
Rett Syndrome: A Focus on Gut Microbiota

 , and
, and
Abstract
: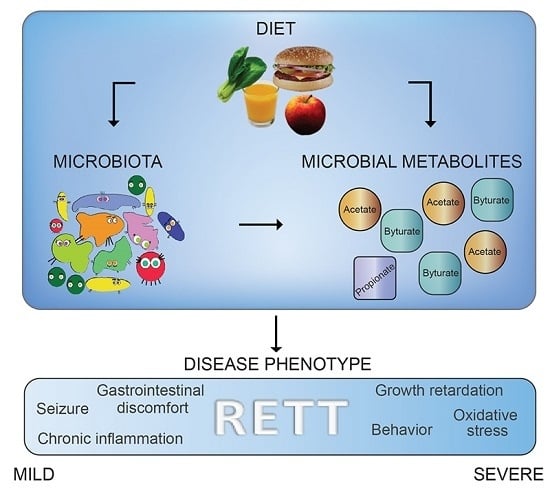
1. Introduction
2. Results
2.1. Cohort Description
2.2. Diet Evaluation
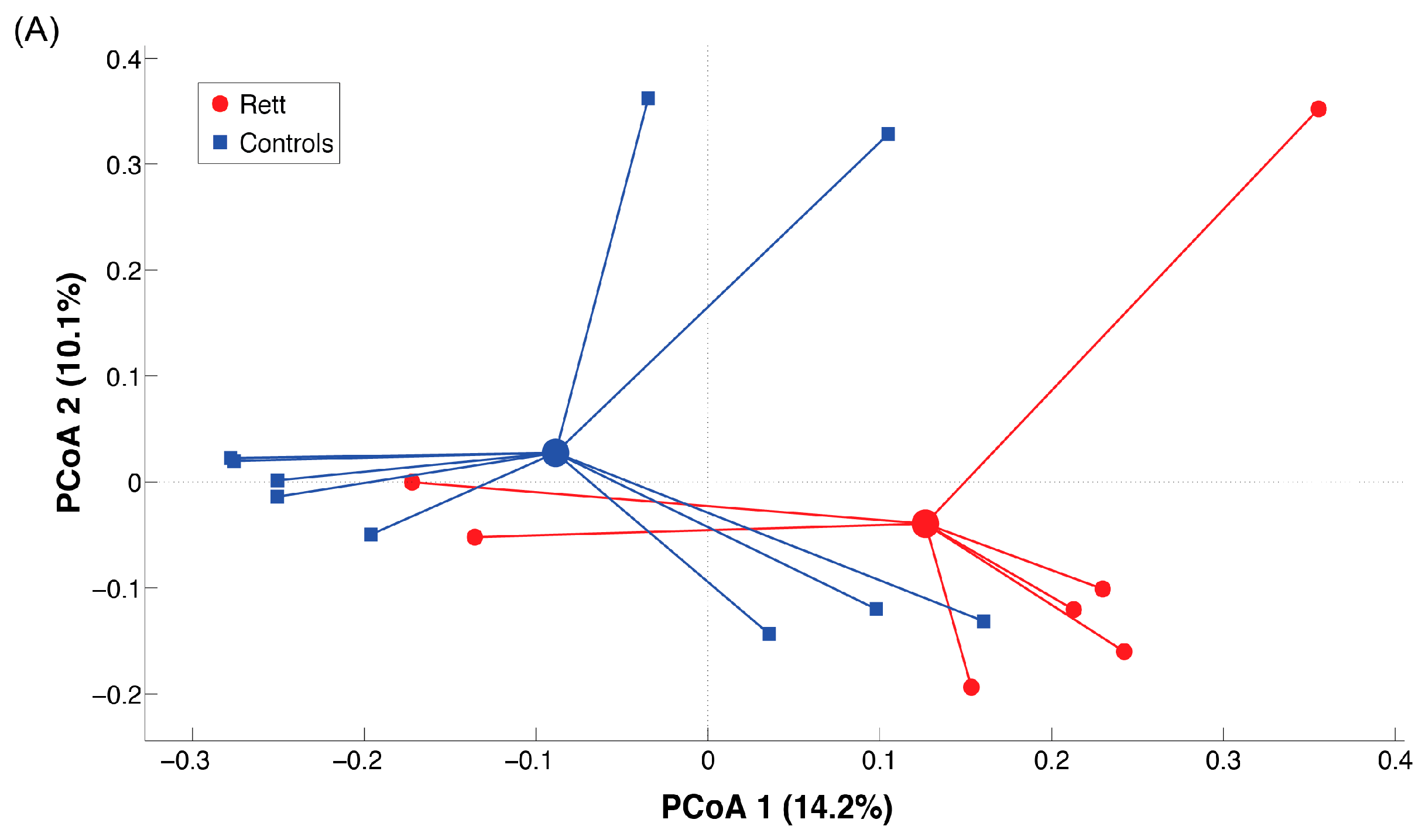
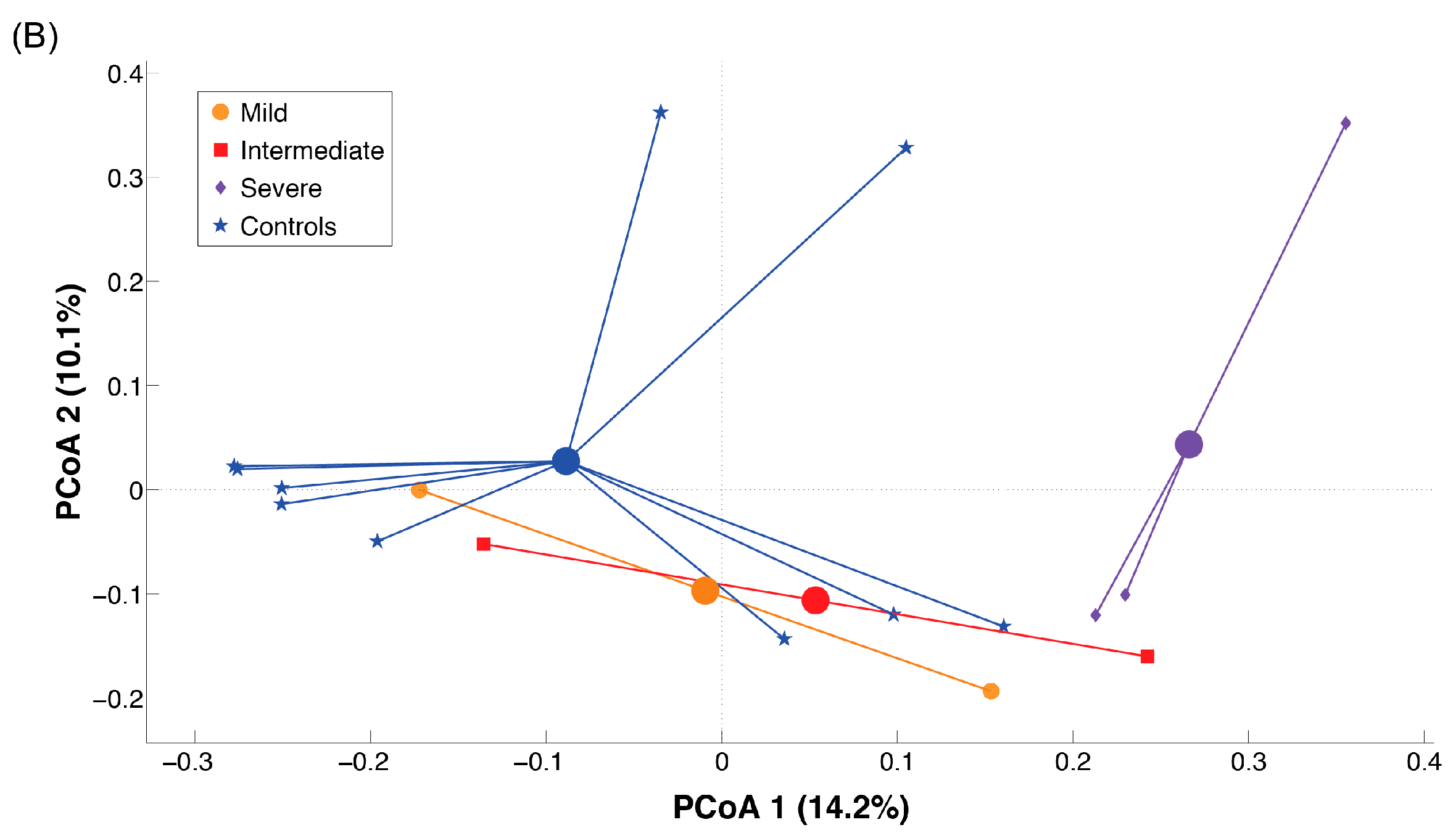
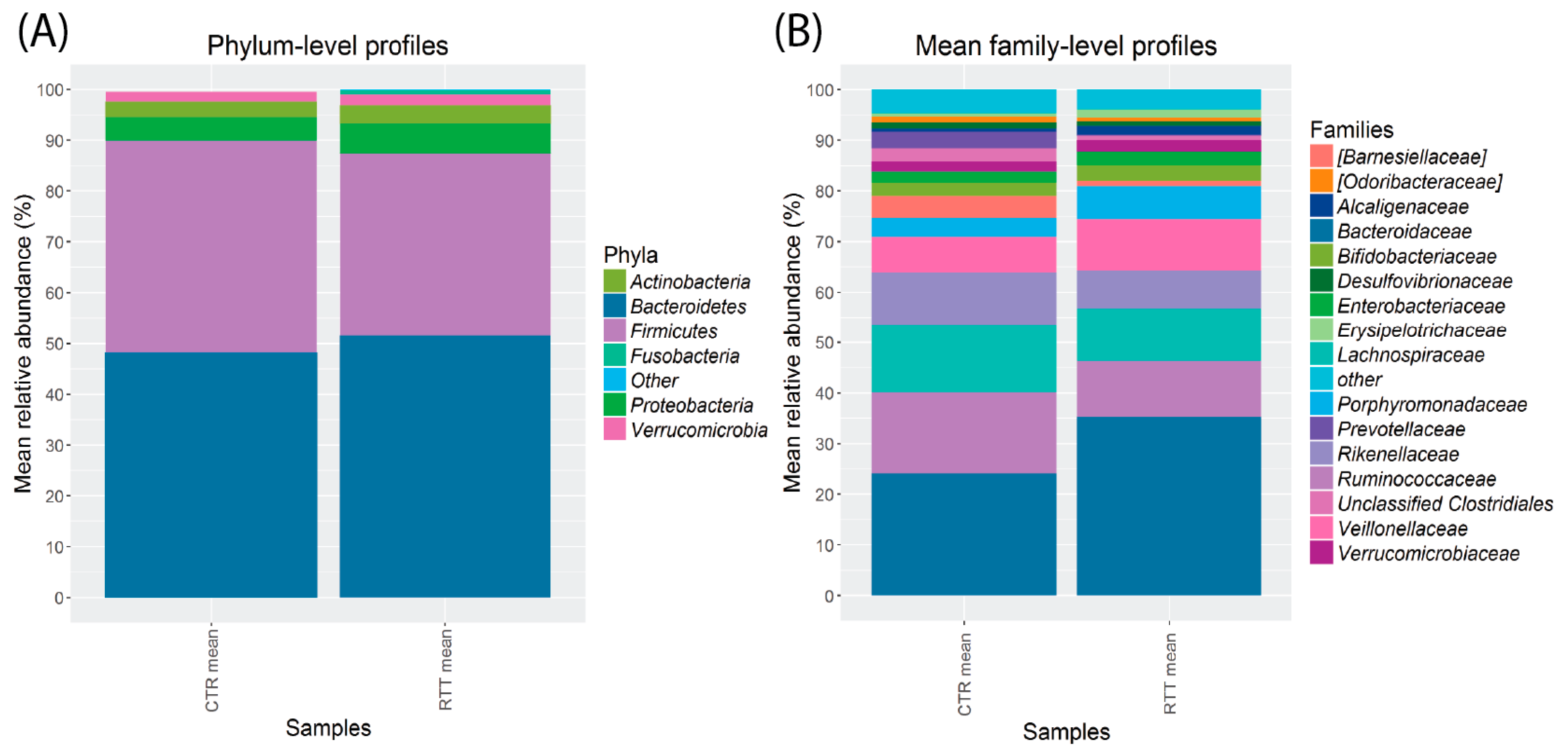
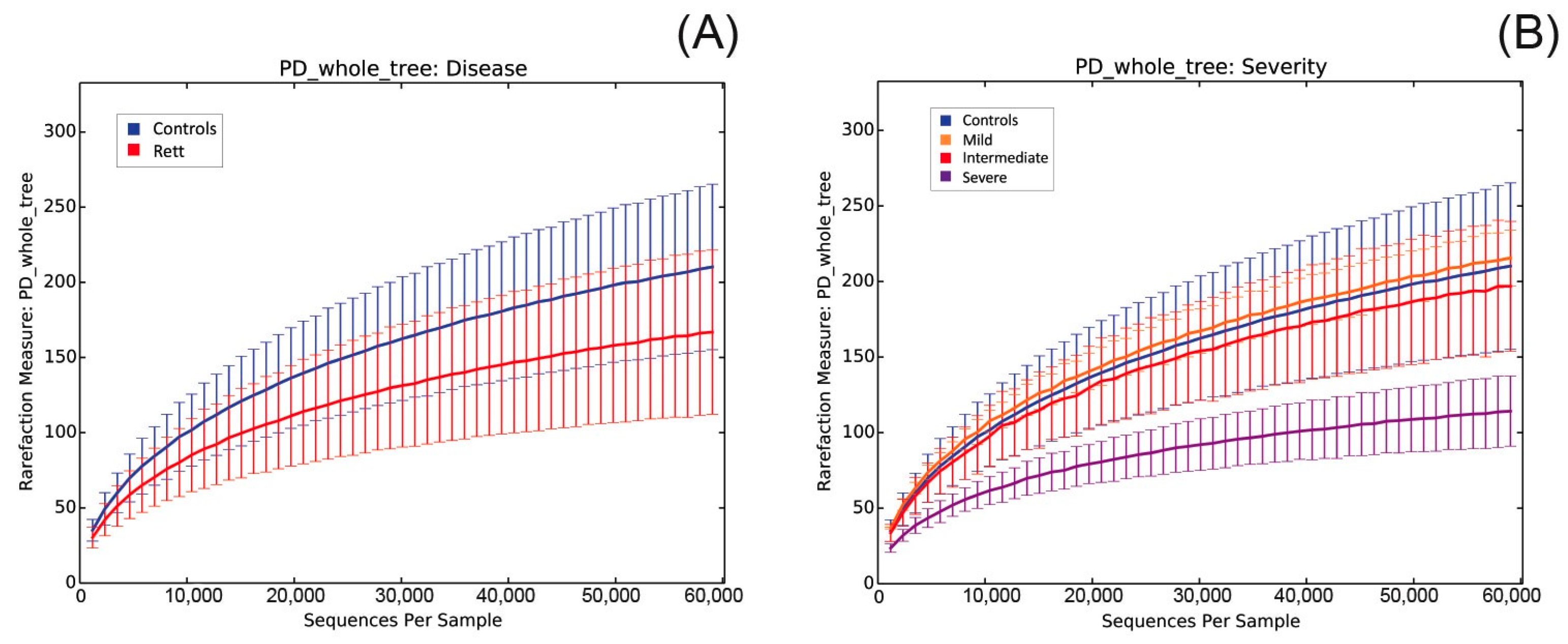
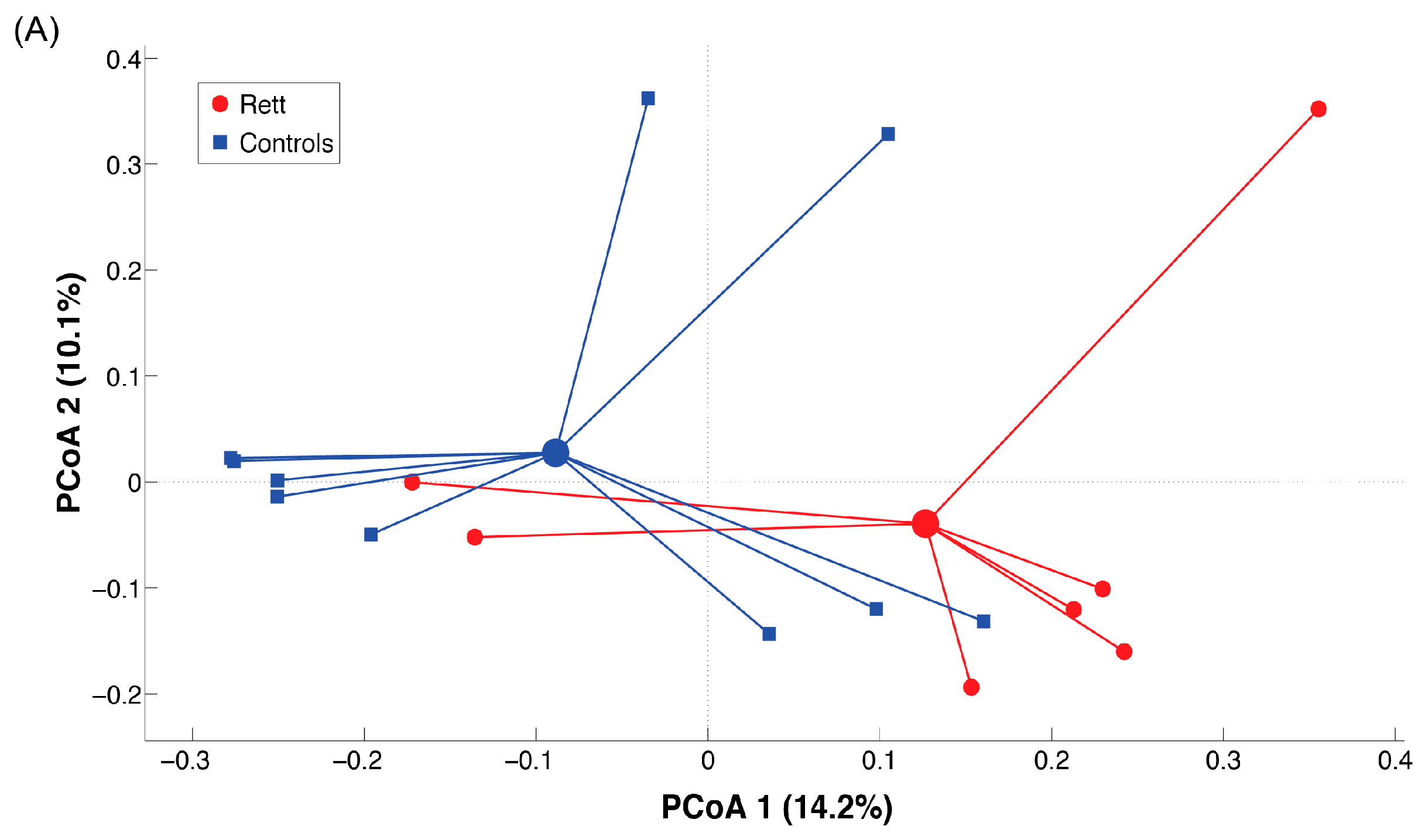
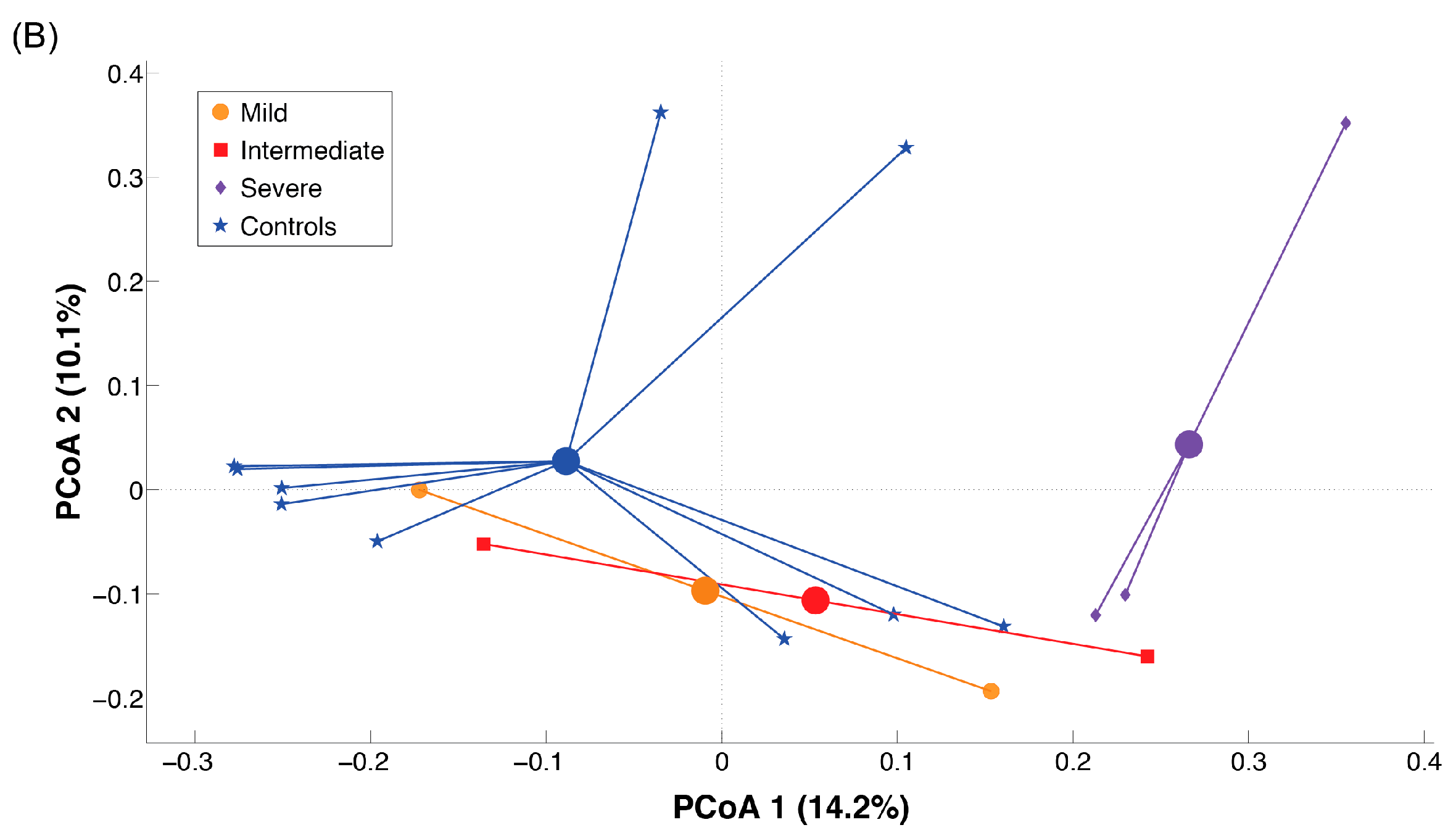
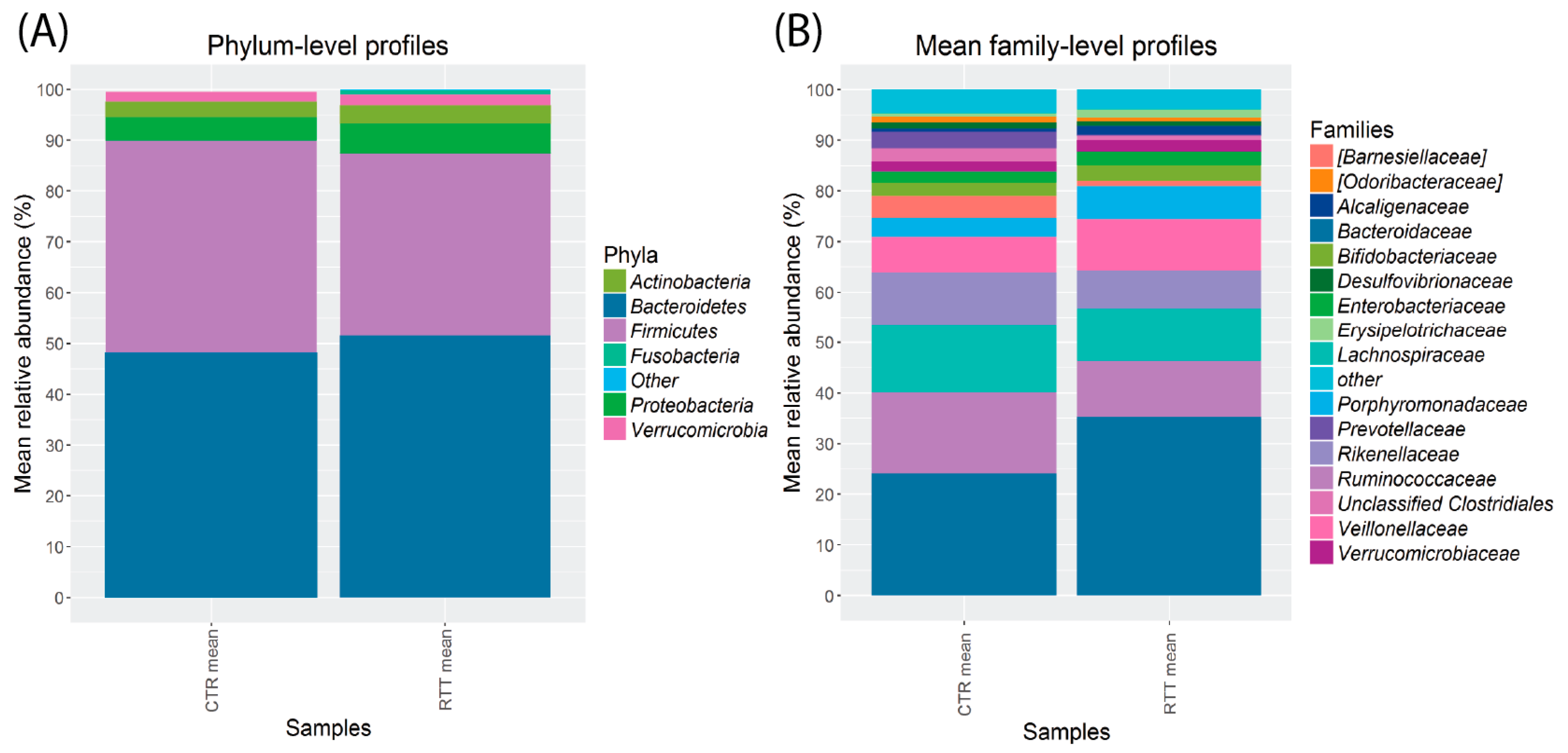
2.3. Microbiota Dysbiosis in Rett syndrome (RTT) Patients Is Related to Disease Severity
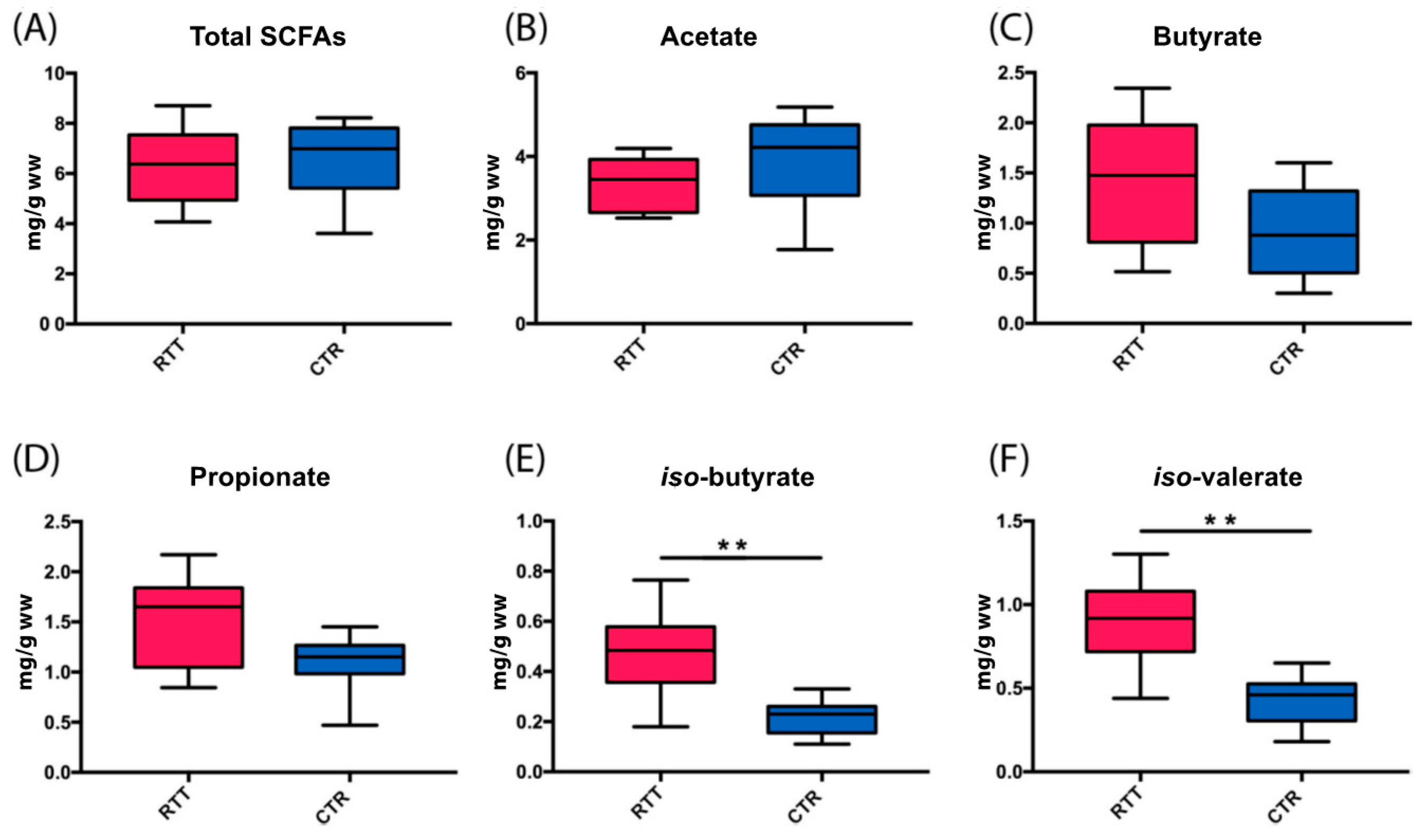
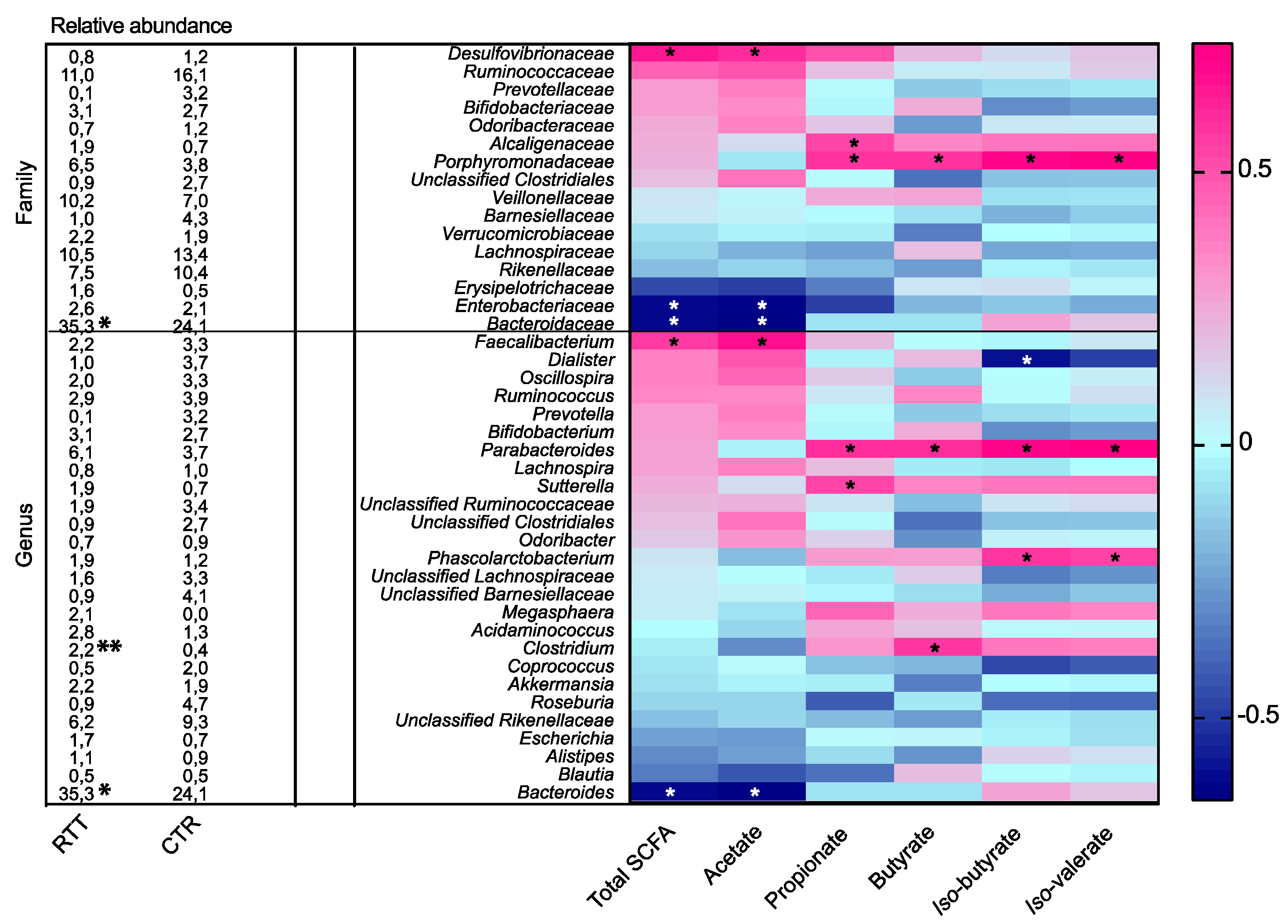
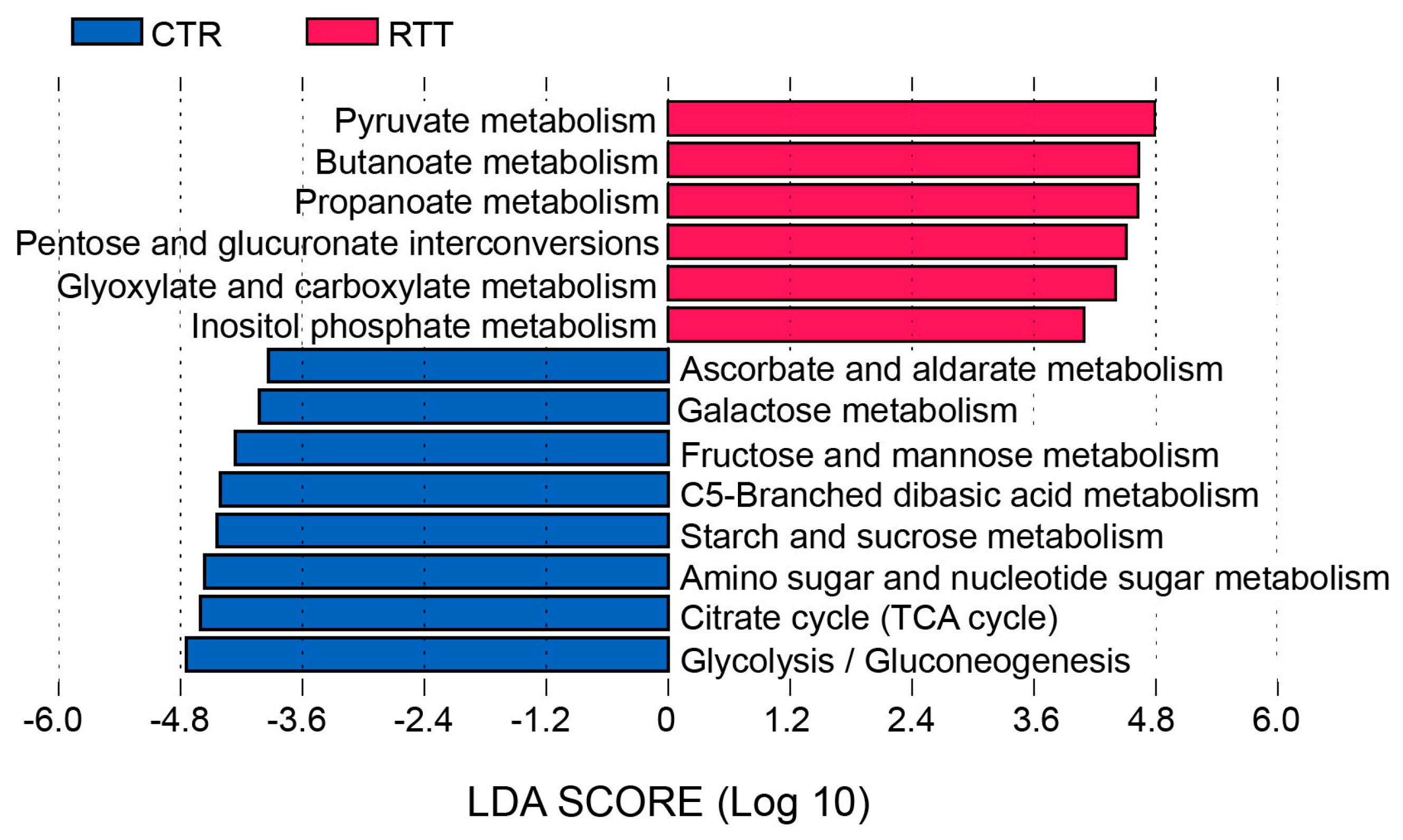
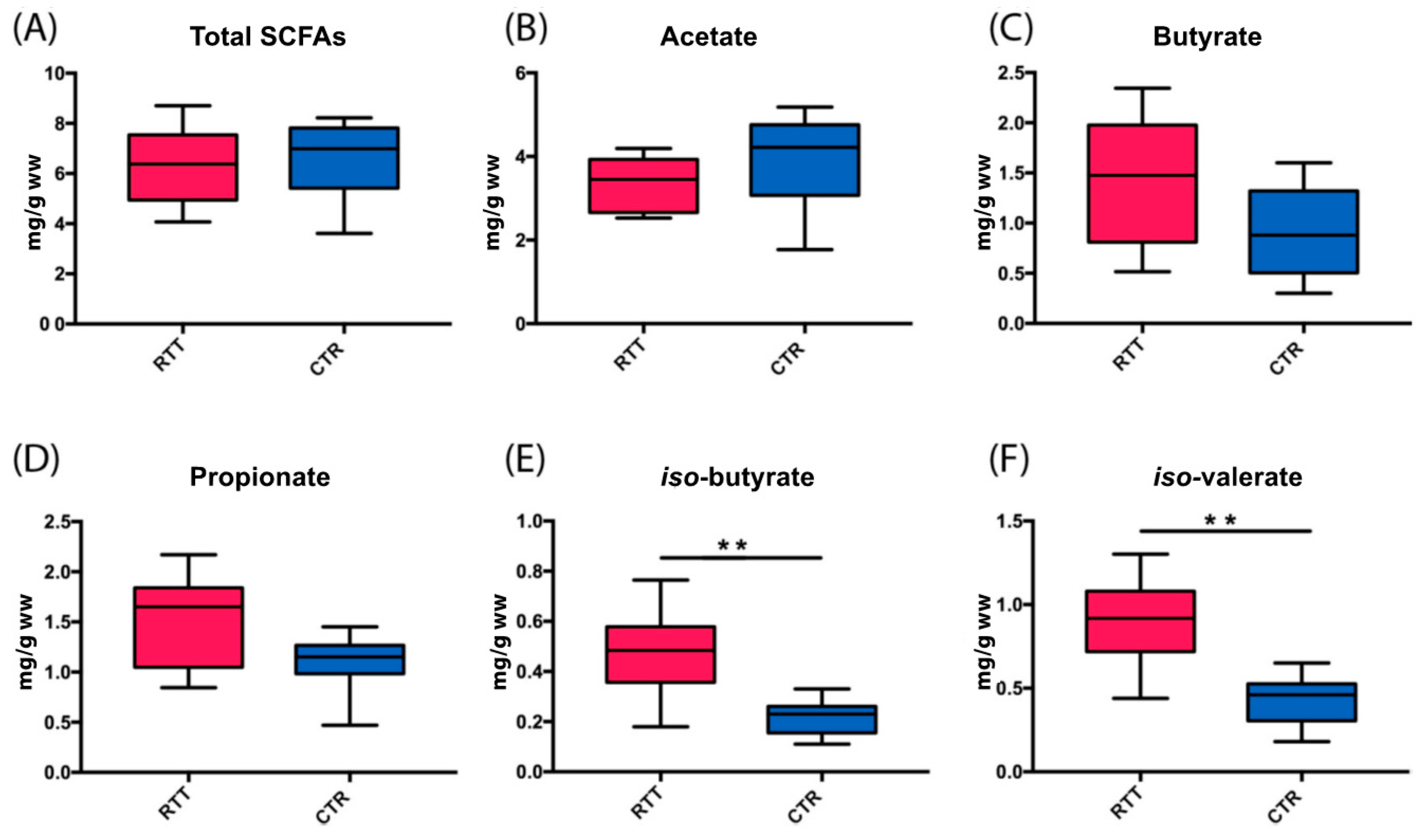
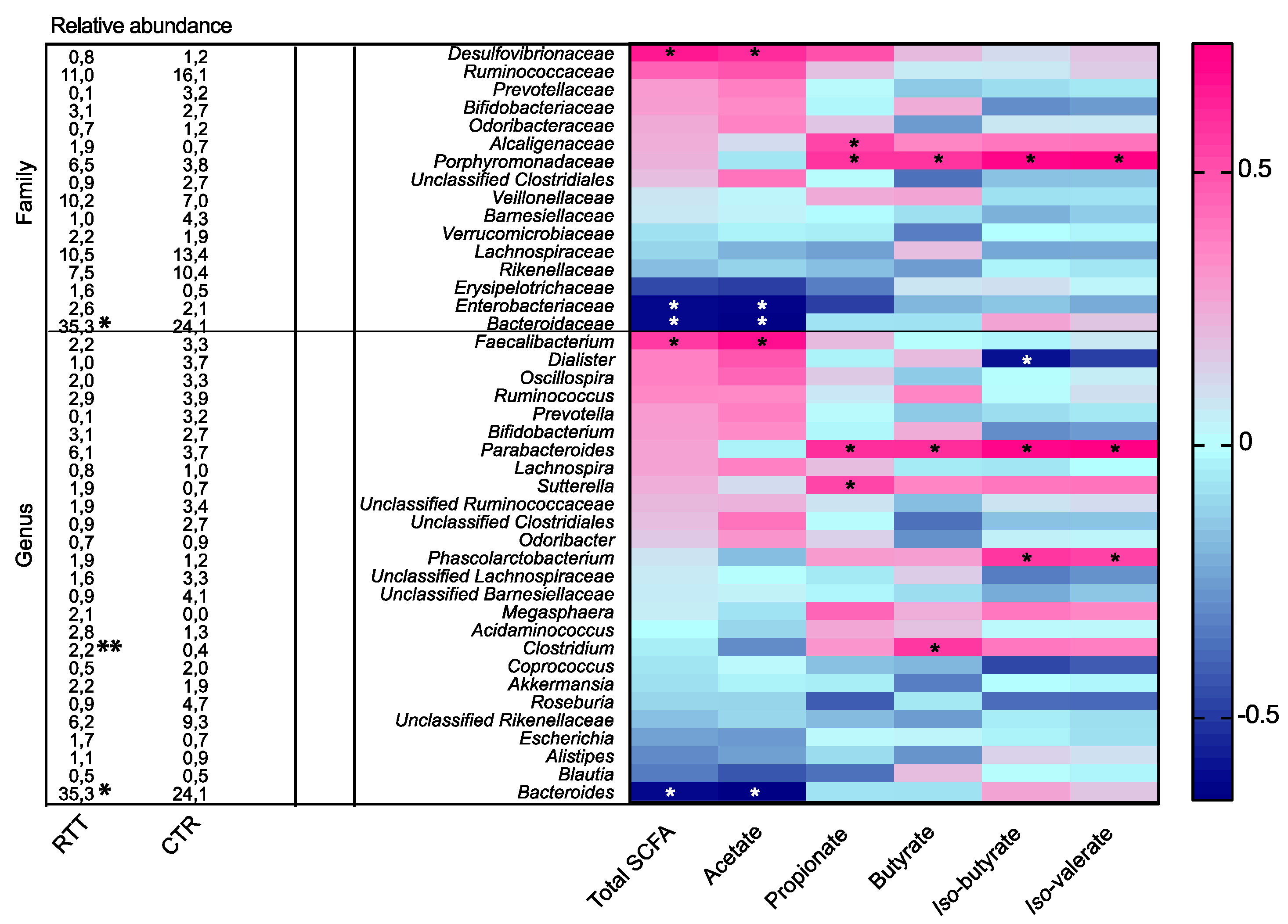
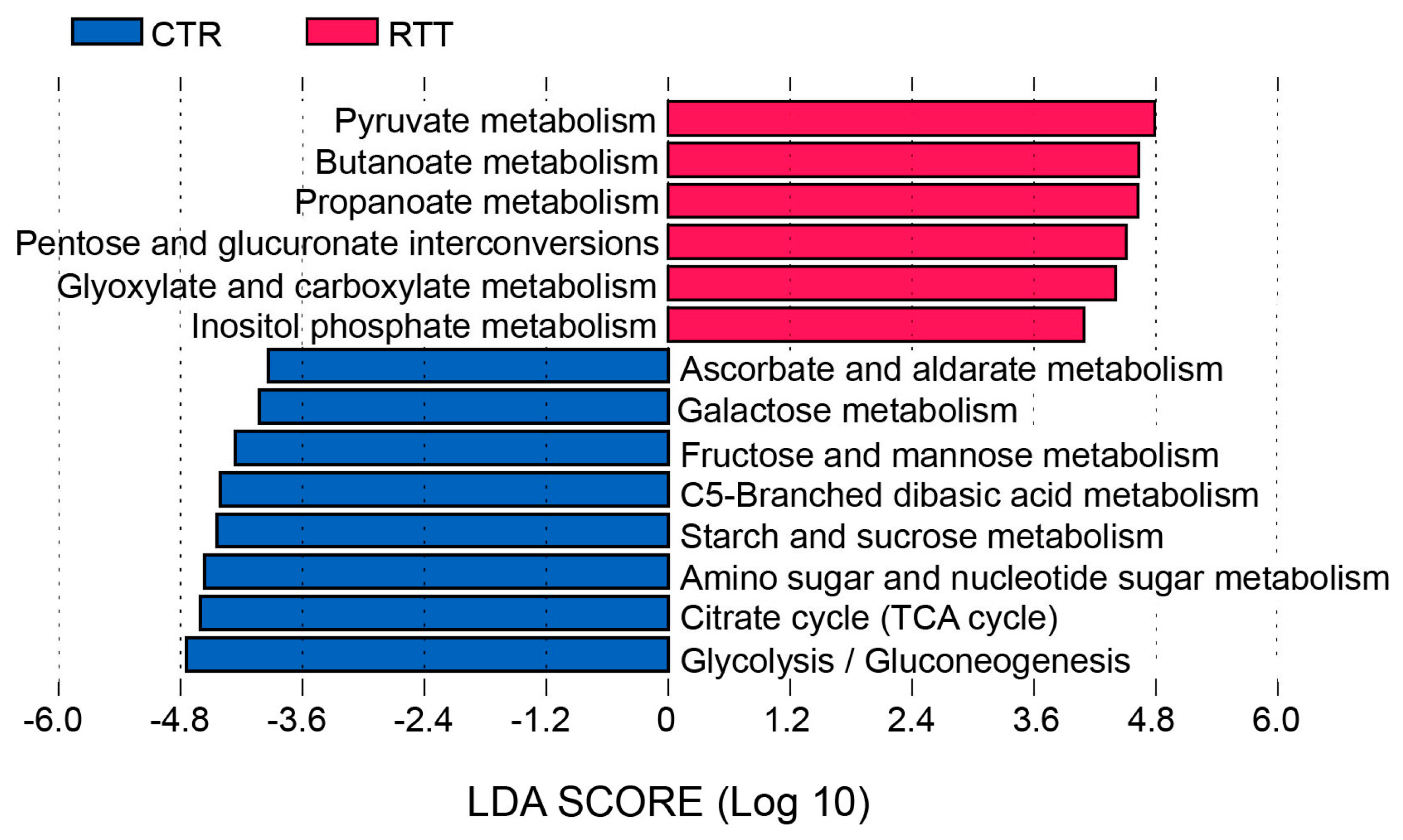
2.4. Microbial Metabolites Are Influenced by Diet and by a Shift in Some Microbial Populations
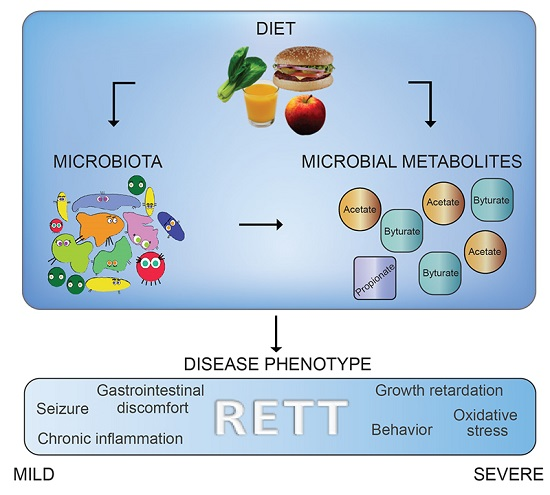
3. Discussion
4. Materials and Methods
4.1. Subject Recruitment
4.2. DNA Extraction and Preparation of 16s rRNA Gene Amplicon Libraries
4.3. Sequencing via an Illumina MiSeq Platform
4.4. Short Chain Fatty Acid (SCFA) Measurement
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AED | Antiepileptic drug |
| ASD | Autism spectrum disorders |
| BCFA | Branched-chain fatty acids |
| BMI | Body mass index |
| CNS | Central nervous system |
| CTR | Controls |
| ENS | Enteric nervous system |
| LDA | Linear discriminant analysis |
| MeCP2 | Methyl CpG binding protein 2 |
| OTU | Operational taxonomic units |
