1. Introduction
Alzheimer’s disease (AD) is the most widespread neurodegenerative disorder in the elderly, characterized by irreversible loss of cortical neurons associated with the accumulation of β-amyloid deposits and neurofibrillary tangles (NFTs) formed by aggregates of hyperphosphorylated Tau protein [
1,
2]. It has been recently shown that oxidative stress and multi-faceted dysfunction of mitochondria take part in the pathogenesis of neurodegeneration [
3,
4]. Indeed, impaired energy metabolism with decreased activity of mitochondrial electron transport chain enzymes (mainly complex I and IV) and citric acid cycle enzymes (pyruvate dehydrogenase complex, α-ketoglutarate dehydrogenase complex) as well as an increased number of mitochondrial DNA mutations, accompanied by augmented mitochondrial reactive oxygen species (ROS) formation have been observed in animal models of neurodegeneration [
4] and in AD patients [
5,
6]. It has been shown that the ROS-dependent lipid peroxidation results in the generation of electrophilic aldehydes such as 4-hydroxy-2-nonenal (4-HNE), malondialdehyde (MDA), and acrolein, which may fervently react with thiols and amino acid residues in proteins. Importantly, it has been observed, that 4-HNE could mediate oxidation of major mitochondrial proteins (e.g., electron transport chain and citric acid enzymes or its cofactors such as lipoic acid) [
7]. Moreover, 4-HNE and accumulation of its protein adducts were implicated in mitochondrial dysfunction and have been postulated to play a role in neurodegenerative processes [
8,
9,
10].
Mitochondrial aldehyde dehydrogenase (ALDH2) plays a main role in the degradation of aldehydes to corresponding non-toxic acids [
11].
N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichloro -benzamide (Alda-1) is a cell-permeable, small molecular weight activator of ALDH2 [
12]. It has been observed that Alda-1 administration shows beneficial action in diseases linked to mitochondrial dysfunction (e.g., acute ischemic injury in the heart [
13], atherosclerosis and hepatic steatosis [
14], diabetes-induced myocardial dysfunction [
15], and parkinsonism [
16]). Recently, we have demonstrated attenuation of depressive- and anxiety-like behaviors by Alda-1 in rat model of depression [
17]. Interestingly, it has also been reported that pharmacological activation of ALDH2 prevents dysfunction of endothelial cells induced by β-amyloid in vitro [
18]. However, there is no information whether prolonged ALDH2 activation by Alda-1 may offer any benefits in animal models of neurodegeneration.
Apolipoprotein E (apoE) is a glycoprotein mainly expressed by brain, liver, spleen, lung, adrenal, ovary, kidney, and muscle. In the brain it is predominantly synthetized by astrocytes and microglia, and involved in cholesterol transport as well as in the development and regeneration of the central nervous system (CNS) [
19]. The presence of epsilon4 allele of apoE (apoE4) was found to predispose to the development of AD, possibly by impairing proteolytic break-down of β-amyloid [
20]. Interestingly, it has also been shown that apoE4 may augment 4-HNE-dependent modification of mitochondrial proteins and cause mitochondrial dysfunction [
21,
22]. The apoE knockout mice (apoE
−/−) are primarily used in atherosclerosis studies since they spontaneously develop hypercholesterolemia, dyslipidemia, and arterial lesions [
23,
24]. They are not considered as a model of full-blown Alzheimer’s disease, however, they have been shown to develop several neurodegenerative changes relevant to the early stages of AD, such as low-grade synaptic and dendrite loss as well as behavioral alterations and deficits in long-term potentiation (LTP) [
25,
26]. Importantly, despite such delicate morphological alterations, some biochemical disturbances—increased ROS formation and lipid peroxidation—have been clearly demonstrated in brains of apoE
−/− mice [
27].
Proteomics represents a valuable tool for the study of mitochondrial pathobiology and may contribute to the better understanding of mitochondrial mechanisms of neurodegenerative disorders [
28]. However, so far the proteomic research has not been used to evaluate the changes in the brain mitochondria in apoE
−/− mice. Thus, the goal of our study was to apply a differential proteomics approach in concert with molecular and morphological techniques to elucidate the changes in the mitochondria-enriched fractions isolated from the frontal cortex and hippocampus of apoE
−/− mice as compared to age-matched wild type animals and in apoE
−/− mice upon treatment with Alda-1.
