
Exome Sequencing in a Family with Luminal-Type Breast Cancer Underpinned by Variation in the Methylation Pathway

Abstract
:1. Introduction
2. Results
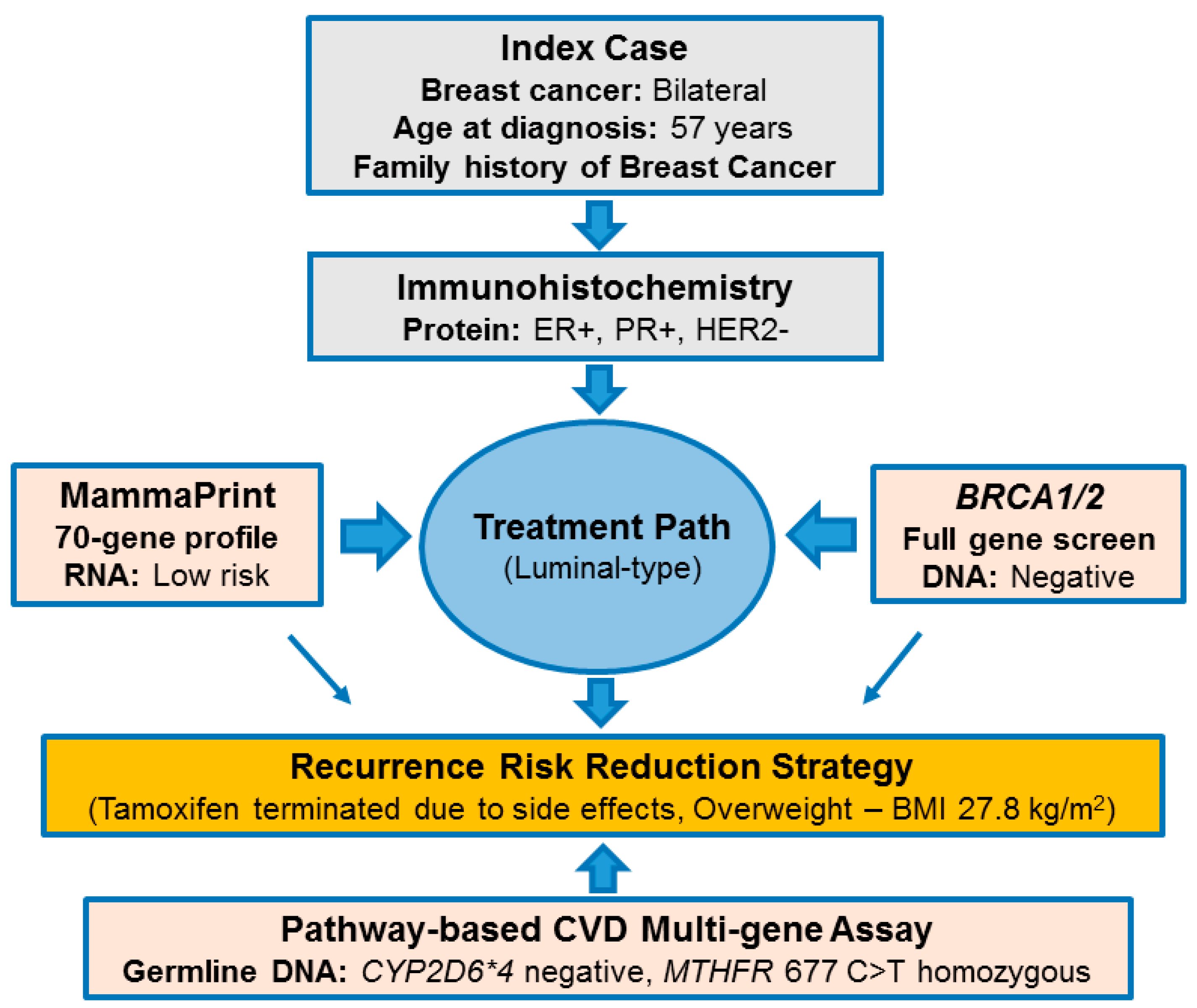
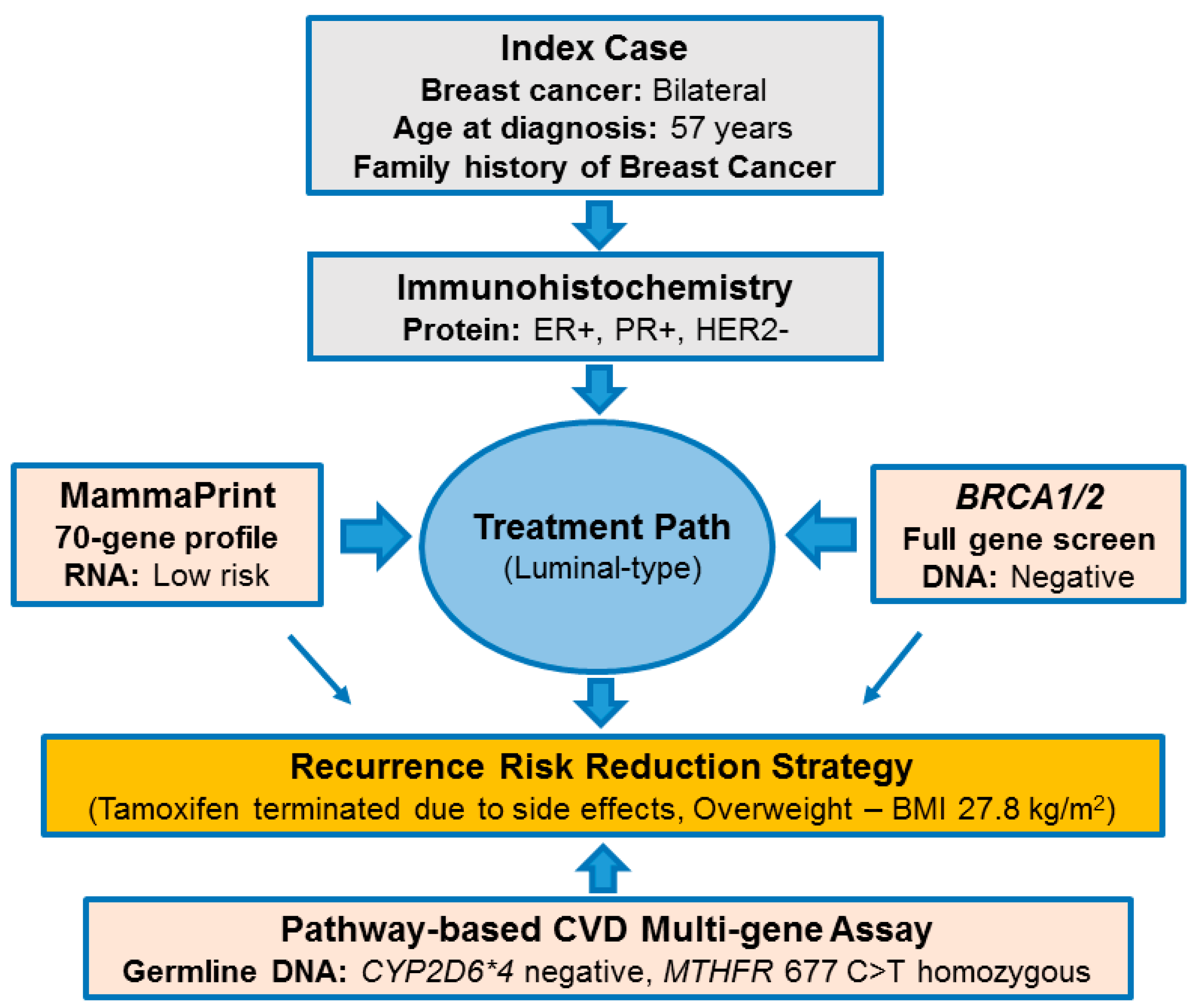
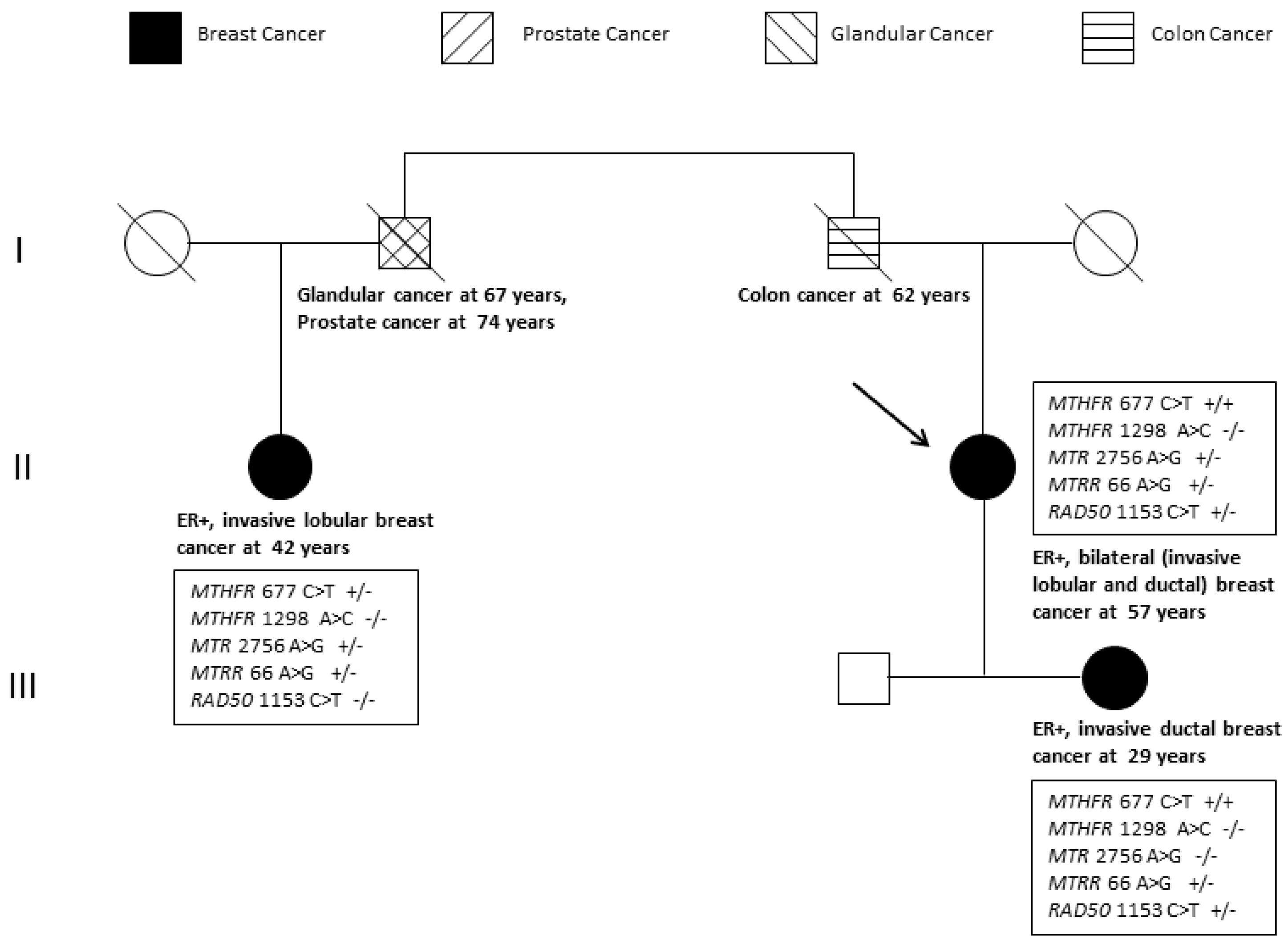
2.1. Pathology-Supported Genetic Testing (PSGT)
2.2. Comprehensive Cancer Panel Screen Using Whole Exome Sequencing
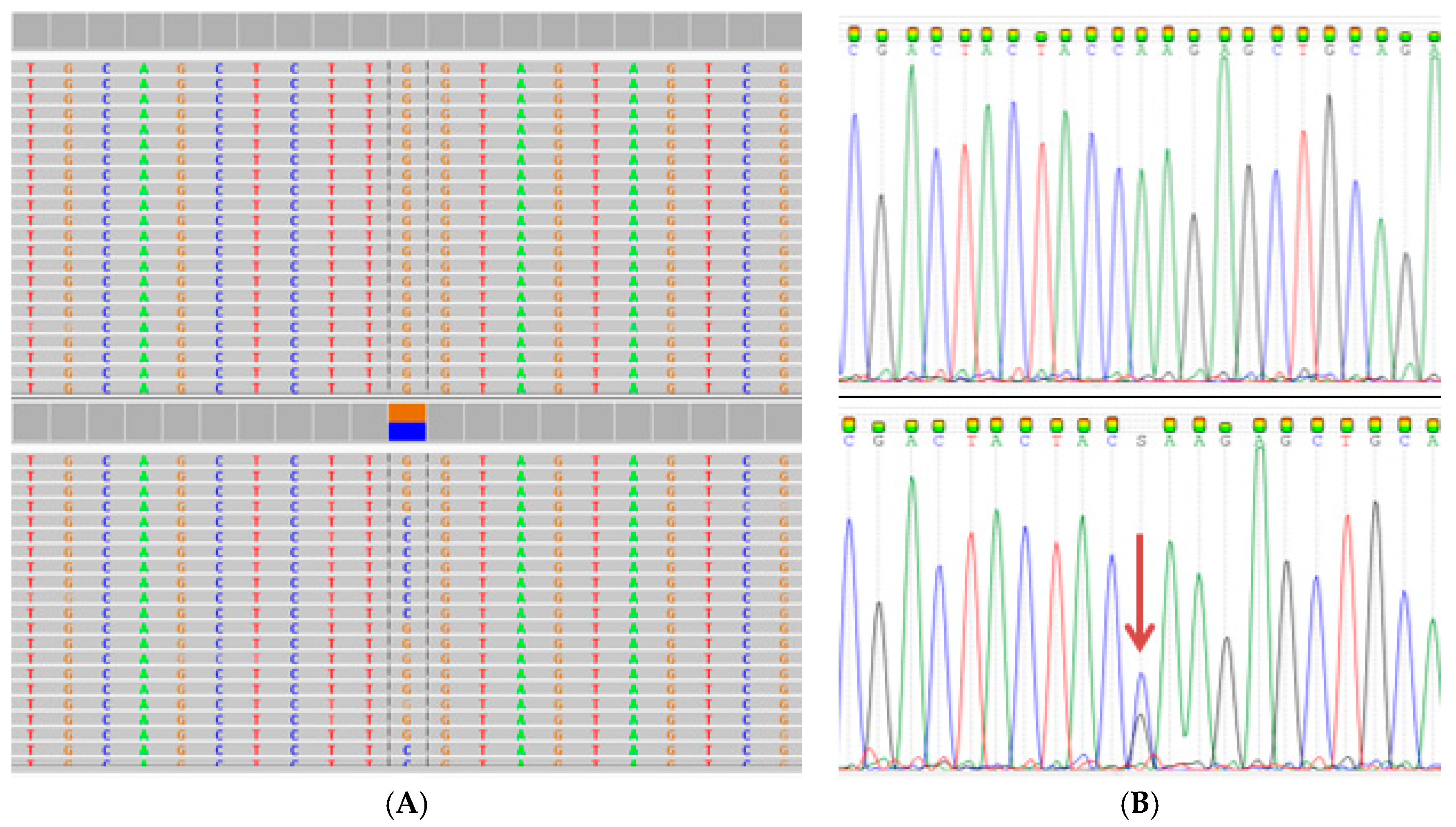
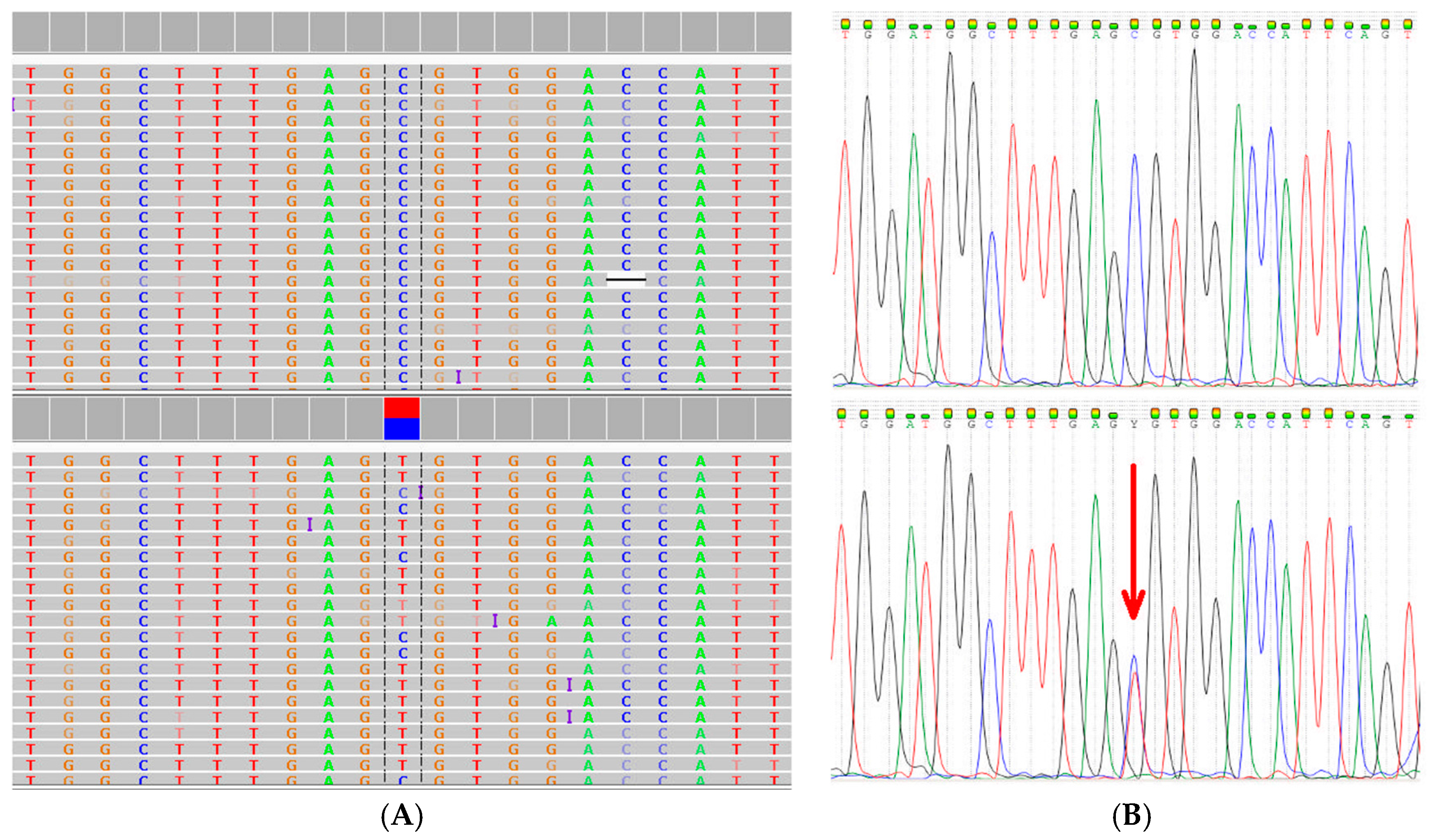
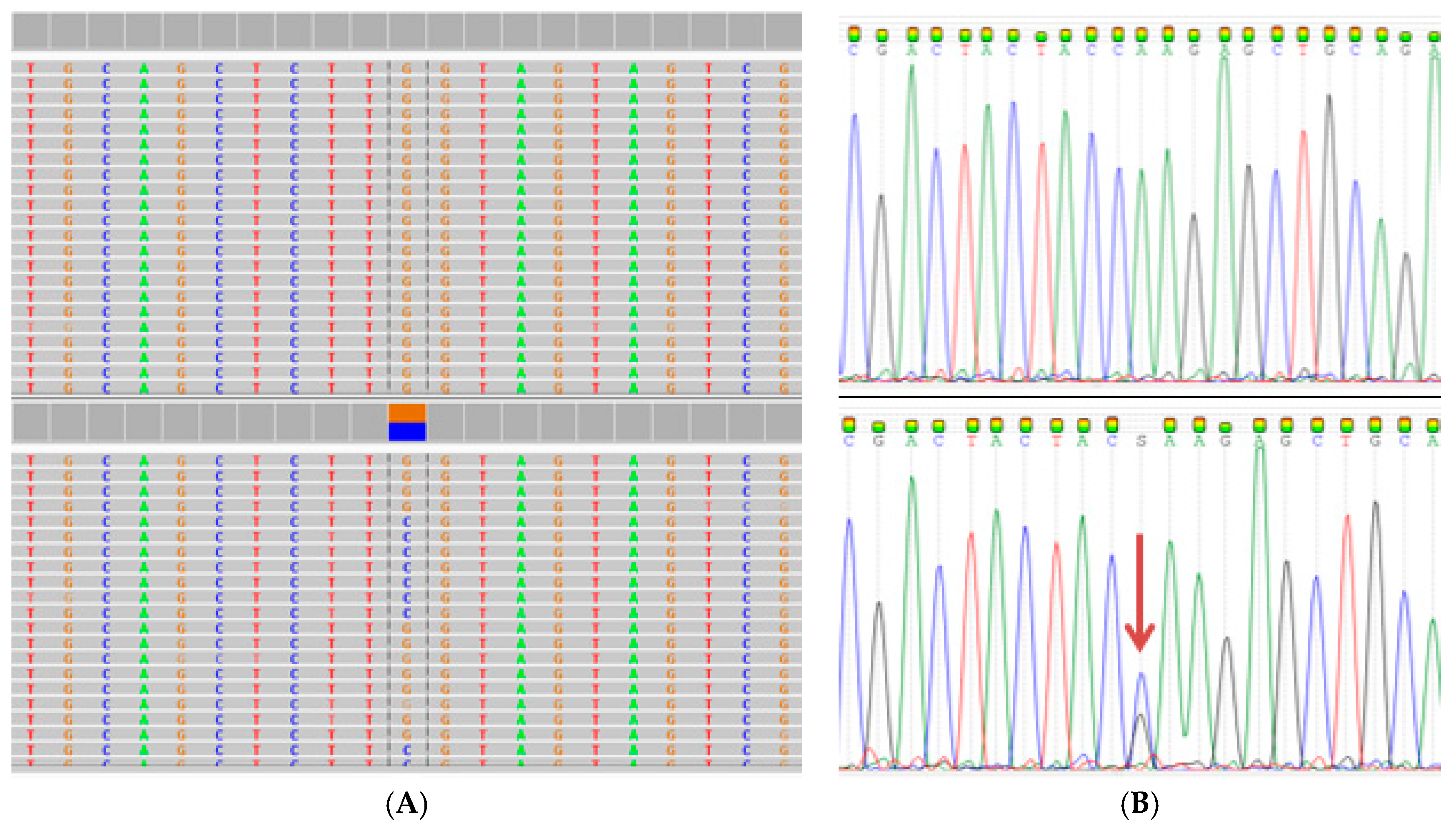
2.3. Extended Mutation Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Study Population
4.3. DNA Extraction
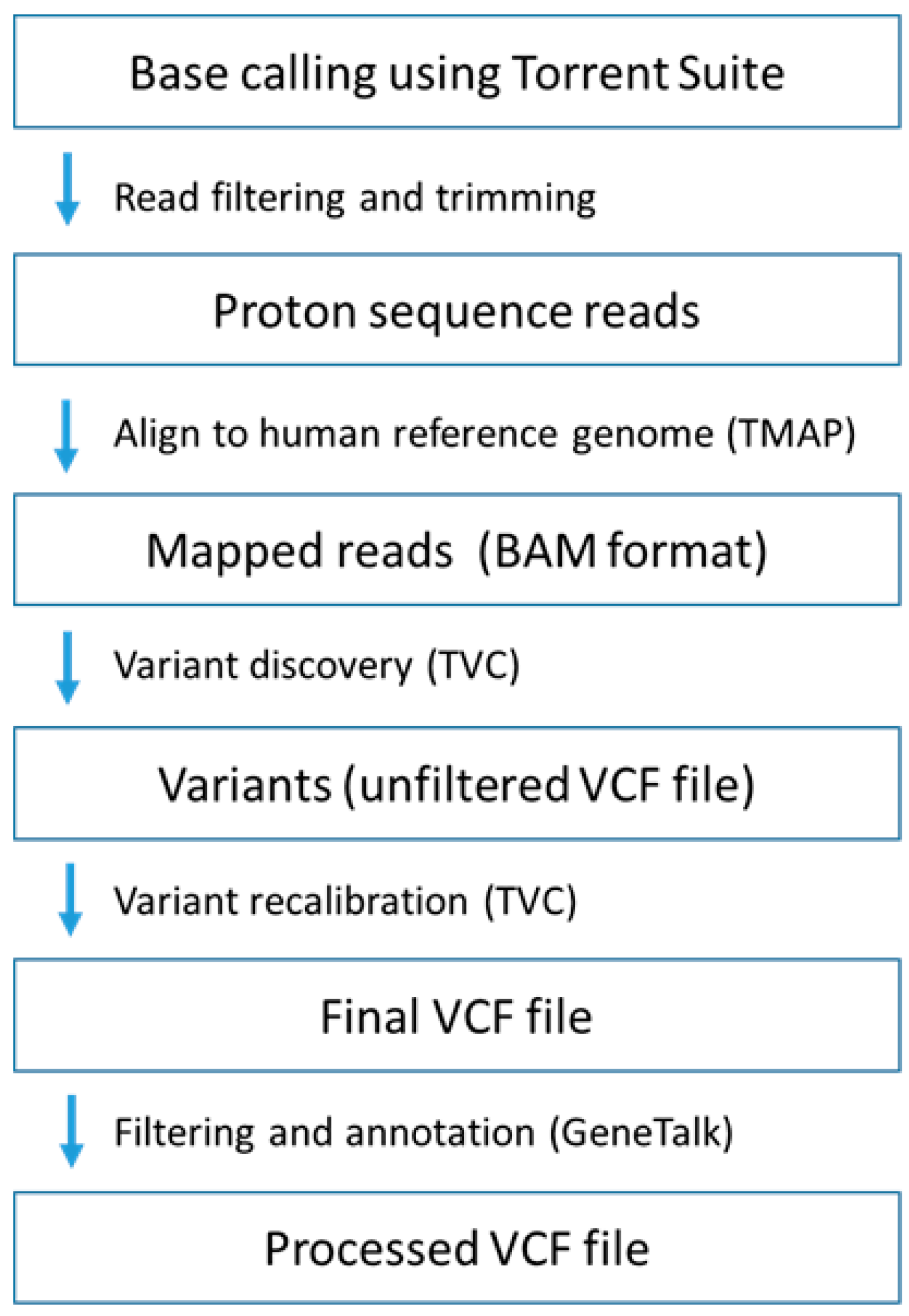
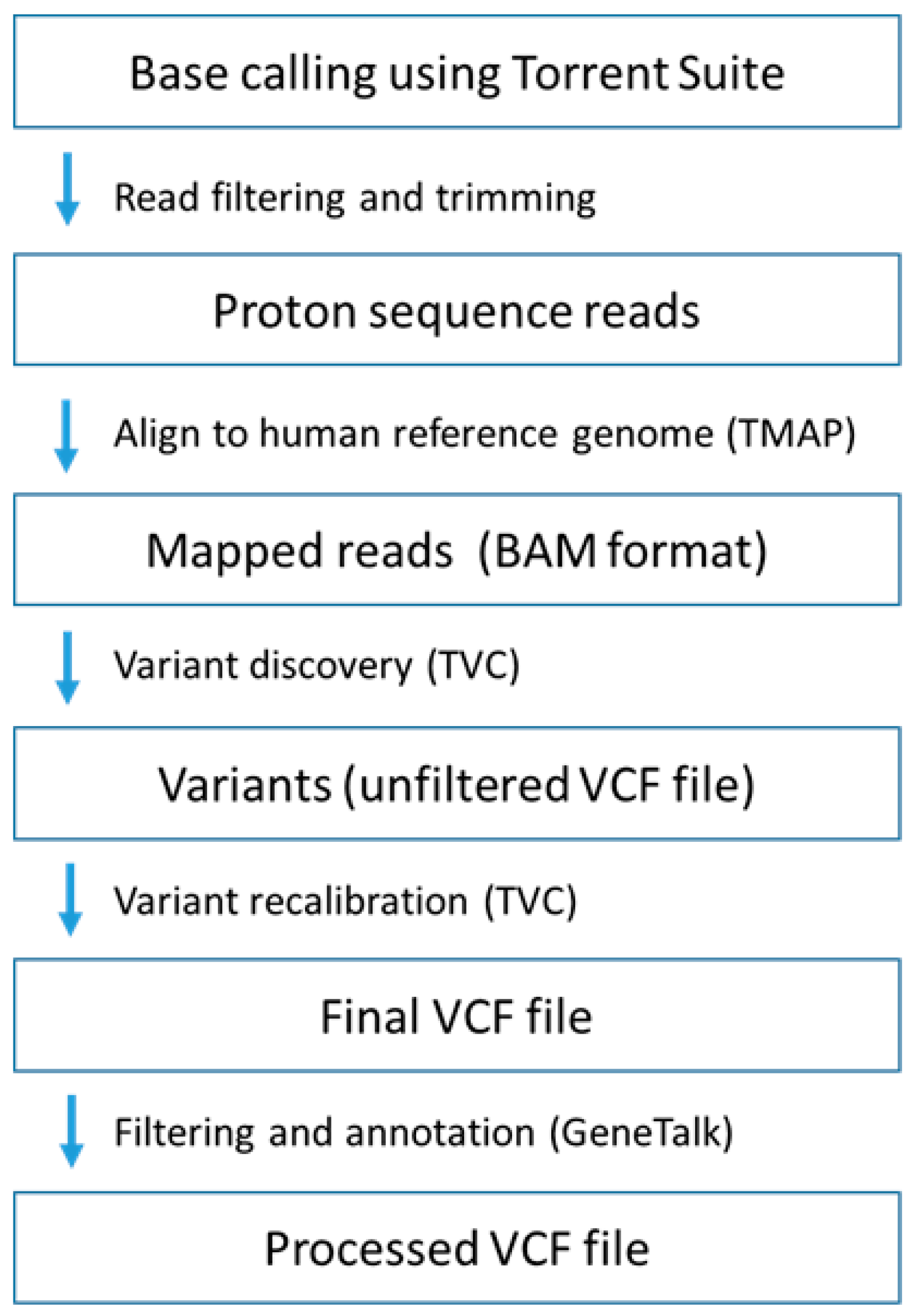
4.4. Whole Exome Sequencing
4.5. Sanger Sequencing and Real-Time Polymerase Chain Reaction (PCR)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABI | Applied Biosystems |
| BAM | Binary alignment/map |
| BMI | Body mass index |
| CEU-MARS | Caucasian major allele reference sequence |
| CVD | Cardiovascular disease |
| CYP | Cytochrome P450 |
| dnSNP | Database of Single Nucleotide Polymorphisms |
| ER | Estrogen receptor |
| hg | Human reference genome |
| HRT | Hormone replacement therapy |
| IGV | Integrative Genome Viewer |
| MINDACT | Microarray In Node negative Disease may Avoid Chemo Therapy |
| MRC | Medical Research Council |
| MRE11 | Meiotic recombination 11 |
| MTHFR | Methylene tetrahydrofolate reductase |
| MTR | Methionine synthase |
| MTRR | Methionine synthase reductase |
| MUC1 | Mucin 1 |
| NBN | Nibrin |
| NGS | Next generation sequencing |
| NRF | National Research Foundation |
| PCR | Polymerase chain reaction |
| PR | Progesterone receptor |
| PSGT | Pathology-supported genetic testing |
| RAD50 | Double strand break repair protein |
| SAM | Sequence alignment/map |
| SHIP | Strategic Health Innovation Partnerships |
| SNPs | Single nucleotide polymorphisms |
| TMAP | Torrent mapping alignment |
| TVC | Torrent Variant Caller |
| VCF | Variant call files |
| VUS | Variant of uncertain clinical significance |
| WES | Whole exome sequencing |
References
- Nordgard, S.H.; Alnaes, G.I.; Hihn, B.; Lingjaerde, O.C.; Liestøl, K.; Tsalenko, A.; Sørlie, T.; Lønning, P.E.; Børresen-Dale, A.L.; Kristensen, V.N. Pathway based analysis of SNPs with relevance to 5-FU therapy: Relation to intratumoral mRNA expression and survival. Int. J. Cancer 2008, 123, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Babyshkina, N.; Malinovskaya, E.; Nazarenko, M.; Koval, M.; Gervas, P.; Potapova, O.; Slonimskaya, E.; Cherdyntseva, N. The