| RTT | Rett syndrome |
| SCFA | Short-chain fatty acids |
| SGS | Severity Global Score |
References
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar]
- Marschik, P.B.; Kaufmann, W.E.; Sigafoos, J.; Wolin, T.; Zhang, D.; Bartl-Pokorny, K.D.; Pini, G.; Zappella, M.; Tager-Flusberg, H.; Einspieler, C.; et al. Changing the perspective on early development of Rett syndrome. Res. Dev. Disabil. 2013, 34, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Romell, M. Rett females: Patterns of characteristic side-asymmetric neuroimpairments at long-term follow-up. Neuropediatrics 2002, 33, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Carolyn Schanen, N.; Olson, C.O.; et al. Mice with an isoform-ablating MECP2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Cheval, H.; Selfridge, J.; Bird, A. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 2011, 27, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; Lasalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Grillo, E.; Lo Rizzo, C.; Bianciardi, L.; Bizzarri, V.; Baldassarri, M.; Spiga, O.; Furini, S.; de Felice, C.; Signorini, C.; Leoncini, S.; et al. Revealing the complexity of a monogenic disease: Rett syndrome exome sequencing. PLoS ONE 2013, 8, e56599. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Pillai, R.B.; Shekar, K.V.; Lane, J.B.; Motil, K.J.; Skinner, S.A.; Tarquinio, D.C.; Glaze, D.G.; McGwin, G.; Kaufmann, W.E.; et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J. Med. Genet. 2014, 51, 152–158. [Google Scholar] [CrossRef]