3. Discussion
Several lines of evidence suggest that altered functioning of apoE may aggravate aldehyde modification of mitochondrial proteins, which may result in mitochondrial dysfunction and neurodegeneration [
5,
6,
21]. In the present work, we focused on the effect of Alda-1 activation of ALDH2—an enzyme responsible for the detoxification of toxic aldehydes—on the protein content of mitochondria-enriched fractions isolated from the frontal cortex and hippocampus of apolipoprotein E knockout mice (apoE
−/−). Moreover, the proteomic approach was supported by molecular and morphological techniques. This is the first report about the molecular and proteomic effects of pharmacological activation of ALDH2 in CNS structures in apoE
−/− mice. The main finding of our study is that Alda-1 administration led to the beneficial changes in the expression of genes and proteins related to neuroplasticity and mitochondrial function.
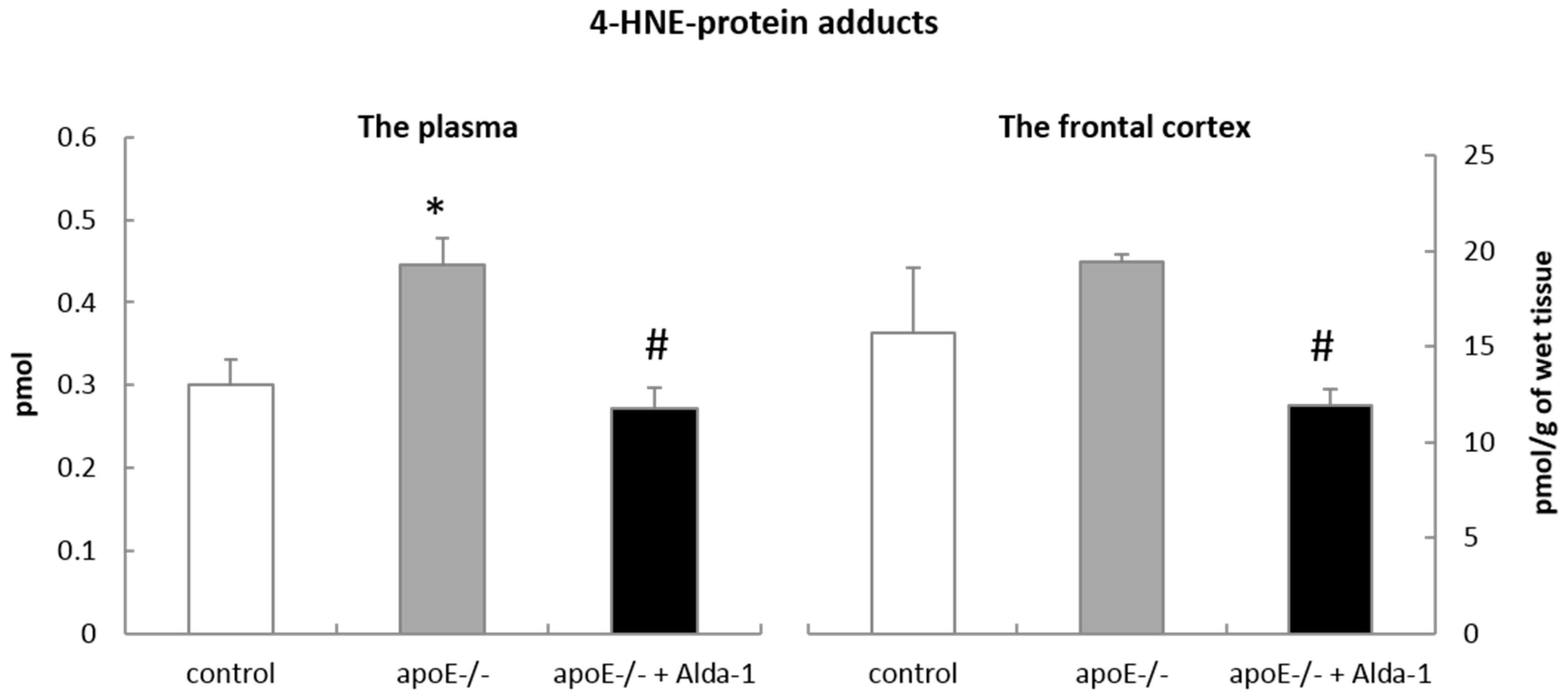
Although the most recognized function of mitochondrial ALDH2 is the degradation of acetaldehyde in the metabolism of ethanol, it can also efficiently oxidize other reactive aldehydes, especially 4-HNE, to non-toxic acids. Indeed, in our hands, Alda-1 caused a significant decrease in 4-HNE-protein content in the plasma and the frontal cortex of apoE−/− mice, which might depend on ALDH2 activation. However, the exact confirmation of Alda-1 influence on ALDH2 activity regarding the metabolism of 4-HNE to related acids does require further investigations including direct measurements of ALDH2 activity and tissue levels of corresponding acids.