effect of folate-related SNPs on clinicopathological features, response to neoadjuvant treatment and survival in pre- and postmenopausal breast cancer patients. Gene 2013, 518, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Seymour, H.J.; Wainstein, T.; Macaulay, S.; Haw, T.; Krause, A. Breast cancer in high-risk Afrikaner families: Is BRCA founder mutation testing sufficient? S. Afr. Med. J. 2016, 106, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Kotze, M.J. Application of advanced molecular technology in the diagnosis and management of genetic disorders in South Africa. S. Afr. Med. J. 2016, 106, S114–S118. [Google Scholar] [CrossRef] [PubMed]
- Bahassi, E.M.; Stambrook, P.J. Next-generation sequencing technologies: Breaking the sound barrier of human genetics. Mutagenesis 2014, 29, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Masser, D.R.; Stanford, D.R.; Freeman, W.M. Targeted DNA methylation analysis by next-generation sequencing. J. Vis. Exp. 2015, 96, 52488. [Google Scholar] [CrossRef] [PubMed]
- Kotze, M.J.; Lückhoff, H.K.; Peeters, A.V.; Baatjes, K.; Schoeman, M.; van der Merwe, L.; Grant, K.A.; Fisher, L.R.; van der Merwe, N.; Pretorius, J.; et al. Genomic medicine and risk prediction across the disease spectrum. Crit. Rev. Clin. Lab. Sci. 2015, 52, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Van der Groep, P.; van der Wall, E.; van Diest, P.J. Pathology of hereditary breast cancer. Cell. Oncol. 2011, 34, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.Y.; Mozersky, J.; Cook-Deegan, R. Variants of uncertain significance in BRCA: A harbinger of ethical and policy issues to come? Genome Med. 2014, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.A.; Apffelstaed, J.P.; Wright, C.; Myburgh, E.; Pienaar, R.; De Klerk, M.; Kotze, M.J. MammaPrint pre-screen algorithm (MPA) reduces chemotherapy in patients with early-stage breast cancer. S. Afr. Med. J. 2013, 103, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.Y.; Brain, E.; Causeret, S.; DeLorenzi, M.; et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.J.; Thomassen, M.; Tan, Q.; Lænkholm, A.V.; Bak, M.; Sørensen, K.P.; Andersen, M.K.; Kruse, T.A.; Gerdes, A.M. RNA profiling reveals familial aggregation of molecular subtypes in non-BRCA1/2 breast cancer families. BMC Med. Genom. 