Expressed Sequence Tags (EST)
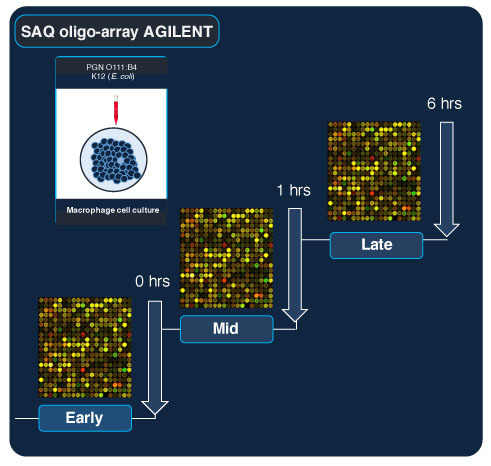
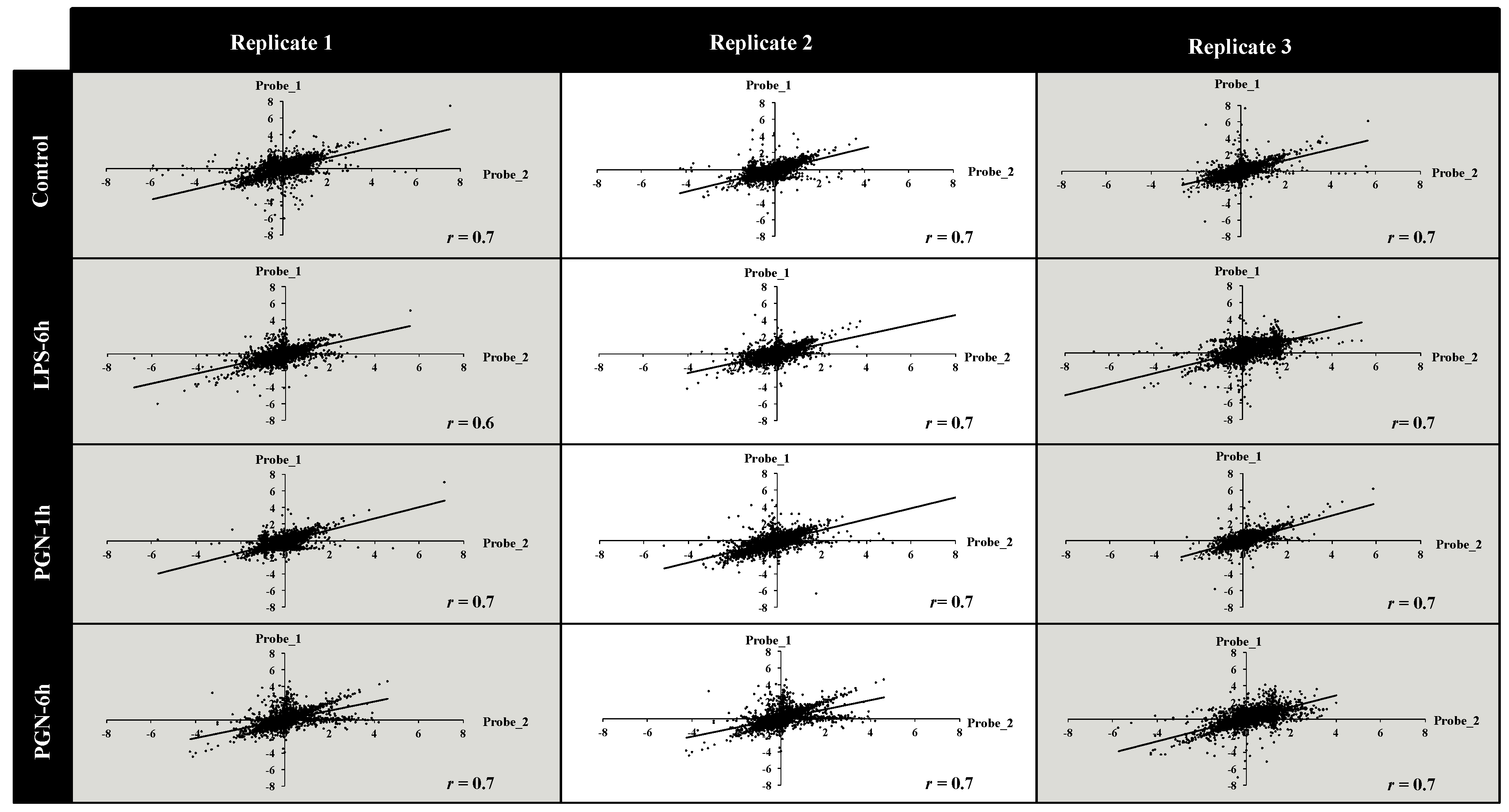
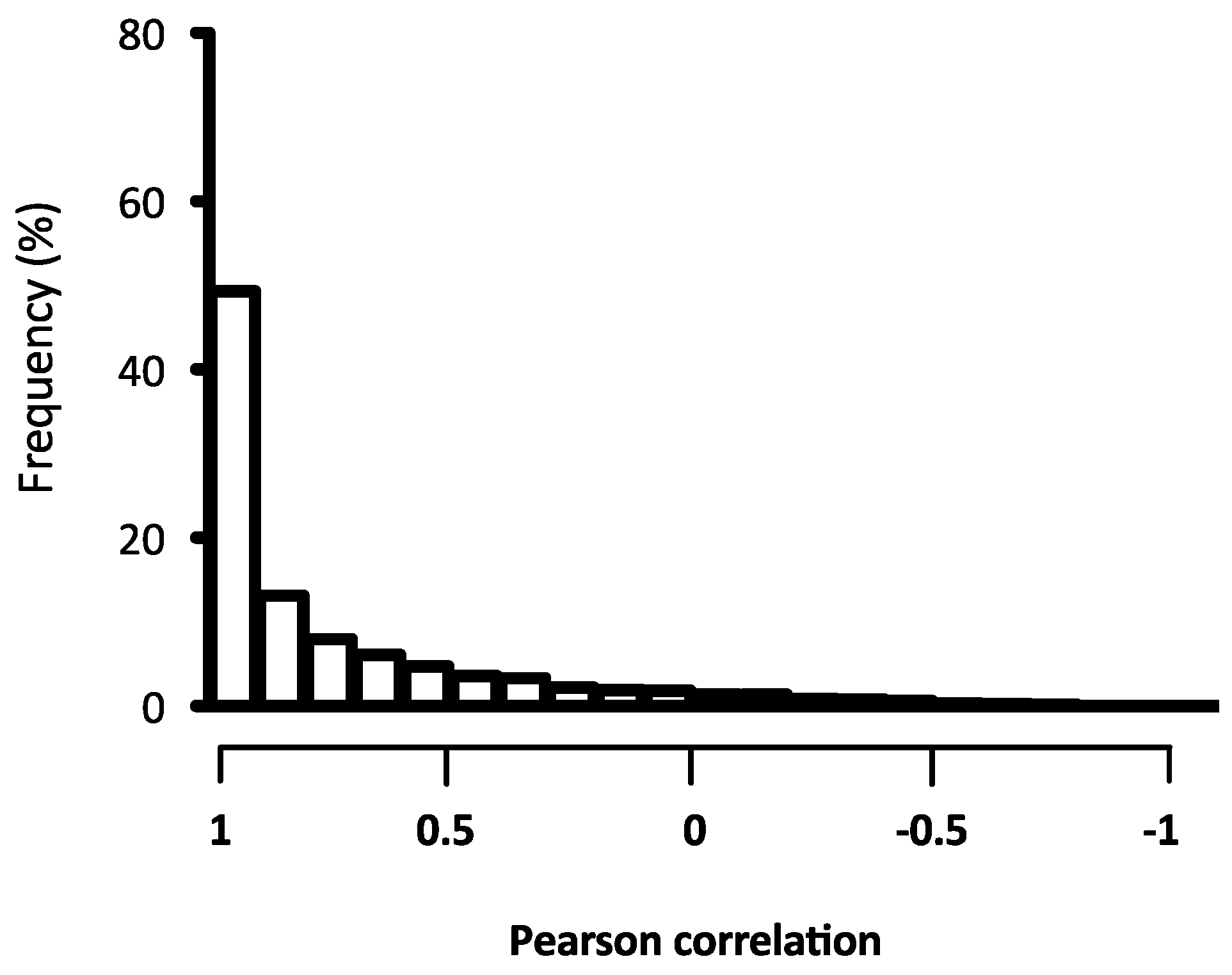
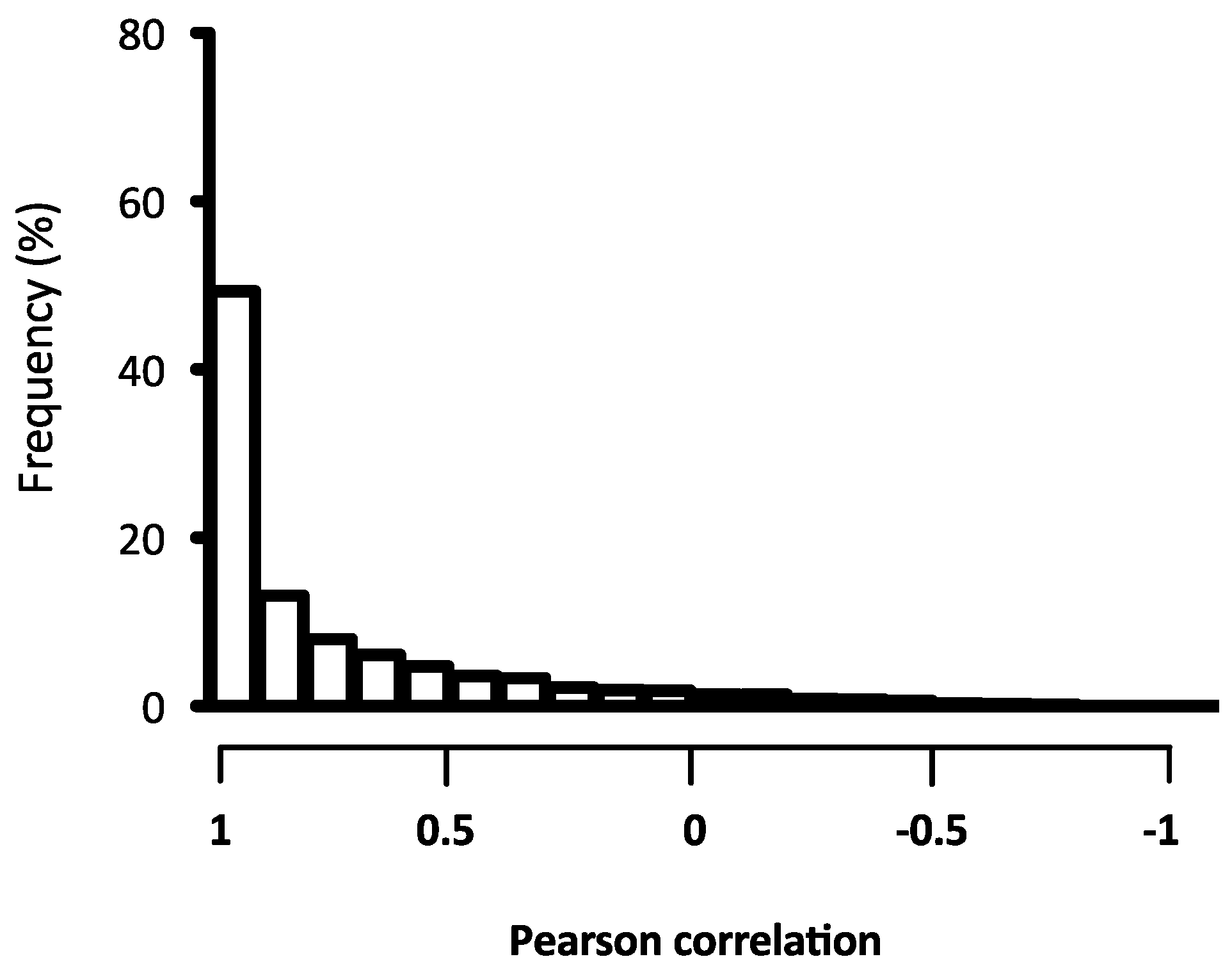
2.2. Quality Assessment of the Microarray Hybridisation
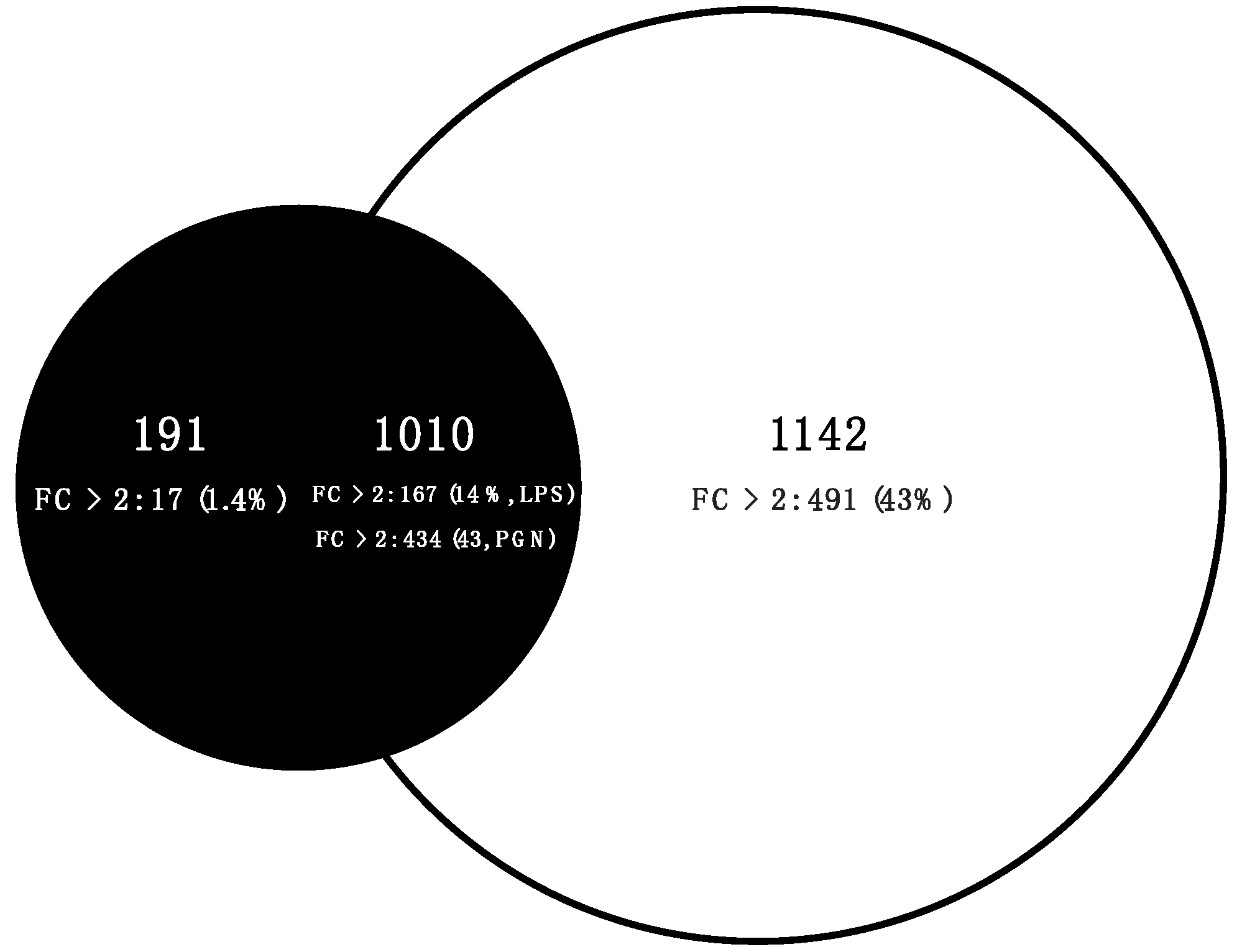
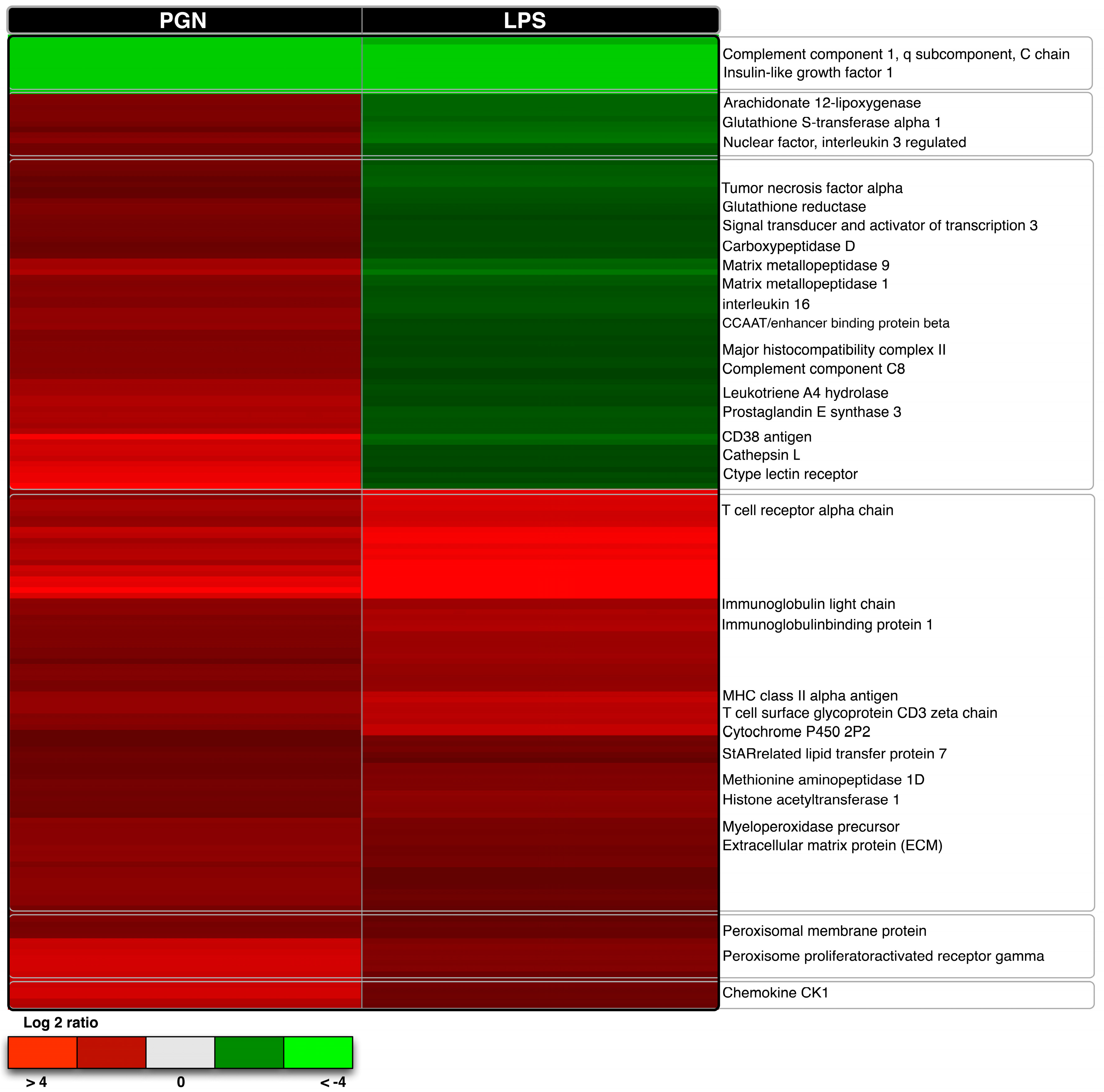
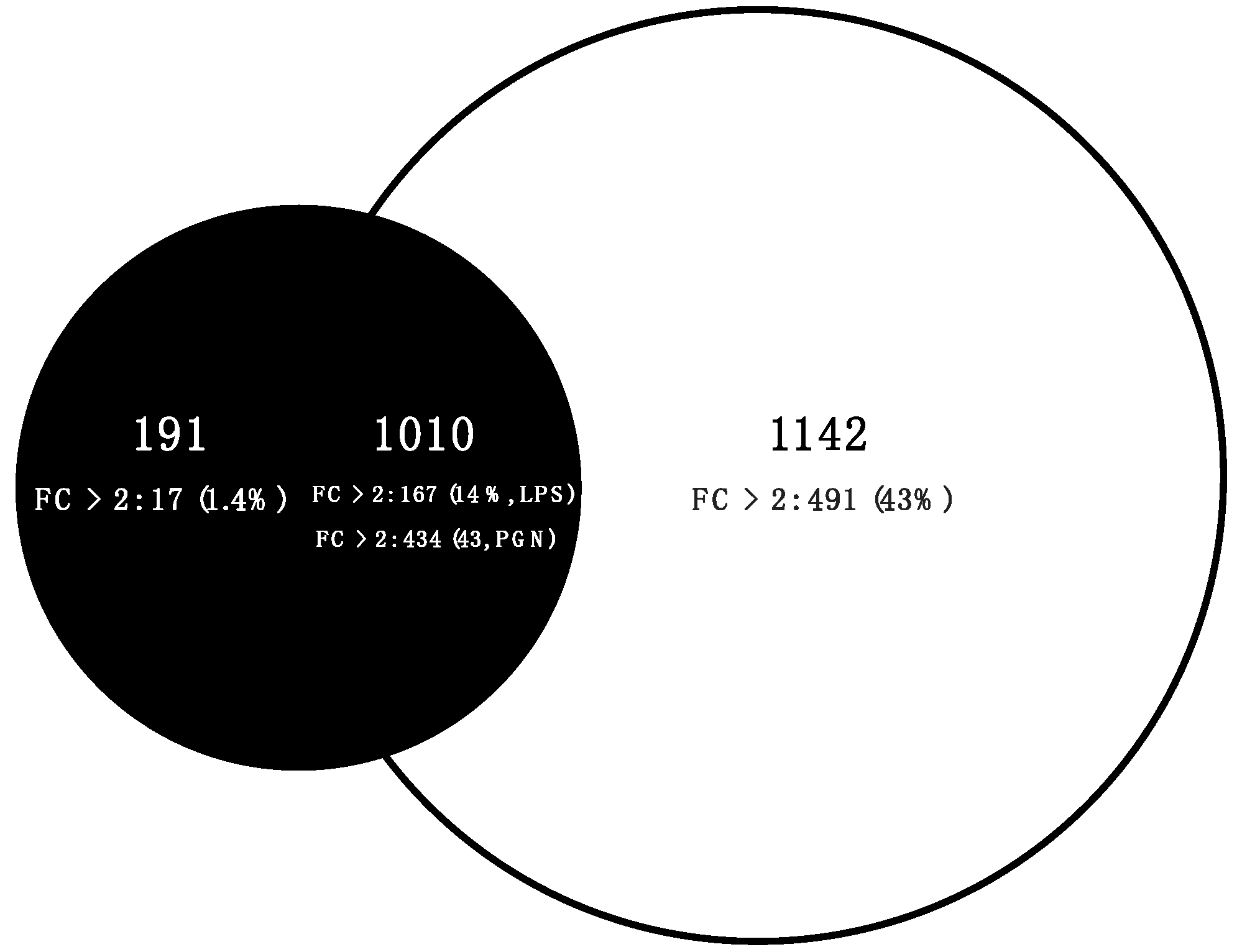
2.3. Comparative Transcriptome Analysis
3. Discussion
3.1. Identification of Immune-Related Transcripts in S. aurata
3.2. Development of S. aurata ONM
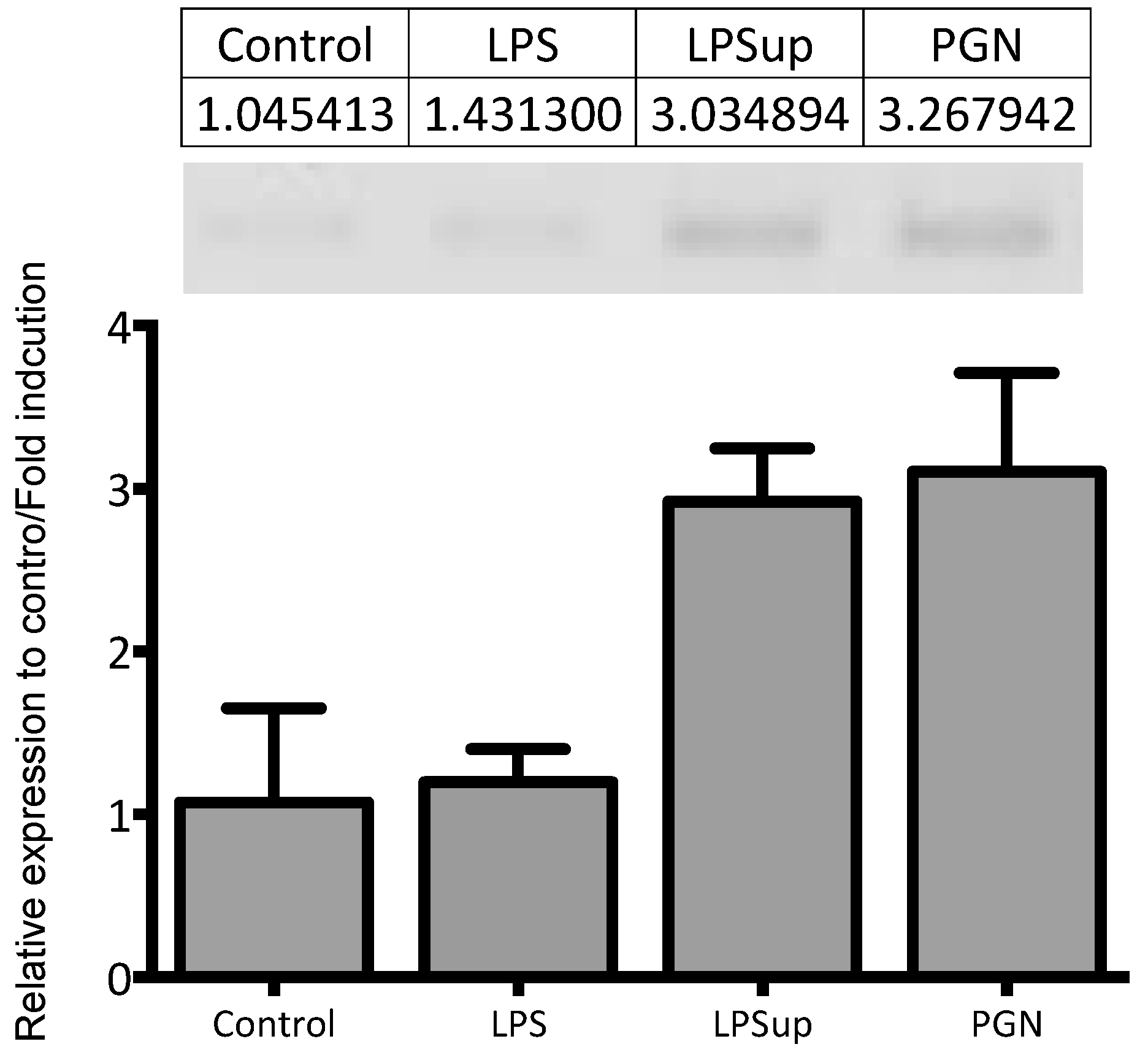
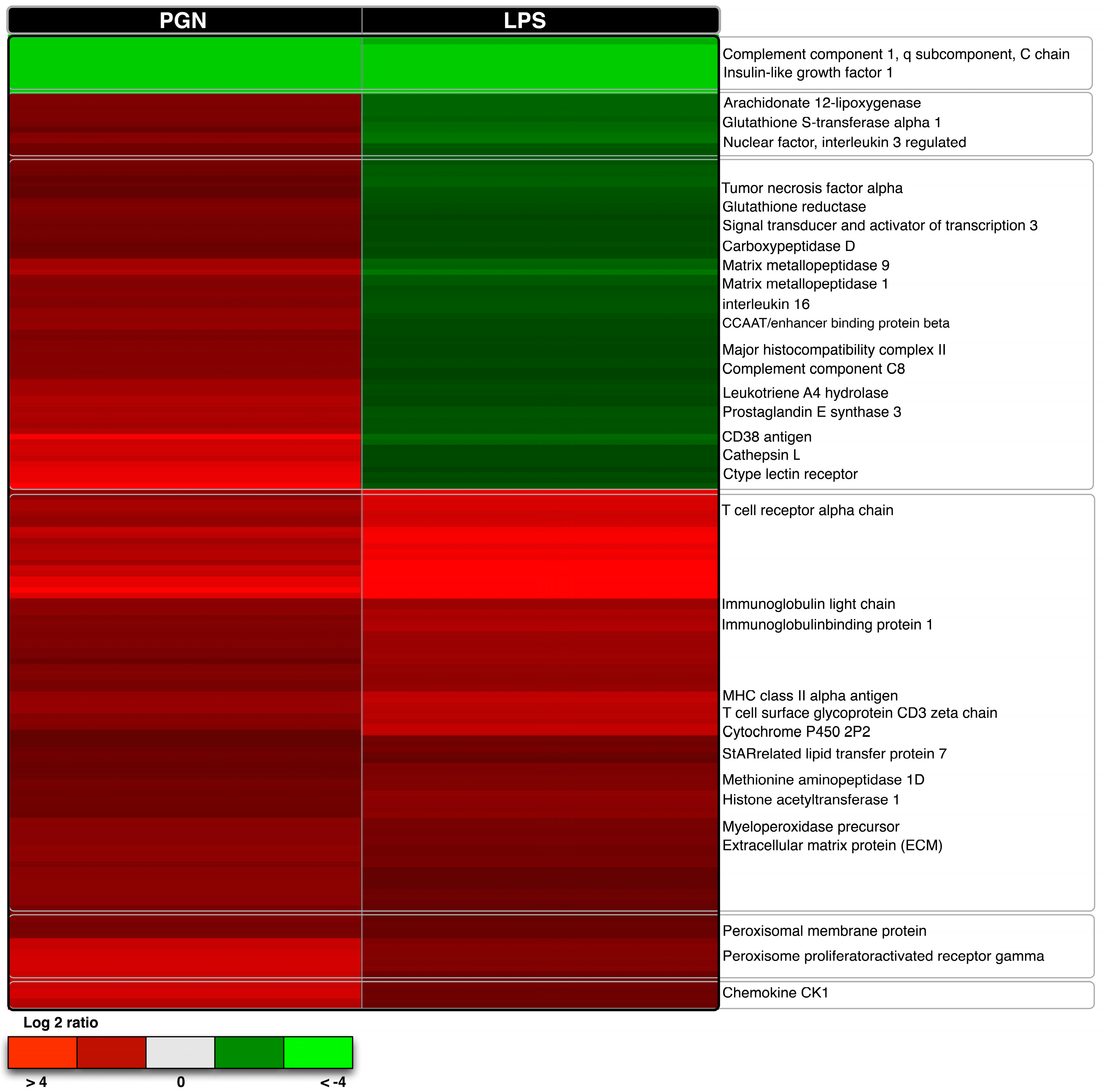
3.3. Qualitative Comparisons of Transcriptional Modulations in Response to LPS-PGN
4. Materials and Methods
4.1. Experimental Cell Culture Setup and Materials
4.2. cDNA Library Construction and Sequencing
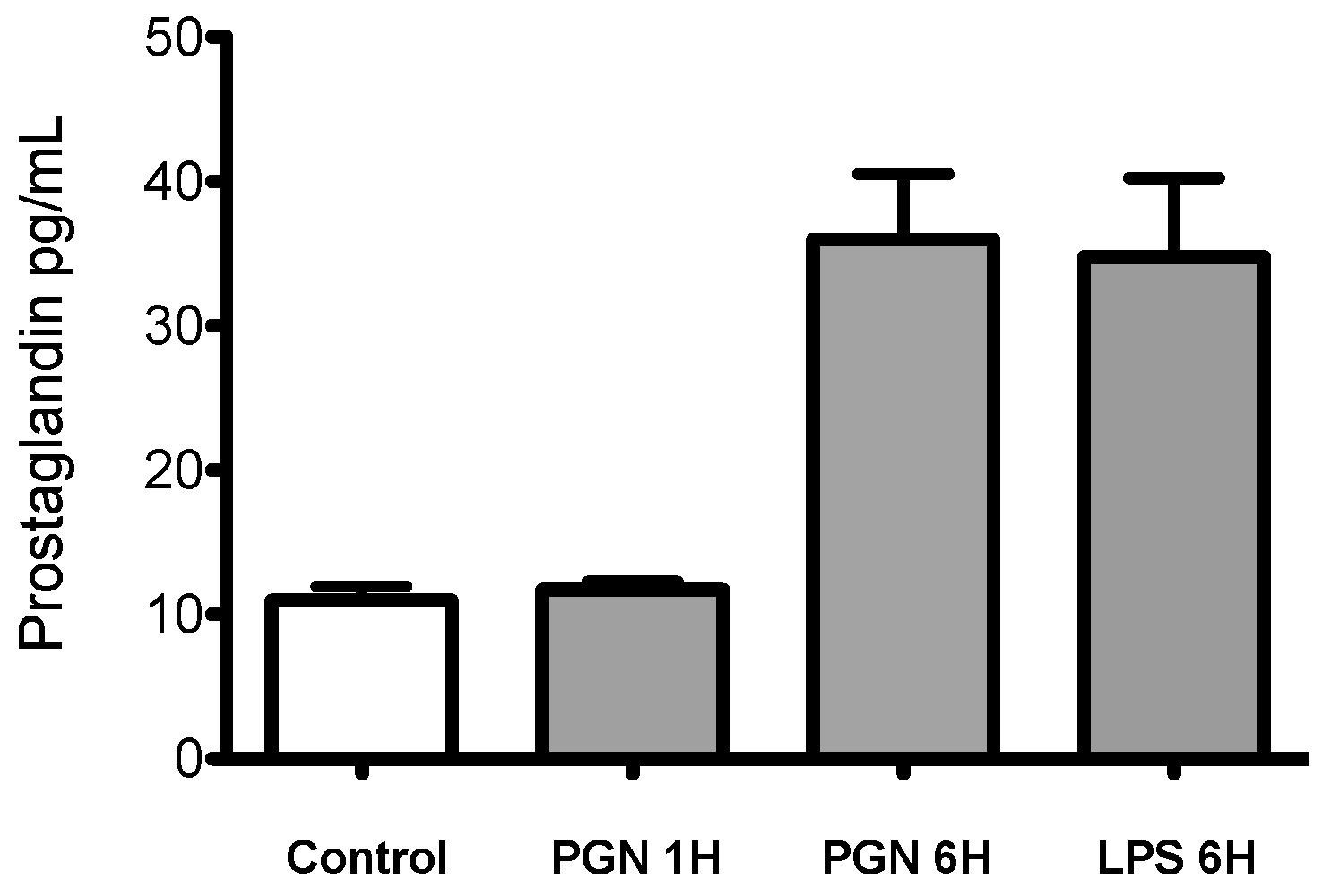
4.3. Measurement of PGE2 and PGD2 Levels
4.4. RNA Isolation and Complementary DNA (cDNA) Synthesis
4.5. RNA Labelling and Hybridisation
4.6. Gene Ontology (GO-DAVID Analysis)
4.7. Gene-Level and Quality Signal Analysis
4.8. Microarray Hybridisation
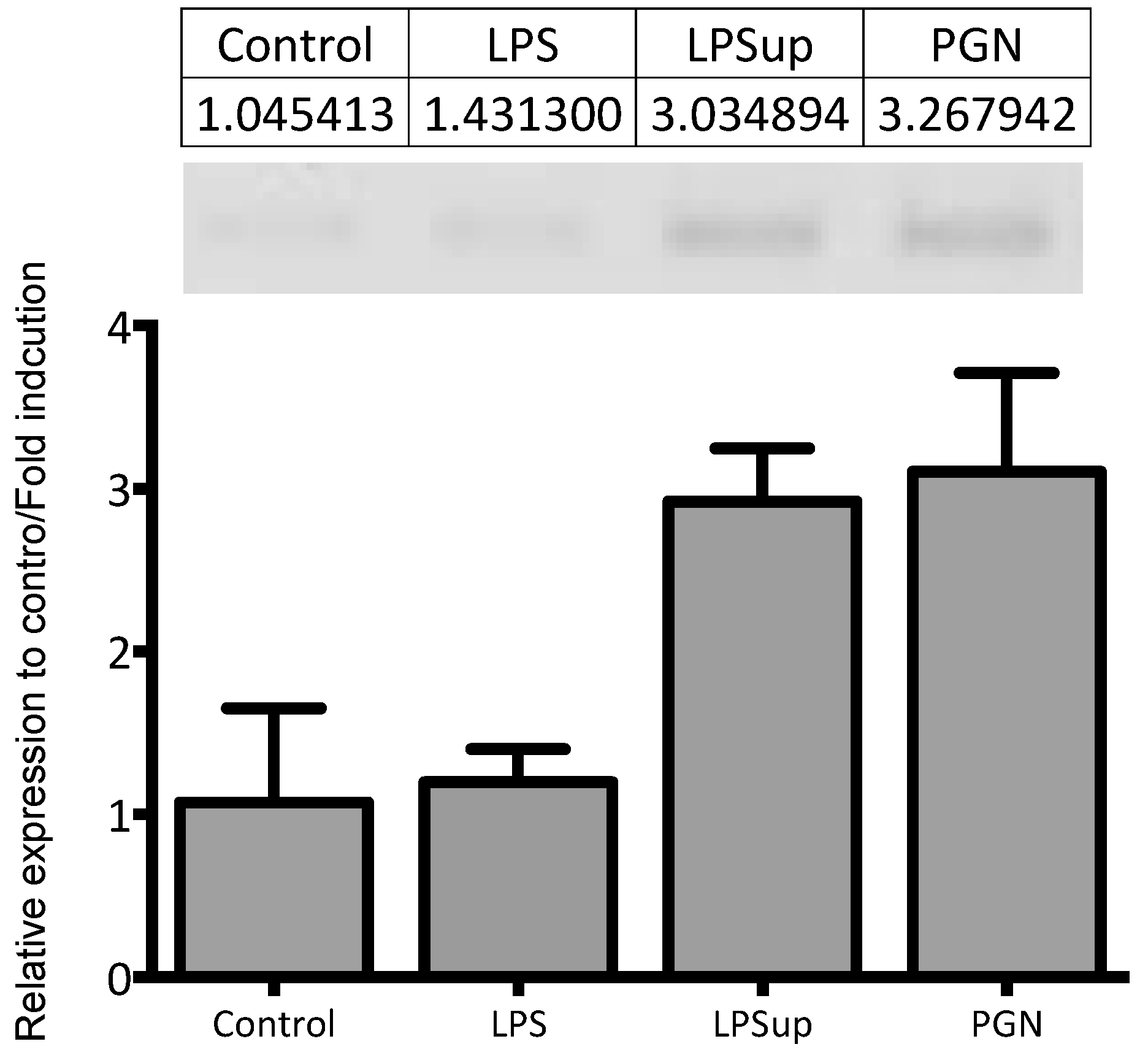
4.9. Real-Time Quantitative PCR and Validation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LPS | lipopolysaccharide |
| PAMPs | pathogen-associated molecular patterns |
| PGN | peptidoglycans |
| PGRPs | peptidoglycan recognition proteins |
| GDE | differentially expressed genes |
| COX-2 | cyclooxygenase 2 |
| PTGDS | prostaglandin D synthase |
| ONM | oligonucleotide microarray |
| SAQ | Aquagenomic Sparus aurata oligo-nucleotide microarray |
References
- Cerda, J.; Manchado, M. Advances in genomics for flatfish aquaculture. Genes Nutr. 2013, 8, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, H.; Pehkonen, P.; Vehniäinen, E.; Krasnov, A.; Rexroad, C.; Afanasyev, S.; Mölsa, H.; Oikari, A. Response of rainbow trout transcriptome to model chemical contaminants. Biochem. Biophys. Res. Commun. 2004, 320, 745–753. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.; Balasch, J.C.; Novoa, B.; Ribas, L.; Roher, N.; Krasnov, A.; Figueras, A. Comparative analysis of the acture response of the trout, O. mykiss, head kidney to in vivo challenge with virulent and attenuated infectious hematopoietic necrosis virus and LPS-induced inflammation. BMC Genom. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taggart, J.B.; Bron, J.E.; Martin, S.A.; Seear, P.J.; Høyheim, B.; Talbot, R.; Carmichael, S.N.; Villeneuve, L.A.; Sweeney, G.E.; Houlihan, D.F.; et al. A description of the origins, design and performance of the TRAITS-SGP Atlantic salmon Salmo salar L. cDNA microarray. J. Fish Biol. 2008, 72, 2071–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnov, A.; Timmerhaus, G.; Afanasyev, S.; Jørgensen, S.M. Development and assessment of oligonucleotide microarrays for Atlantic salmon (Salmo salar L.). Comp. Biochem. Physiol. Part D 2010, 6, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sarropoulou, E.; Kotoulas, G.; Power, D.M.; Geisler, R. Gene expression profiling of gilthead sea bream during early development and detection of stress-related genes by the application of cDNA microarray technology. Physiol. Genom. 