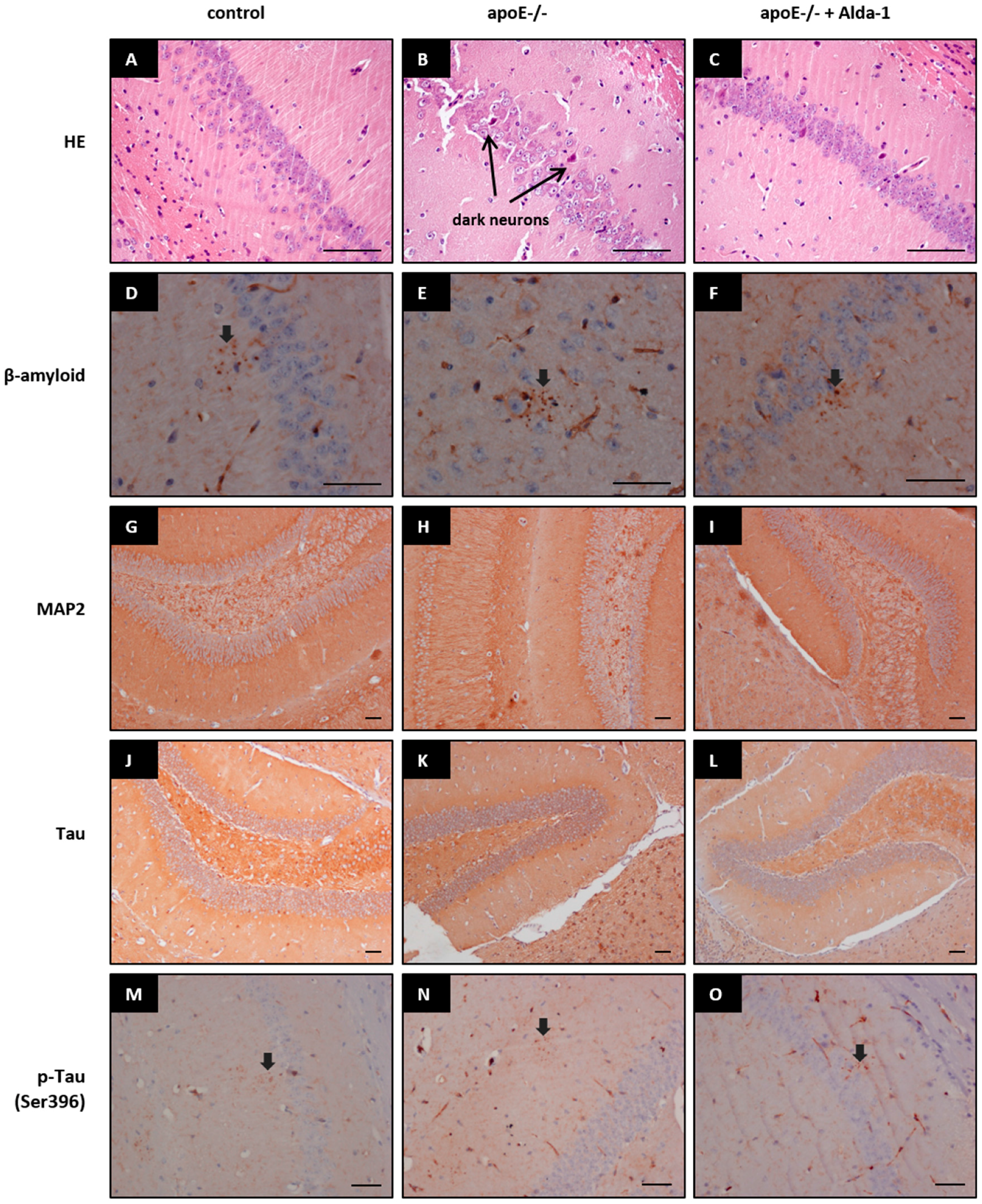
In line with some previous reports [
25,
26], we observed delicate signs of the nonspecific mild tissue injury in brains of apoE
−/− mice. The number of shrunken and deeply stained neurons (“dark neurons”) was higher in brains apoE
−/− mice. It should be noted however, that the presence of such changes may depend on the lot of animals, as they were absent in apoE
−/− mice studied by Anderson et al. [
29]. Moreover, according to some reports, the dark neurons may rather reflect histological artifacts than trustworthy markers of tissue injury [
30]. In keeping with the majority of reports [
25,
26], we did not reveal any changes in tissue contents of β-amyloid, Tau, p-Tau (Ser396), and microtubule-associated protein 2 (MAP2). In our study, we focused on the changes on the levels of the gene/protein expression occurring in the absence of morphologically overt tissue injury.
A growing amount of evidence points to the impairment of neuroplasticity in the pathogenesis of Alzheimer's disease [
31,
32,
33]. Importantly, mitochondria perform a key role in the regulation of neuroplasticity and maintenance of cellular calcium homeostasis [
34]. Brain-derived neurotrophic factor (BDNF) is a main regulator of synaptic plasticity, long-term memory, as well as neuronal survival and differentiation [
35,
36]. Moreover, BDNF is able to streamline mitochondrial respiratory coupling and increase ATP synthesis [
37]. Interestingly, it has been observed that lower levels of BDNF in the plasma and the cerebrospinal fluid were presented in the early stages of AD and mild cognitive impairment (MCI) [
38]. In keeping with these reports, we showed diminished
Bdnf mRNA expression in the FCx and Hp of apoE