2014, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.; Goldschmidt, R.; Turk, M.; Wesseling, J.; Stork-Sloots, L.; de Snoo, F.; Cristofanilli, M. Molecular subtyping improves diagnostic stratification of patients with primary breast cancer into prognostically defined risk groups. Breast Cancer Res. Treat. 2015, 154, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Myburgh, E.J.; Langenhoven, L.; Grant, K.A.; van der Merwe, L.; Kotze, M.J. Clinical overestimation of HER2 positivity in early estrogen and progesterone receptor–positive breast cancer and the value of molecular subtyping using BluePrint. J. Glob. Oncol. 2016. [Google Scholar] [CrossRef]
- Naushad, S.M.; Pavani, A.; Rupasree, Y.; Divyya, S.; Deepti, S.; Digumarti, R.R.; Gottumukkala, S.R.; Prayaga, A.; Kutala, V.K. Association of aberrations in one-carbon metabolism with molecular phenotype and grade of breast cancer. Mol. Carcinog. 2012, 51, E32–E41. [Google Scholar] [CrossRef] [PubMed]
- Ghoussaini, M.; Pharoah, P.D.P.; Easton, D.F. Inherited genetic susceptibility to breast cancer—The beginning of the end or the end of the beginning? Am. J. Pathol. 2013, 183, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, L.; Panchal, S.; Goodwin, P. Prognosis of BRCA-associated breast cancer: A summary of evidence. Breast Cancer Res. Treat. 2010, 119, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Bijl, M.J.; van Schaik, R.H.; Lammers, L.A.; Hofman, A.; Vulto, A.G.; van Gelder, T.; Stricker, B.H.; Visser, L.E. The CYP2D6*4 polymorphism affects breast cancer survival in tamoxifen users. Breast Cancer Res. Treat. 2009, 18, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Umegaki, K.; Higuchi, M.; Thomas, P.; Fenech, M. Methylenetetrahydrofolate reductase C677T polymorphism, folic acid and riboflavin are important determinants of genome stability in cultured human lymphocytes. J. Nutr. 2004, 134, 48–56. [Google Scholar] [PubMed]
- Xu, X.; Gammon, M.D.; Wetmur, J.G.; Bradshaw, P.T.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M.; Chen, J. B-vitamin intake, one-carbon metabolism, and survival in a population-based study of women with breast cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.F.; Avila, S.; Fraga, M.F.; Pollan, M.; Capella, G.; Peinado, M.A.; Sanchez-Cespedes, M.; Herman, J.G.; Esteller, M. Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in normal tissues and human primary tumours. Cancer Res. 2002, 62, 4519–4524. [Google Scholar] [PubMed]