2005, 23, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Sarropoulou, E.; Franch, R.; Louro, B.; Power, D.M.; Bargelloni, L.; Magoulas, A.; Senger, F.; Tsalavouta, M.; Patarnello, T.; Galibert, F.; et al. A gene-based radiation hybrid map of the gilthead sea bream Sparus aurata refines and exploits conserved synteny with Tetraodon nigroviridis. BMC Genom. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Franch, R.; Louro, B.; Tsalavouta, M.; Chatziplis, D.; Tsigenopoulos, C.S.; Sarropoulou, E.; Antonello, J.; Magoulas, A.; Mylonas, C.C.; Babbucci, M.; et al. A genetic link-age map of the hermaphrodite teleost fish Sparus aurata L. Genetics 2006, 174, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Senger, F.; Priat, C.; Hitte, C.; Sarropoulou, E.; Franch, R.; Geisler, R.; Bargelloni, L.; Power, D.M.; Galibert, F. The first radiation hybrid map of a perch-like fish: The gilthead seabream (Sparus aurata L.). Genomics 2006, 87, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Ferraresso, S.; Vitulo, N.; Mininni, A.N.; Romualdi, C.; Cardazzo, B.; Negrisolo, E.; Reinhardt, R.; Canario, A.V.M.; Patarnello, T.; Bargelloni, L. Development and validation of a gene expression oligo microarray for the gilthead sea bream (Sparus aurata). BMC Genom. 2008. [Google Scholar] [CrossRef] [PubMed]
- Emrich, S.J.; Barbazuk, W.B.; Li, L.; Schnable, P.S. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 2007, 17, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Sequencing goes 454 and takes large-scale genomics into the wild. Mol. Ecol. 2008, 17, 1629–1631. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.R.; Mao, M.; Jones, A.R.; Burchard, J.; Marton, M.J.; Shannon, K.W.; Lefkowitz, S.M.; Ziman, M.; Schelter, J.M.; Meyer, M.R. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 2001, 19, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Cawley, S.; Bekiranov, S.; Ng, H.H.; Kapranov, P.; Sekinger, E.A.; Kampa, D.; Piccolboni, A.; Sementchenko, V.; Cheng, J.; Williams, A.J.; et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 2004, 116, 499–509. [Google Scholar] [CrossRef]
- Mockler, T.C.; Ecker, J.R. Applications of DNA tiling arrays for whole-genome analysis. Genomics 2005, 85, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Skugor, S.; Jørgensen, S.M.; Gjerde, B.; Krasnov, A. Hepatic gene expression profiling reveals protective responses in Atlantic salmon vaccinated against furunculosis. BMC Genom. 2009. [Google Scholar] [CrossRef]