- Wahba, G.; Schock, S.C.; Cudd, S.; Grynspan, D.; Humphreys, P.; Staines, W.A. Activity and MeCP2-dependent regulation of nNOS levels in enteric neurons. Neurogastroenterol. Motil. 2016, 28, 1723–1730. [Google Scholar] [CrossRef]
- Symons, F.J.; Byiers, B.; Tervo, R.C.; Beisang, A. Parent-reported pain in Rett syndrome. Clin. J. Pain 2013, 29, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Cavalieri, D.; Albanese, D.; de Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Pindo, M.; Renzi, D.; et al. Altered gut microbiota in Rett syndrome. Microbiome 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, J.M. The microbiota-gut-brain axis and its potential therapeutic role in autism spectrum disorder. Neuroscience 2016, 324, 131–139. [Google Scholar] [CrossRef]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O'Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; de Felice, C.; Leoncini, S.; Durand, T.; Galano, J.M.; Cortelazzo, A.; Zollo, G.; Guerranti, R.; Gonnelli, S.; Caffarelli, C.; et al. Altered erythrocyte membrane fatty acid profile in typical Rett syndrome: Effects of ω-3 polyunsaturated fatty acid supplementation. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Tabbaa, M.; Golubic, M.; Roizen, M.F.; Bernstein, A.M. Docosahexaenoic acid, inflammation, and bacterial dysbiosis in relation to periodontal disease, inflammatory bowel disease, and the metabolic syndrome. Nutrients 2013, 5, 3299–3310. [Google Scholar] [CrossRef] [PubMed]