- Wishart, D.S. Is cancer a genetic disease or a metabolic disease? EBioMedicine 2015, 2, 478–479. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.B. Folate, methyl-related nutrients, alcohol, and the MTHFR 677 C>T polymorphism affect cancer risk: Intake recommendations. J. Nutr. 2003, 133, 3748S–3753S. [Google Scholar] [PubMed]
- McEligor, A.J.; Ziogas, A.; Pfeiffer, C.M.; Fazil, Z.; Anton-Culver, H. The association between circulating total folate and folate vitamers with overall survival after postmenopausal breast cancer diagnosis. Nutr. Cancer 2015, 67, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Delport, D.; Schoeman, R.; van der Merwe, N.; van der Merwe, L.; Fisher, L.R.; Geiger, D.; Kotze, M.J. Significance of dietary folate intake, homocysteine levels and MTHFR 677 C>T genotyping in South African patients diagnosed with depression: Test development for clinical application. Metab. Brain Dis. 2014, 29, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, N.; Bouwens, C.S.; Pienaar, R.; van der Merwe, L.; Yako, Y.Y.; Geiger, D.H.; Kotze, M.J. CYP2D6 genotyping and use of antidepressants in breast cancer patients: Test development for clinical application. Metab. Brain Dis. 2012, 27, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.D.; Yawitch, T.M.; van der Merwe, N.C.; van den Berg, H.J.; Dreyer, G.; van Rensburg, E.J. BRCA1 mutations in South African breast and/or ovarian cancer families: Evidence of a novel founder mutation in Afrikaner families. Int. J. Cancer 2004, 110, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, N.C.; Hamel, N.; Schneider, S.R.; Apffelstaedt, J.P.; Wijnen, J.T.; Foulkes, W.D. A founder BRCA2 mutation in non-Afrikaner breast cancer patients of the Western Cape of South Africa. Clin. Genet. 2012, 81, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, N. Development and Application of a Pathology Supported Pharmacogenetic Test for Improved Clinical Management of South African Patients with Breast Cancer and Associated Comorbidities. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, 2016. [Google Scholar]
- Phipps, A.I.; Buist, D.S.; Malone, K.E.; Barlow, W.E.; Porter, P.L.; Kerlikowske, K.; Li, C.I. Family history of breast cancer in first-degree relatives and triple-negative breast cancer risk. Breast Cancer Res. Treat. 2011, 126, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.I.; Buist, D.S.; Malone, K.E.; Barlow, W.E.; Porter, P.L.; Kerlikowske, K.; O’Meara, E.S.; Li, C.I. Breast density, body mass index, and risk of tumour marker-defined subtypes of breast cancer. Ann. Epidemiol. 