- Khaitovich, P.; Weiss, G.; Lachmann, M.; Hellmann, I.; Enard, W.; Muetzel, B.; Wirkner, U.; Ansorge, W.; Paabo, S. A neutral model of transcriptome evolution. PLoS Biol. 2004, 2, E132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Or, C.; Bar-Eyal, M.; Gal, T.Z.; Kapulnik, Y.; Czosnek, H.; Koltai, H. Derivation of species-specific hybridization-like knowledge out of cross-species hybridization results. BMC Genom. 2006. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.P.; Broadley, M.R.; Craigon, D.J.; Higgins, J.; Emmerson, Z.F.; Townsend, H.J.; White, P.J.; May, S.T. Using genomic DNA-based probe- selection to improve the sensitivity of high-density oligonucleotide arrays when applied to heterologous species. Plant Methods 2005. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Reid, L.H.; Jones, W.D.; Shippy, R.; Warrington, J.A.; Baker, S.C.; Collins, P.J.; de Longueville, F.; Kawasaki, E.S.; Lee, K.Y.; et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006, 24, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Patterson, T.A.; Lobenhofer, E.K.; Fulmer-Smentek, S.B.; Collins, P.J.; Chu, T.; Bao, W.; Fang, H.; Kawasaki, E.S.; Hager, J.; Tikhonova, I.R.; et al. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) Project. Nat. Biotechnol. 2006, 24, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Boltaña, S.; Reyes-Lopez, F.; Morera, D.; Goetz, F.W.; MacKenzie, S. Divergent responses to peptidoglycans derived from different E. coli serotypes influence inflammatory outcome in trout, Oncorhynchus mykiss, macrophages. BMC Genom. 2011, 12, 2–13. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.; Iliev, D.; Liarte, C.; Koskinen, H.; Planas, J.V.; Goetz, F.W.; Molsa, H.; Krasnov, A.; Tort, L. Transcriptional analysis of LPS-stimulated activation of trout (Oncorhynchus mykiss) monocyte/macrophage cells in primary culture treated with cortisol. Mol. Immunol. 2006, 43, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Blaney, S.C.; Houlihan, D.F.; Secombes, C.J. Transcriptome response following administration of a live bacterial vaccine in Atlantic salmon (Salmo salar). Mol. Immunol. 2006, 43, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Iliev, D.B.; Jørgensen, S.M.; Rode, M.; Krasnov, A.; Harneshaug, I.; Jørgensen, J.B. CpG-induced secretion of MHCIIbeta and exosomes from salmon (Salmo salar) APCs. Dev. Comp. Immunol. 2010, 34, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Goetz, F.W.; Iliev, D.B.; McCauley, L.A.; Liarte, C.Q.; Tort, L.B.; Planas, J.V.; Mackenzie, S. Analysis of genes isolated from lipopolysaccharide-stimulated rainbow trout (Oncorhynchus mykiss) macrophages. Mol. Immunol. 2004, 41, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Kiyoshi, T. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Sepulcre, M.P.; Alcaraz-Pérez, F.; López-Muñoz, A.; Roca, F.J.; Meseguer, J.; Cayuela, M.L.; Mulero, V. Evolution of lipopolysaccharide (LPS) recognition and signaling: Fish TLR4 does not recognize LPS and negatively regulates NF-κB activation. J. Immunol. 