−/− mice, as compared to wild type animals. In turn, noggin (NOG) and bone morphogenetic protein 4 (BMP4) have been recognized as potent regulators of neurogenesis. It is known that BMP4 is related to decreased neurogenesis and increased gliogenesis, while expression of BMP4 inhibitor, NOG, is linked to increased neurogenesis [
39] and therefore plays a significant role in the process of learning and memory [
40]. Disturbances in hippocampal neurogenesis have been connected to memory deficits and cognitive impairment in the early stages of AD [
41]. In this study,
Nog mRNA expression was decreased both in the FCx and Hp of apoE
−/− mice. Importantly, Alda-1 administration led to the increase in
Nog mRNA expression in the Hp and the decrease in
Bmp4 mRNA expression in the FCx.
Proteomic analysis using iTRAQ and mass spectrometry methods also revealed changes in protein expression related to the impairment of neuroplasticity in the Hp of apoE
−/− mice: we observed decreased expression of collapsin response mediator protein 1 (CRMP1), heterogeneous nuclear ribonucleoprotein K (hnRNP K), calcium-dependent secretion activator 1 (CAPS-1), and 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNP). CRMP1 is involved in axonal growth and long-term potentiation (LTP) [
42], whereas hnRNP K regulates neurite outgrowth, dendritic spine density, and synaptic plasticity in hippocampal neurons [
43,
44]. In turn, CAPS-1 is a calcium-binding protein engaged in exocytosis of neurotransmitters and neuropeptides, while CNP is required for proper axoglial interactions [
45]. Decreased CNP expression, observed in entorhinal and auditory cortex of AD patients, might suggest the impairment in myelination with subsequent synaptic and cognition loss [
46]. It is worth highlighting that in our study CNP was a common protein differentially expressed in the Hp of control mice, apoE
−/− mice, and apoE
−/− mice treated with Alda-1. Importantly, Alda-1 administration up-regulated CNP expression, which may denote its beneficial action in the early stages of AD.