- Società Italiana di Nutrizione Umana (Italian Society of Human Nutrition, SINU). Nutrients and Energy Reference Intake Levels for Italian Population, 4th ed.; SICS: Milan, Italy, 2014. [Google Scholar]
- Lewis, S.J.; Heaton, K.W. Stool form scale as a useful guide to intestinal transit time. Scand. J. Gastroenterol. 1997, 32, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Borgo, F.; Lassandro, C.; Verduci, E.; Morace, G.; Borghi, E.; Berry, D. Pediatric obesity is associated with an altered gut microbiota and discordant shifts in Firmicutes populations. Env. Microbiol. 2016, 1–29. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Darzi, Y.; Lima-Mendez, G.; Garcia Yunta, R.; Okuda, S.; Vandeputte, D.; Valles-Colomer, M.; Hildebrand, F.; Chaffron, S.; et al. Species–function relationships shape ecological properties of the human gut microbiome. Nat. Microbiol. 2016, 124, 16088. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of prevotella and other fermenters in intestinal microflora of Autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef] [PubMed]
- Hold, G.L.; Smith, M.; Grange, C.; Watt, E.R.; El-Omar, E.M.; Mukhopadhya, I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: What have we learnt in the past 10 years? World J. Gastroenterol. 2014, 20, 1192–1210. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson's disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Su, W.; Rahat-Rozenbloom, S.; Wolever, T.M.; Comelli, E.M. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr. Diabetes 2014, 4, e121. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; Perdicaro, D.J.; Brown, A.W.; Hammons, S.; Carden, T.J.; Carr, T.P.; Eskridge, K.M.; Walter, J. Diet-induced alterations of host cholesterol metabolism are likely to affect the gut microbiota composition in hamsters. Appl. Environ. Microbiol. 2013, 79, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Voreades, N.; Kozil, A.; Weir, T.L. Diet and the development of the human intestinal microbiome. Front. Microbiol. 2014, 5, 494. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Ribbera, A.; Foroni, E.; van Sinderen, D.; Ventura, M. Human gut microbiota and bifidobacteria: From composition to functionality. Antonie Van Leeuwenhoek 2008, 94, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Gratz, S.W.; Sheridan, P.O.; Flint, H.J.; Duncan, S.H. The influence of diet on the gut microbiota. Pharmacol. Res. 2013, 69, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut bifidobacteria populations in human health and aging. Front. Microbiol. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio 2014, 5, e00889. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [PubMed]
- Soret, R.; Chevalier, J.; De Coppet, P.; Poupeau, G.; Derkinderen, P.; Segain, J.P.; Neunlist, M. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology 2010, 138. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Di Costanzo, M.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Lahti, L.; Salojarvi, J.; Holtrop, G.; Korpela, K.; Duncan, S.H.; Date, P.; Farquharson, F.; Johnstone, A.M.; Lobley, G.E.; et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014, 8, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.K.; Ben Bacha, A.; Kotb, M. Etiology of autistic features: The persisting neurotoxic effects of propionic acid. J. Neuroinflamm. 