2009, 182, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zhu, S.Y.; Wang, Y.J.; Hu, Z. Toll-like receptor 3 regulates Mx expression in rare minnow Gobiocypris rarus after viral infection. Immunogenetics 2008, 60, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R. Recognition of bacterial peptidoglycan by the innate immune system. Cell. Mol. Life Sci. 2003, 60, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R.; Tapping, R.I.; Tobias, P.S. Binding of Bacterial Peptidoglycan to CD14. J. Biol. Chem. 1998, 273, 8680–8690. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Wang, Q.; Vinson, C.H.; Dziarski, R. Bacterial Peptidoglycan Induces CD14-dependent Activation of Transcription Factors CREB/ATF and AP-1. J. Biol. Chem. 1999, 274, 14012–14020. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan-and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17419. [Google Scholar] [CrossRef] [PubMed]
- Gobel, U.; Weber, J.; Ralf, S. Lipoteichoic Acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus Activates Immune Cells via Toll-like Receptor (TLR)-2, Lipopolysaccharide-binding Protein (LBP), and CD14, whereas TLR-4 and MD-2 Are Not Involved. J. Biol. Chem. 2003, 278, 15587–15594. [Google Scholar]
- Mackenzie, S.A.; Roher, N.; Boltaña, S.; Goetz, F.W. Peptidoglycan, not endotoxin, is the key mediator of cytokine gene expression induced in rainbow trout macrophages by crude LPS. Mol. Immunol. 2010, 47, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Roher, N.; Callol, S.; Planas, J.V.; Goetz, F.W.; Mackenzie, S. Endotoxin recognition in fish results in inflammatory cytokine secretion not gene expression. Innate Immun. 2011, 17, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Karaghiosoff, M.; Steinborn, R.; Kovarik, P.; Kriegshäuser, G.; Baccarini, M.; Donabauer, B.; Reichart, U.; Kolbe, T.; Bogdan, C.; Leanderson, T.; et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 2003, 4, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E. Signalling: Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.J.; Jin, X.; Lee, J.J. Zinc-finger protein 91 plays a key role in LIGHT-induced activation of non-canonical NF-κB pathway. Biochem. Biophys. Res. Commun. 2010, 400, 581–586. [Google Scholar]
- Agrawal, A.; Cha-Molstad, H.; Samols, S.; Kushner, I. Transactivation of C-Reactive Protein by IL-6 Requires Synergistic Interaction of CCAAT/Enhancer Binding Protein (C/EBP) and Rel p50. J. Immunol. 2001, 166, 2378–2384. [Google Scholar] [CrossRef] [PubMed]
- Calduch-Giner, J.A.; Davey, G.; Saera-Vila, A.; Houeix, B.; Talbot, A.; Prunet, P.; Cairns, M.T.; Pérez-Sánchez, J. Use of microarray technology to assess the time course of liver stress response after confinement exposure in gilthead sea bream (Sparus aurata L.). BMC Genom. 2010. [Google Scholar] [CrossRef] [PubMed]