2012, 22, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Arieas, E.T.; Zhang, Z.; Defilippis, A.; Tarpinian, K.; Jeter, S.; Nguyen, A.; Henry, N.L.; Flockhart, D.A.; Hayes, D.F.; et al. Comparison of breast cancer recurrence risk and cardiovascular disease incidence risk among postmenopausal women with breast cancer. Breast Cancer Res. Treat. 2012, 131, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Li, C.I.; Daling, J.R.; Porter, P.L.; Tang, M.T.; Malone, K.E. Relationship between potentially modifiable lifestyle factors and risk of second primary contralateral breast cancer among women diagnosed with estrogen receptor-positive invasive breast cancer. J. Clin. Oncol. 2009, 27, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Integrative Genomics Viewer. Available online: http://software.broadinstitute.org/software/igv/ (accessed on 31 July 2015).
- Aloraifi, F.; Boland, M.R.; Green, A.J.; Geraghty, J.G. Gene analysis techniques and susceptibility gene discovery in non-BRCA1/BRCA2 familial breast cancer. Surg. Oncol. 2015, 24, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, W.; Tomlinson, I. Rare genetic variants and the risk of cancer. Curr. Opin. Genet. Dev. 2010, 20, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Damiola, F.; Pertesi, M.; Oliver, J.; Le Calvez-Kelm, F.; Voegele, C.; Young, E.L.; Robinot, N.; Forey, N.; Durand, G.; Vallée, M.P.; et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: Results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 2014, 16, R58. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, K.; Rapakko, K.; Karppinen, S.M.; Erkko, H.; Knuutila, S.; Lundán, T.; Mannermaa, A.; Børresen-Dale, A.L.; Borg, A.; Barkardottir, R.B.; et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 2006, 27, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Tommiska, J.; Oplustilova, L.; Aaltonen, K.; Tamminen, A.; Heikkinen, T.; Mistrik, M.; Aittomäki, K.; Blomqvist, C.; Heikkilä, P. Aberrations of the MRE11–RAD50–NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer predisposing gene. Mol. Oncol. 2008, 2, 296–316. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Nishida, Y.; Tsutsumida, H.; Hamada, T.; Goto, M.; Higashi, M.; Nomoto, M.; Yonezawa, S. MUC1 expression is regulated by DNA methylation and histone H3 lysine 9 modification in cancer cells. Cancer Res. 2008, 68, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.; Gnirke, A.; Jaffe, D.B.; Barešová, V.; Pochet, N.; Blumenstiel, B.; Ye, C.; Aird, D.; Stevens, C.; Robinson, J.T. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat. Genet. 2013, 45, 299–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholtz, C.L.; Odendaal, H.J.; Thiart, R.; Loubser, L.; Hillermann, R.; Delport, R.; Vermaak, W.J.; Kotze, M.J. Analysis of two mutations in the MTHFR gene associated with mild hyperhomocysteinaemia: Heterogeneous distribution in the South African population. S. Afr. Med. J. 2002, 92, 464–467. [Google Scholar] [PubMed]
- Lambrinoudaki, I.; Papadimitriou, D.; Kaparos, G.; Rizos, D.; Panoulis, C.; Deligeoroglou, E.; Alexandrou, A.; Auguolea, A.; Apostolakis, M.; Creatsa, M.; et al. MTHFR C677 T polymorphism modifies the effect of HRT on metabolic parameters in postmenopausal women. Climacteric 2013, 16, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.; Pangilinan, F.; Molloy, A.M.; Shane, B.; Scott, J.M.; Ueland, P.M.; Mills, J.L.; Kirke, P.N.; Sethupathy, P.; Brody, L.C. Bioinformatic and genetic association analysis of microRNA target sites in one-carbon metabolism genes. PLoS ONE 2011, 6, e21851. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Kremer, D.; Tobi, E.W.; Boomsma, D.I.; Slagboom, P.E. Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Hum. Mol. Genet. 2007, 16, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, A.C.; Spurdle, A.B.; Sinilnikova, O.M.; Healey, S.; Pooley, K.A.; Schmutzler, R.K.; Versmold, B.; Engel, C.; Meindl, A.; Arnold, N.; et al. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am. J. Hum. Genet. 2008, 82, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.; Antoniou, A.C.; Easton, D.F.; Ponder, B.A. Polygenes, risk prediction, and targeted prevention of breast cancer. N. Engl. J. Med. 2008, 358, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Ricks-Santi, L.J.; Nie, J.; Marian, C.; Ochs-Balcom, H.M.; Trevisan, M.; Edge, S.B.; Kanaan, Y.; Freudenheim, J.L.; Shields, P.G. BRCA1 Polymorphisms and breast cancer epidemiology in the Western New York Exposures and Breast Cancer [WEB] Study. Genet. Epidemiol. 2013, 37, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Pepe, C.; Guidugli, L.; Sensi, E.; Aretini, P.; D’Andrea, E.; Montagna, M.; Manoukian, S.; Ottini, L.; Radice, P.; Viel, A.; et al. Methyl group metabolism gene polymorphisms as modifiers of breast cancer risk in Italian BRCA1/2 carriers. Breast Cancer Res. Treat. 2007, 103, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Easton, D.F. Models of genetic susceptibility to breast cancer. Oncogene 2006, 25, 5898–5905. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Aznarez, F.J.; Fernandez, V.; Pita, G.; Peterlongo, P.; Dominguez, O.; de la Hoya, M.; Duran, M.; Osorio, A.; Moreno, L.; Gonzalez-Neira, A.; et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS ONE 2003, 8, e55681. [Google Scholar] [CrossRef] [PubMed]