2012, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Kurth, M.J.; McGilligan, V.; McGlynn, H.; Rowland, I. Effect of colonic bacterial metabolites on Caco-2 cell paracellular permeability in vitro. Nutr. Cancer 2008, 60, 259–266. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; de Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M. Desulfovibrio species are potentially important in regressive autism. Med. Hypotheses 2011, 77, 270–274. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington County, VA, USA, 2013. [Google Scholar]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Cui, J.Y. Review: Mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids. Drug Metab. Dispos. 2015, 43, 1505–1521. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108, 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- De Theije, C.G.M.; Wopereis, H.; Ramadan, M.; van Eijndthoven, T.; Lambert, J.; Knol, J.; Garssen, J.; Kraneveld, A.D.; Oozeer, R. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain. Behav. Immun. 2014, 37, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Mellott, T.J.; Berger-Sweeney, J.E. Effects of postnatal dietary choline supplementation on motor regional brain volume and growth factor expression in a mouse model of Rett syndrome. Brain Res. 2008, 1237, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Aja, S.; Li, Q.; Degano, A.L.; Penati, J.; Zhuo, J.; Roe, C.R.; Ronnett, G.V. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE 2014, 9, e109527. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Weaver, G.A.; Tangel, C.T.; Krause, J.A.; Parfitt, M.M.; Jenkins, P.L.; Rader, J.M.; Lewis, B.A.; Miller, T.L.; Wolin, M.J. Acarbose enhances human colonic butyrate production. J. Nutr. 1997, 27, 717–723. [Google Scholar]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.; Clemente, J.; Burkepile, D.; Vega Thurber, R.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | MECP2 Mutation | Type of Mutation | Disease Phenotype | SGS |
|---|---|---|---|---|
| R1 | p. (R255 *) | Nonsense | Classic | 12 |
| R2 | p. (L383Pfs) | Frame-shift | Classic | 7 |
| R3 | c. (27 + 1_28 − 1)_(1461_?)del | Intragenic deletion | Classic | 5 |
| R4 | p. (R133C) | Missense | Classic | 5 |
| R5 | p. (R270Efs) | Frame-shift | Classic | 10 |
| R6 | p. (R306C) | Missense | Classic | 6 |
| R7 | p. (R294 *) | Nonsense | Classic | 8 |
| R8 | p. (R106W) | Missense | Congenital | 12 |
| Variable | RTT Mean ± SD | Control Mean ± SD | p-Value | Reference Values (LARN) |
|---|---|---|---|---|
| Energy | ||||
| kcals | 1739 ± 680 | 1493 ± 202 | 0.291 | Women: 1790–2320 kcal (AR) |
| Proteins | ||||
| g | 70.3 ± 21.8 | 51.5 ± 12.2 | 0.033 * | 43–50 g (AR) |
| g/kg weight | 2.0 ± 0.6 | 1.0 ± 0.3 | <0.001 *** | 0.71 g/kg bw (AR) |
| % E | 16 ± 4 | 14 ± 3 | 0.087 | 12%–15% E (RI) |
| Animal proteins | ||||
| g | 46.3 ± 19.8 | 31.3 ± 11.9 | 0.062 | |
| g/kg weight | 1.3 ± 0.5 | 0.6 ± 0.3 | <0.001 *** | |