- Soanes, K.H.; Ewart, K.V.; Mattatall, N.R. Recombinant production and characterization of the carbohydrate recognition domain from Atlantic salmon C-type lectin receptor C (SCLRC). Protein Expr. Purif. 2008, 59, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xin, J.L.; Yang, H.; Shi, Y.H. An interaction between a C-type lectin receptor and leukocyte cell-derived chemotaxin 2 of ayu, Plecoglossus altivelis. Fish Shellfish Immunol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Piesco, N.P.; Johns, L.P.; Riccelli, A.E. Differential Expression of IL-1β, TNF-α, IL-6, and IL-8 in Human Monocytes in Response to Lipopolysaccharides from Different Microbes. J. Dent. Res. 1995, 74, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Harada, A.; Matsushima, K. Interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF/ MCP-1), chemokines essentially involved in in ammatory and immune reactions. Cytokine Growth Factor Rev. 1998, 9, 9–23. [Google Scholar] [CrossRef]
- Cook, D.N. The role of MIP-1 alpha in inflammation and hematopoiesis. J. Leukoc. Biol. 1996, 59, 61–66. [Google Scholar] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jehanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zaehringer, U.; et al. NOD1 Detects a Unique Muropeptide from Gram-Negative Bacterial Peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Nunez, G. NODs: Intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 2003, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.G.; Scange, C.A.; Collazo-Custodio, C.M.; Cheever, A.W.; Hieny, S.; Caspar, P.; Sher, A. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J. Immunol. 2003, 171, 4758–4764. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, R.; Xu, X.M.; Wu, K.K.; Wang, L.H. Characterization of the Genomic Structure, Chromosomal Location and Promoter of Human Prostaglandin H Synthase-2 Gene. Biochem. Biophys. Res. Commun. 1994, 203, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Hayaishi, O. Prostaglandin D synthase: Structure and function. Vitam. Horm. 2000, 58, 89–120. [Google Scholar] [PubMed]
- Sarropoulou, E.; Sepulcre, P.; Poisa-Beiro, L.; Mulero, V.; Meseguer, J.; Figueras, A.; Novoa, B.; Terzoglou, V.; Reinhardt, R.; Magoulas, A.; et al. Profiling of infection specific mRNA transcripts of the European seabass Dicentrarchus labrax. BMC Genom. 2009. [Google Scholar] [CrossRef] [PubMed]
- Castellana, B.; Iliev, D.B.; Sepulcre, M.P.; MacKenzie, S.; Goetz, F.W.; Mulero, V.; Planas, J.V. Molecular characterization of interleukin-6 in the gilthead seabream (Sparus aurata). Mol. Immunol. 2008, 45, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- López-Castejón, G.; Sepulcre, M.P.; Roca, F.J.; Castellana, B.; Planas, J.V.; Meseguer, J.; Mulero, V. The type II interleukin-1 receptor (IL-1RII) of the bony fish gilthead seabream Sparus aurata is strongly induced after infection and tightly regulated at transcriptional and post-transcriptional levels. Mol. Immunol. 2007, 44, 2772–2780. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.M.; Afanasyev, S.; Krasnov, A. Gene expression analyses in Atlantic salmon challenged with infectious salmon anemia virus reveal differences between individuals with early, intermediate and late mortality. BMC Genom. 2008. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Jentsch, M.; Rasche, A.; Hummel, M.; Leser, U. Algorithms for differential splicing detection using exon arrays: A comparative assessment. BMC Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, M.A.; Gumenscheimer, M.T.; Kalis, C.; Landmann, R.; Galanos, C. Role of lipopolysaccharide susceptibility in the innate immune response to Salmonella typhimurium infection: LPS, a primary target for recognition of Gram-negative bacteria. Microbes Infect. 2001, 3, 1213–1222. [Google Scholar] [CrossRef]