It is well known that a major hallmark of Alzheimer’s disease is the loss of short and long-term memory [
13]. Interestingly, we noted down-regulation of calcium/calmodulin-dependent protein kinase type IV (CaMK IV) and basigin (CD147) in the Hp and FCx of apoE
−/− mice, respectively. CaMK IV has been demonstrated to participate in the activation of CREB transcription factor, thereby regulating genes responsible for memory and neuronal survival [
47]. It can also perform a protective role against neuronal injury [
48,
49]. In turn, glycoprotein CD147 may regulate the activity of β-amyloid processing enzyme, γ-secretase. It was shown that mice deficient in CD147 showed deficits in memory and spatial learning [
50]. It might well be that the decreased expression of the aforementioned proteins could be linked to learning and memory impairment observed in apoE
−/− mice in the Morris water maze test [
51]. In our case, treatment with Alda-1 resulted in the increase in carbonic anhydrase 2 (CA2) expression in the Hp of apoE
−/− mice. Interestingly, CA2 has been postulated to modulate hippocampal CA1 neuronal network activity and its downregulation may result in impaired cognition. Moreover, use of CA2 activators has been shown to improve learning and memory [
52]. In this regard, Alda-1 activity toward increase of CA2 might represent promising area of research.
A growing amount of evidence points to the presence of mitochondrial dysfunction manifested by energy metabolism impairment and decreased activity of oxidative phosphorylation (OXPHOS) and Krebs cycle enzymes both in animal models of AD and in patients [
5,
6,
53]. Similarly, mostly in the FCx of apoE
−/− mice, we have observed diminished expression of OXPHOS proteins (NADH dehydrogenase flavoprotein 1, cytochrome C oxidase subunit 7A2), proteins supporting OXPHOS (mitochondrial aspartate glutamate carrier 1), and proteins involved in Krebs cycle (succinyl-CoA synthetase subunit α, fumarate hydratase). On the other hand, proteins participating in glycolysis (6-phosphofructokinase type A, hexokinase-1) have been upregulated in the Hp and FCx of apoE
−/− mice. However, we have also revealed changes, which may be considered as contradictory: increased expression of ATP synthase subunit δ and cytochrome b-c1 complex subunit 7 in the Hp and FCx of apoE
−/− mice, respectively. Clearly, further investigations are required to clarify an interpretation of above effects.
Interestingly, in the FCx of apoE
−/− mice we have found decreased mRNA expression of nuclear respiratory factor 1 (
Nrf1), which is a transcription factor responsible for the activation of expression of many factors involved in energy metabolism, cellular respiration, and replication and transcription of mitochondrial DNA [
54]. Diminished expression of peroxisome proliferator-activated receptor gamma coactivator 1-α (
PGC-1α) and downregulation of
Nrf1 mRNA have been attributed to the impairment of mitochondrial biogenesis in transgenic mouse model of AD [
53]. In our case, Alda-1 administration led to the increased expression of proteins that participated in OXPHOS at gene (mitochondrially encoded NADH dehydrogenase 1 (
ND1), cytochrom b (
CYTB)), and protein level (NADH dehydrogenase 1α subcomplex subunit 10) in the FCx of apoE
−/− mice, which may suggest the stimulation of mitochondrial biogenesis.