| Vegetal proteins | ||||
| g | 17.9 ± 6 | 20.2 ± 3.3 | 0.151 | |
| g/kg weight | 0.5 ± 0.2 | 0.4 ± 0.1 | 0.180 | |
| Total Carbohydrates | ||||
| g | 181.5 ± 65.9 | 209.0 ± 25.9 | 0.242 | |
| g/1000 kcal | 107.0 ± 22.0 | 140.7 ± 13.9 | 0.001 ** | |
| % E | 43 ± 9 | 56 ± 6 | 0.001 ** | 45%–60% E (RI) |
| Sugars | ||||
| % E | 13 ± 3 | 15 ± 4 | 0.302 | <15% E (SDT) |
| Fiber | ||||
| g | 11.3 ± 5.4 | 14.5 ± 5.3 | 0.225 | |
| g/1000 kcal | 6.5 ± 1.6 | 9.9 ± 3.9 | 0.036 * | 8.4–12.6 g/1000 Kcal |
| Fats | ||||
| g | 65.5 ± 15.0 | 52.9 ± 12.8 | 0.243 | |
| g/1000 kcal | 37.3 ±7.8 | 35.2 ± 5.2 | 0.488 | |
| % E | 33 ± 7 | 32 ± 5 | 0.488 | 20%–35% E (RI) |
| Saturated Fats | ||||
| % E | 8 ± 3 | 11 ± 4 | 0.117 | <10% E (SDT) |
| Cholesterol | ||||
| mg | 244 ± 97 | 156 ± 66 | 0.035 * | <300 mg·day |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borghi, E.; Borgo, F.; Severgnini, M.; Savini, M.N.; Casiraghi, M.C.; Vignoli, A. Rett Syndrome: A Focus on Gut Microbiota. Int. J. Mol. Sci. 2017, 18, 344. https://doi.org/10.3390/ijms18020344
Borghi E, Borgo F, Severgnini M, Savini MN, Casiraghi MC, Vignoli A. Rett Syndrome: A Focus on Gut Microbiota. International Journal of Molecular Sciences. 2017; 18(2):344. https://doi.org/10.3390/ijms18020344
Chicago/Turabian StyleBorghi, Elisa, Francesca Borgo, Marco Severgnini, Miriam Nella Savini, Maria Cristina Casiraghi, and Aglaia Vignoli. 2017. "Rett Syndrome: A Focus on Gut Microbiota" International Journal of Molecular Sciences 18, no. 2: 344. https://doi.org/10.3390/ijms18020344