- Freudenberg, M.A.; Kalis, C.; Chvatchko, Y.; Merlin, T.; Gumenscheimer, M.; Galanos, C. Role of interferonsin LPS hypersensitivity. J. Endotoxin Res. 2003, 9, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, S.; Planas, J.V.; Goetz, F.W. LPS-stimulated expression of a tumor necrosis factor-α mRNA in primary trout monocytes and in vitro differentiated macrophages. Dev. Comp. Immunol. 2003, 27, 393–400. [Google Scholar] [CrossRef]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Nunez, G. NOD2, a NOD1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Koseki, T.; del Peso, L.; Hu, Y.; Yee, C.; Chen, S.; Carrio, R.; Merino, J.; Liu, D.; Ni, J.; et al. NOD1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J. Biol. Chem. 1999, 274, 14560–14567. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Q.; Xu, Q.Q.; Chang, M.X.; Zou, J.; Secombes, C.J.; Peng, K.M.; Nie, P. Molecular characterization and expression analysis of the IFN-γ related gene (IFN-γ rel) in grass carp Ctenopharyngodon idella. Vet. Immunol. Immunopathol. 2010, 134, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Q.; Xu, Q.Q.; Chang, M.X.; Nie, P.; Peng, K.M. Molecular characterization and expression analysis of nuclear oligomerization domain proteins NOD1 and NOD2 in grass carp Ctenopharyngodon idella. Fish Shellfish Immunol. 2009, 28, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Kirkland, T.N.; Viriyakosol, S.; Dziarski, R. CD14 is a cell-activating receptor for bacterial peptidoglycan. J. Biol. Chem. 1996, 271, 23310–23316. [Google Scholar] [PubMed]
- Wang, Q.; Dziarski, R.; Kirsching, C.; Muzio, M.; Gupta, D. Micrococci and Peptidoglycan Activate TLR2 MyD88 IRAK TRAF NIK IKK NF-κB Signal Transduction Pathway That Induces Transcription of Interleukin-8. Infect. Immun. 2001, 69, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Hermsen, T.; Taverne-Thiele, A.J.; Savelkoul, H.; Wiegertjes, G.F. Evolution of Recognition of Ligands from Gram-Positive Bacteria: Similarities and Differences in the TLR2-Mediated Response between Mammalian Vertebrates and Teleost Fish. J. Immunol. 2010, 184, 2355–2368. [Google Scholar] [CrossRef] [PubMed]