- Macis, D.; Maisonneuve, P.; Johansson, H.; Bonanni, B.; Botteri, E.; Iodice, S.; Santillo, B.; Penco, S.; Gucciardo, G.; D’Aiuto, G.; et al. Methylenetetrahydrofolate reductase (MTHFR) and breast cancer risk: A nested-case-control study and a pooled meta-analysis. Breast Cancer Res. Treat. 2007, 106, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Weiwei, Z.; Liping, C.; Dequan, L. Association between dietary intake of folate, vitamin B6, B12 & MTHFR, MTR Genotype and breast cancer risk. Pak. J. Med. Sci. 2014, 30, 106–110. [Google Scholar] [PubMed]
- Sangrajrang, S.; Sato, Y.; Sakamoto, H.; Ohnami, S.; Khuhaprema, T.; Yoshida, T. Genetic polymorphisms in folate and alcohol metabolism and breast cancer risk: A case-control study in Thai women. Breast Cancer Res. Treat. 2010, 123, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Chen, Y.; Ayub, Q.; Huang, N.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Stenson, P.D.; Cooper, D.N.; et al. Deleterious- and disease-allele prevalence in healthy individuals: Insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012, 91, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Kotze, M.J.; van Velden, D.P.; Botha, K.; Badenhorst, C.H.; Avenant, H.J.; van Rensburg, S.; Cronje, F. Pathology-supported genetic testing directed at shared disease pathways for optimized health in later life. Personal. Med. 2013, 10, 497–507. [Google Scholar] [CrossRef]
- Kotze, M.J.; Thiart, R. Genetics of dyslipidaemia. CME J. 2003, 21, 399–402. [Google Scholar]
- Github. Available online: https://github.com/iontorrent/TS/tree/master/Analysis/TMAP (accessed on 7 April 2015).
- Dewey, F.E.; Chen, R.; Cordero, S.P.; Ormond, K.E.; Caleshu, C.; Karczewski, K.J.; Whirl-Carrillo, M.; Wheeler, M.T.; Dudley, J.T.; Byrnes, J.K.; et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011, 7, e1002280. [Google Scholar] [CrossRef] [PubMed]
- GeneTalk. Available online: https://www.gene-talk.de/ (accessed on 12 April 2015).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioinformatics Tool Applied | Predicted Biological Effect |
|---|---|
| Sorting Intolerant From Tolerant (SIFT) | Damaging (rank score = 0.7209) |
| Polyphen | Benign (score = 0.339) |