Recent reports emphasize the role of glutamate-mediated excitotoxicity in the pathogenesis of neurodegenerative diseases, such as Alzheimer’s disease, Huntington disease, and amyotrophic lateral sclerosis (ALS). Our results indicate that proline-rich transmembrane protein 2 (PRRT2) was down-regulated in the FCx of apoE
−/− mice. PRRT2 is a transmembrane protein found in glutamatergic neurons, where it interacts with synaptosomal-associated protein 25 (SNAP25), which inhibits glutamate release and neuronal hyperexcitability. Thus, diminished expression of PRRT2 may lead to decreased interactions with SNAP25, which could potentially result in the increased release of glutamate [
55]. Importantly, Alda-1 administration led to the upregulation of excitatory amino acid transporter 2 (EAAT2) both in the Hp and FCx of apoE
−/− mice. EAAT2 is crucial for tuning of the glutamate neurotransmission by its rapid removal from the synaptic cleft, thus maintaining the levels of glutamate within safe range [
56]. Moreover, it has been shown that reduced EAAT2 function observed in AD is associated with cognitive decline and increased amyloid β production, thus restoring EAAT2 protein function could represent a potential therapeutic approach in AD [
57]. It is tempting to speculate that upregulation of EAAT2 by Alda-1 may denote its beneficial action in the early stages of AD.
It is well recognized that oxidative stress plays a crucial role in the etiology and progression of AD [
58]. We have observed diminished expression of antioxidant proteins, such as glutathione S-transferase Mu 1 (GST 1-1) and thioredoxin (TRX) in the Hp of apoE
−/− mice. GST 1-1 is best known for its ability to detoxify xenobiotics, including 4-HNE, by their conjugation with the reduced form of glutathione (GSH). In turn, TRX acts as an antioxidant by maintaining the reduced form of cellular thiols. Our results are in keeping with previous reports: it has been observed that the lack of TRX2 impairs mitochondrial redox homeostasis and leads to early-onset neurodegeneration [
59]; the decreased activity of GST 1-1 has been detected in the Hp of AD patients [
60].
Interestingly, some changes observed in apoE
−/− mice may represent compensatory mechanisms, for example, Parkinson disease protein 7 homolog (PARK7) was upregulated in the Hp of apoE
−/− mice. PARK7 plays multiple cellular roles as a modulator of gene transcription, an antioxidant protein, and a regulator of mitochondrial functions. It can also regulate the activity of complex I in mitochondria and exert a mitoprotective effect [
61,
62]. Mutations in PARK7 gene are associated with hereditary Parkinson’s disease (PD) [
63]. In addition, the increased level of oxidized (inactive) form of PARK7 has been detected in the brain of AD and PD patients [
64]. The upregulation of PARK7 in the Hp of apoE
−/− mice might be interpreted as a compensatory mechanism, to counteract accelerated oxidative stress and mitochondrial dysfunction presented in the early stages of AD. Similarly, the increase in protein required for synaptic plasticity associated with NMDA receptor signaling-postsynaptic density protein 95 (PSD-95) might represent a compensatory mechanism, supporting synaptic plasticity in the Hp of apoE
−/− mice [
65].
It should also be noted that not all actions of Alda-1 could be interpreted unambiguously in terms of stimulation of neurogenesis, neuroplasticity, and mitogenesis—for example, in our case, Alda-1 upregulated glycogen synthase kinase 3β (
Gsk3b) and proapoptotic factor
Bax mRNA expression in the Hp of apoE
−/− mice. It has been shown that increased expression of the active GSK3B form was associated with the formation of senile plaques and neurofibrillary tangles in AD patients [
66]. Clearly, further research is needed to clarify the physiological meaning of Alda-1 effects on mRNA and protein of
Gsk3b and
Bax in the brain of apoE
−/− mice. The main aim of our study was to elucidate the proteomic and molecular changes in the FCx and Hp of apoE
−/− mice upon treatment with Alda-1. However, our findings cannot determine whether the changes in gene or protein expression are due to direct or indirect effects of Alda-1 administration in apoE
−/− mice. Moreover, the obvious limitation of our study is a lack of corresponding behavioral data, which would significantly increase the impact of the biochemical/molecular and proteomic findings. Clearly, correlation between molecular/proteomic and functional data requires further research.


 ,
, 


{kind=link}
{kind=link}
{kind=link}