- Purcell, M.K.; Smith, K.D.; Hood, L.; Winton, J.R.; Roach, J.C. Conservation of Toll-Like Receptor Signaling Pathways in Teleost Fish. Comp. Biochem. Physiol. Part D Genom. Proteom. 2006, 1, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liao, C.C.; Hsu, M.J.; Liao, Y.T.; Lin, C.C.; Sheu, J.R.; Lin, C.H. Peptidoglycan-Induced IL-6 Production in RAW 264.7 Macrophages Is Mediated by Cyclooxygenase-2, PGE2/PGE4 Receptors, Protein Kinase A, IkB Kinase, and NF-κB. J. Immunol. 2006, 177, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Kang, J.C.; Luc, Y.T.; Hsub, M.J.; Liao, C.C.; Chiu, W.T.; Yeh, F.L.; Lin, C.H. Rac1 regulates peptidoglycan-induced nuclear factor-kB activation and cyclooxygenase-2 expression in RAW 264.7 macrophages by activating the phosphatidylinositol 3-kinase/Akt pathway. Mol. Immunol. 2009, 46, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Hinson, R.M.; Williams, J.A.; Shacter, E. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: Possible role of cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 1996, 93, 4885–4890. [Google Scholar] [CrossRef] [PubMed]
- Ogle, C.K.; Guo, X.; Szczur, K.; Hartmann, S.; Ogle, J.D. Production of tumor necrosis factor, interleukin-6 and prostaglandin E2 by LPS-stimulated rat bone marrow macrophages after thermal injury: Effect of indomethacin. Inflammation 1994, 18, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 45–53. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
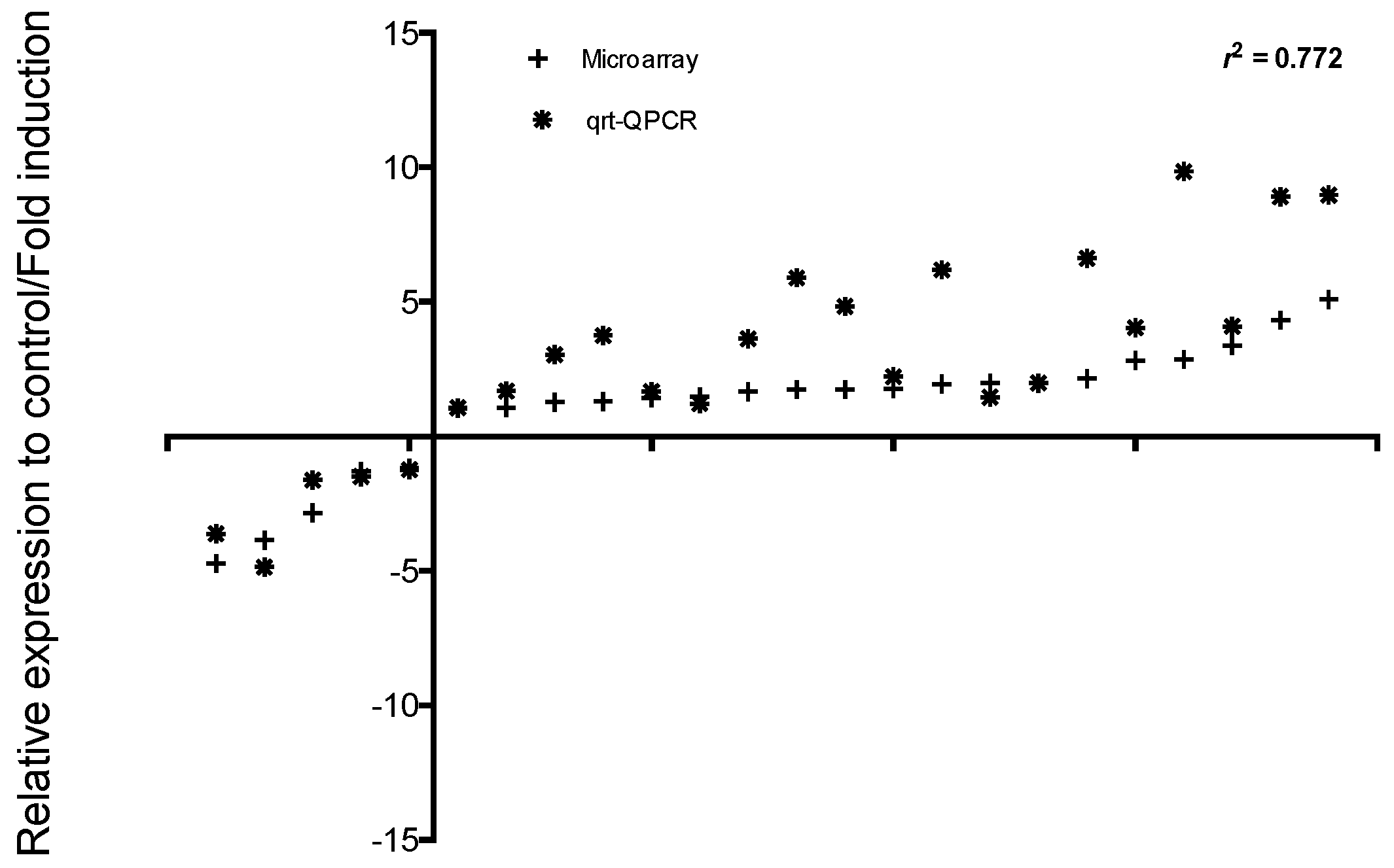{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Microarray-LPS | Microarray-PGN | PCR-LPS | PCR-PGN |
|---|---|---|---|---|
| MX | −4.724382 | 1.739085 | −3.62258 | 4.83517 |
| CD8 | −3.8668 | −3.8668 | −4.85774 | −4.85774 |
| T-cell recpetor | 1.470813 | −1.21089 | 2.851373 | −1.624026667 |
| CD83 | 1.3097895 | 1.4322673 | 1.979696667 | 3.03769 |
| GR | −1.189567 | 1.051587 | −1.233847 | 1.05836 |
| Troponin T2 | 1.9828836 | 1.2732812 | 1.979696667 | 3.03769 |
| G6PC | 1.295544 | 1.738823 | 3.751023 | 5.89183 |
| MHCI | 1.656065 | 1.934764 | 3.62856 | 6.18487 |
| IL6 | 2.864684 | 2.1441178 | 9.840256667 | 6.62607 |
| IL1 | 4.3157287 | 5.093681 | 8.908796667 | 8.970556667 |
| LIPO | 1.9686182 | 7.5816884 | 1.45541 | 3.483463333 |
| MHCII | 2.8191724 | 3.3714116 | 4.03926 | 4.083143 |
| (a) | ||||
| Description | Corrected p-Value | FC LPS | FC PGN | Common Regulation |
| Stat3 | 0.03 | 2.34 | 2.96 | up |
| Tumour necrosis factor alpha (TNF-α) | 0.01 | 2.71 | 3.13 | up |
| Serine/threonineprotein kinase (TBK1) | 0.03 | 1.54 | 1.75 | up |
| NF-κB inhibitor | 0.01 | 2.04 | 1.97 | up |
| Ankyrin repeat and zinc finger | 0.02 | 1.87 | 2.12 | up |
| CCAAT/enhancer binding protein beta | 0.03 | 2.97 | 2.36 | up |
| Extracellular matrix protein (ECM) | 0.05 | 1.43 | 3.79 | down |
| Matrix metalloproteinase 9 | 0.03 | 1.98 | 3.18 | up |
| Matrix metalloproteinase 1 | 0 | 2.33 | 2.67 | up |
| Ctype lectin receptor | 0.03 | 1.76 | 1.23 | up |
| p67phox | 0.04 | 2.89 | 2.09 | up |
| Myeloperoxidase precursor | 0.01 | 1.87 | 1.88 | down |
| (b) | ||||
| Gene Ontology Class | No. Genes | Corrected p-Value | ||
| Cellular defence response | 3 | 0.02 | ||
| Detection of bacteria | 1 | 0.02 | ||
| CC chemokine | 2 | 0.01 | ||
| Activation of JAK/STAT | 7 | 0.01 | ||
| Cell homeostasis | 13 | 0.04 | ||
| NF-κB pathway | 4 | 0.05 | ||
| (a) | |||