| Variant Effect Predictor | Moderate impact |
| MutationTaster | Disease causing (rank score = 0.8103) |
| MetalR | Tolerated (rank score = 0.2311) |
| Provean | Neutral (rank score = 0.4681) |
| Likelihood Ratio Test | Deleterious (rank score = 0.5373) |
| Variables | ER-Negative | ER-Positive | |
|---|---|---|---|
| Median (Range) | Number | 49 | 115 * |
| Age (median, range) | 164 | 49 (30–77) | 54 (31–83) |
| Body mass index | 146 | 25 (17–47) | 26 (17–41) |
| Count (Percentage) | |||
| Ethnicity † | 164 | 22 (45) | 38 (33) |
| Family history of cancer | 163 | 30 (61) | 59 (52) |
| Hormone replacement therapy | 163 | 9 (19) | 15 (13) |
| Oral contraceptives | 163 | 20 (42) | 32 (28) |
| Current smoking | 160 | 17 (35) | 34 (30) |
| High alcohol consumption | 156 | 25 (52) | 62 (57) |
| Gene | dbSNP ID# | Primer | Oligonucleotide Primers for Sanger Sequencing | Size (bp) | TaqMan Assay ID Numbers |
|---|---|---|---|---|---|
| MTHFR | rs1801133 | Forward | ATCCCTCGCCTTGAACA | 256 | C_1202889_20 |
| Reverse | TCACCTGGATGGGAAAGAT | ||||
| MTR | rs1805087 | Forward | GAACATCCCAAGCCCAC | 595 | C_12005959_10 |
| Reverse | CACCTGTTTCCCTGCTG | ||||
| MTRR | rs1801394 | Forward | GTTTCATTCGTACACTCTCC | 616 | C_3068176_10 |
| Reverse | CAGCATATGCTACTTCTGTC | ||||
| RAD50 | rs139372231 | Forward | ATCCACATGCTCAGGGGTAC | 528 | C_171053490_10 |
| Reverse | GCCAAAATGGAGTCCAACC | ||||
| MUC1 | rs773704188 | Forward | ATTCCCAGCCACCACTCTGA | 493 | Not available |
| Reverse | CCCAACCTTAAGTGCACCAGT |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Merwe, N.; Peeters, A.V.; Pienaar, F.M.; Bezuidenhout, J.; Van Rensburg, S.J.; Kotze, M.J. Exome Sequencing in a Family with Luminal-Type Breast Cancer Underpinned by Variation in the Methylation Pathway. Int. J. Mol. Sci. 2017, 18, 467. https://doi.org/10.3390/ijms18020467
Van der Merwe N, Peeters AV, Pienaar FM, Bezuidenhout J, Van Rensburg SJ, Kotze MJ. Exome Sequencing in a Family with Luminal-Type Breast Cancer Underpinned by Variation in the Methylation Pathway. International Journal of Molecular Sciences. 2017; 18(2):467. https://doi.org/10.3390/ijms18020467
Chicago/Turabian StyleVan der Merwe, Nicole, Armand V. Peeters, Fredrieka M. Pienaar, Juanita Bezuidenhout, Susan J. Van Rensburg, and Maritha J. Kotze. 2017. "Exome Sequencing in a Family with Luminal-Type Breast Cancer Underpinned by Variation in the Methylation Pathway" International Journal of Molecular Sciences 18, no. 2: 467. https://doi.org/10.3390/ijms18020467