| Description | Corrected p-Value | FC | Regulation |
| Interleukin 8 receptor CXCR1 | 0.00 | 2.27 | up |
| Interleukin 8 like protein | 0.03 | 1.97 | up |
| CC chemokine receptor 3 | 0.02 | 3.04 | down |
| Chemokine CK1 | 0.01 | 3.03 | up |
| Allograft inflammatory factor1 (AIF1) | 0.00 | 3.51 | up |
| (b) | |||
| Description | Corrected p-Value | FC | Regulation |
| NOD receptor C | 0.02 | 2.41 | up |
| Myeloid differentiation factor 88 | 0.02 | 2.48 | up |
| TRAF-6 | 0.02 | 3.75 | up |
| Serine/threonineprotein kinase (TBK1) | 0.03 | 2.22 | up |
| Leukotriene A4 hydrolase | 0.05 | 1.63 | down |
| Prostaglandin E synthase 3 | 0.02 | 1.50 | up |
| Prostaglandin F receptor | 0.02 | 2.28 | up |
| Prostaglandin transporter | 0.02 | 2.01 | up |
| COX isoform 2 | 0.03 | 2.78 | up |
| Microsomal glutathione S transferase 2 | 0.04 | 2.28 | up |
| 15hydroxyprostaglandin dehydrogenase | 0.03 | 1.87 | down |
| CC chemokine receptor3 | 0.03 | 4.07 | down |
| (c) | |||
| Gene Ontology Class | No. Genes | Corrected p-Value | |
| MHC class I receptor activity | 3 | 0.0015 | |
| Antigen presentation | 3 | 0.0004 | |
| Antigen processing | 3 | 0.0041 | |
| NF-κB cascade | 7 | 0.0617 | |
| Regulation of NF-κB cascade | 6 | 0.0698 | |
| Cell adhesion | 11 | 0.0407 | |
| Eicosanoid synthesis | 6 | 0.00148 | |
| G-protein chemoattractant receptor activity | 2 | 0.0263 | |
| Chemokine receptor activity | 2 | 0.0263 | |
| CC chemokine binding | 2 | 0.0066 | |
| Protein-tyrosine kinase activity | 0 | 0.0343 | |
| Activation of JNK | 4 | 0.0221 | |
| Activation of MAPK | 4 | 0.0438 | |
| Protein kinase cascade | 27 | 0.0417 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boltaña, S.; Castellana, B.; Goetz, G.; Tort, L.; Teles, M.; Mulero, V.; Novoa, B.; Figueras, A.; Goetz, F.W.; Gallardo-Escarate, C.; et al. Extending Immunological Profiling in the Gilthead Sea Bream, Sparus aurata, by Enriched cDNA Library Analysis, Microarray Design and Initial Studies upon the Inflammatory Response to PAMPs. Int. J. Mol. Sci. 2017, 18, 317. https://doi.org/10.3390/ijms18020317
Boltaña S, Castellana B, Goetz G, Tort L, Teles M, Mulero V, Novoa B, Figueras A, Goetz FW, Gallardo-Escarate C, et al. Extending Immunological Profiling in the Gilthead Sea Bream, Sparus aurata, by Enriched cDNA Library Analysis, Microarray Design and Initial Studies upon the Inflammatory Response to PAMPs. International Journal of Molecular Sciences. 2017; 18(2):317. https://doi.org/10.3390/ijms18020317
Chicago/Turabian StyleBoltaña, Sebastian, Barbara Castellana, Giles Goetz, Lluis Tort, Mariana Teles, Victor Mulero, Beatriz Novoa, Antonio Figueras, Frederick W. Goetz, Cristian Gallardo-Escarate, and et al. 2017. "Extending Immunological Profiling in the Gilthead Sea Bream, Sparus aurata, by Enriched cDNA Library Analysis, Microarray Design and Initial Studies upon the Inflammatory Response to PAMPs" International Journal of Molecular Sciences 18, no. 2: 317. https://doi.org/10.3390/ijms18020317