
Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions

Abstract
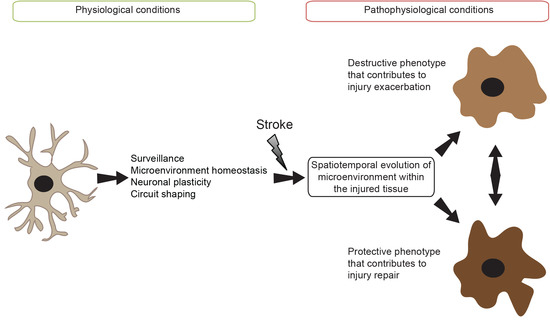
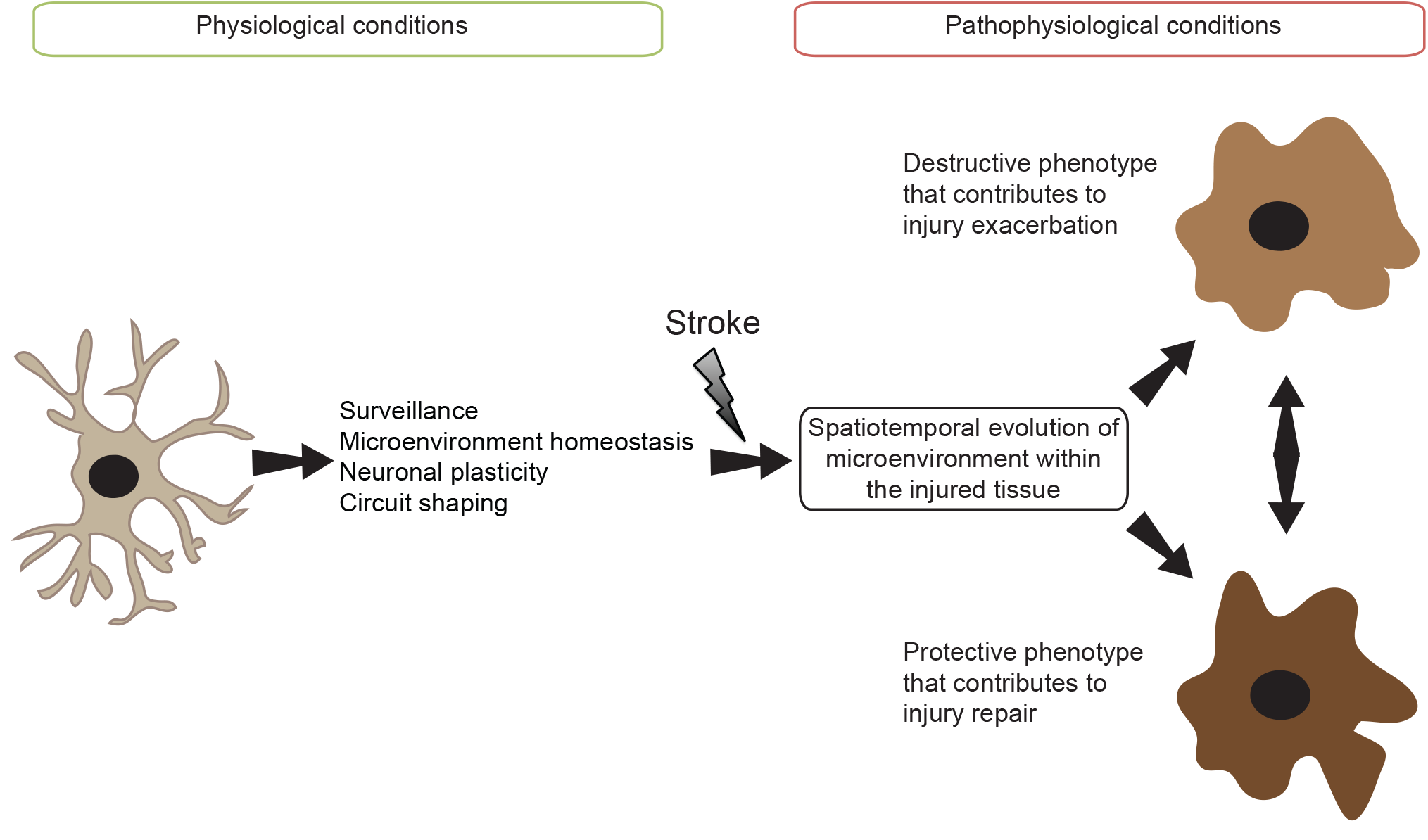
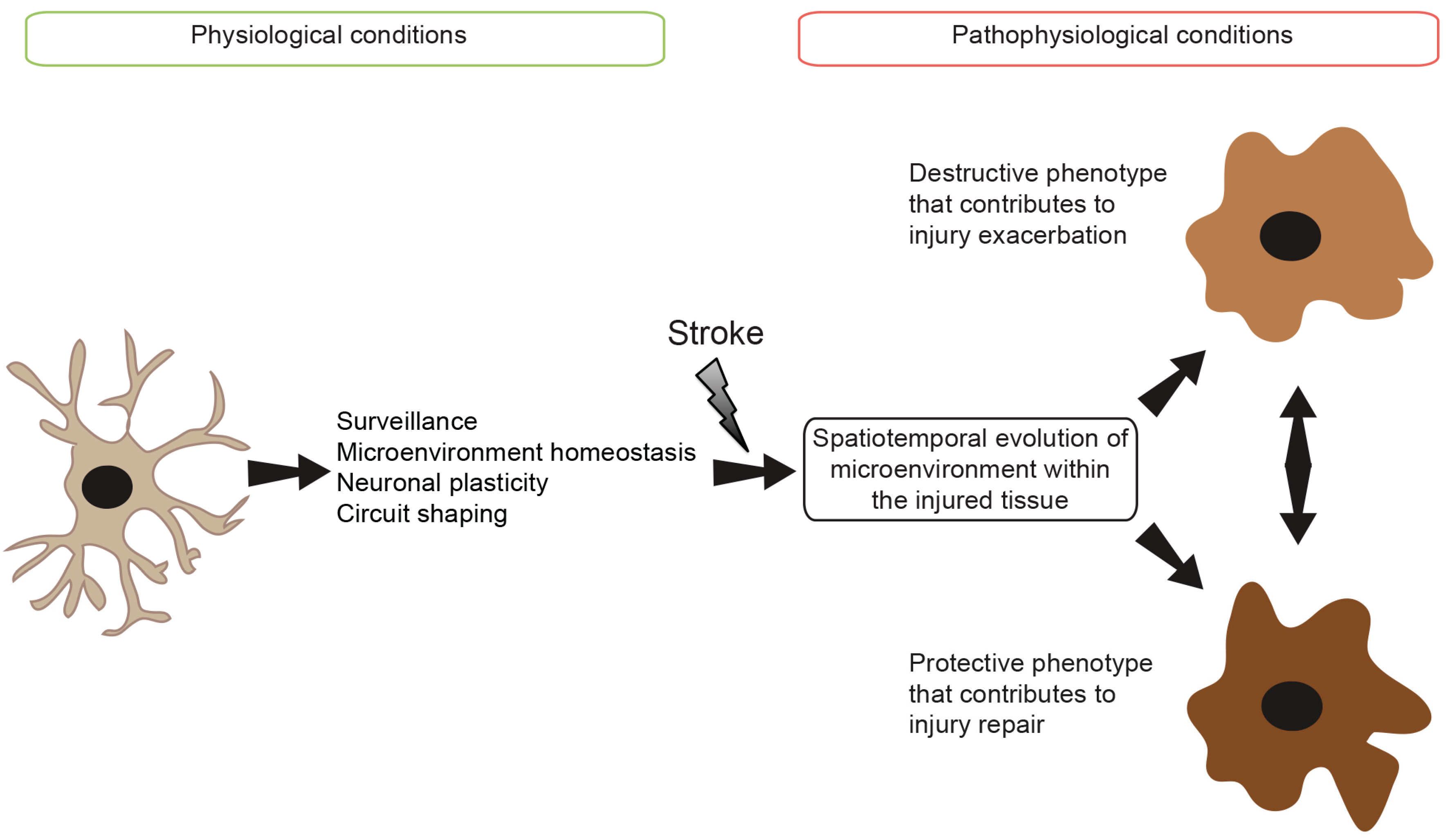
:
{kind=link}
{kind=link}
1. Introduction
2. Origin and Physiological Roles of Microglia
3. Microglial Cell Signaling
3.1. Pattern Recognition Receptor (PRR) Signaling
3.2. Cytokine Receptor Signaling
3.3. Chemokine Receptor Signaling
3.4. Neurotransmitter Receptor Signaling
3.5. Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) Receptor Signaling
3.6. Phosphatidylserine (PS) Receptor Signaling
3.7. Scavenger Receptor (SRs) Signaling
3.8. Other Receptor-Mediated Signaling
3.9. Microglial Cell Markers
4. Microglial Cell Responses Following Ischemic Stroke
4.1. Resident Microglia
4.2. Monocyte-Derived Macrophages (MDMs)
4.3. Novel Sources of Microglial-Like Cells
5. Conclusive Summary and Therapeutic Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A. The implication of neurovascular unit signaling in controlling the subtle balance between injury and repair following ischemic stroke. Neural Regen. Res. 2016, 11, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med. 2008, 14, 497–500. [Google Scholar] [CrossRef]
- ElAli, A.; Jean LeBlanc, N. The role of monocytes in ischemic stroke pathobiology: New avenues to explore. Front. Aging Neurosci. 2016, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Besselmann, M.; Leonhard, C.; Mueller, M.; Ringelstein, E.B.; Kiefer, R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: A study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2003, 183, 25–33. [Google Scholar] [CrossRef]
- Chiba, T.; Umegaki, K. Pivotal roles of monocytes/macrophages in stroke. Mediat. Inflamm. 2013, 2013, 759103. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Komine-Kobayashi, M.; Mochizuki, H.; Yamada, M.; Furuya, T.; Migita, M.; Shimada, T.; Mizuno, Y.; Urabe, T. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience 2003, 117, 531–539. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Benakis, C.; Garcia-Bonilla, L.; Iadecola, C.; Anrather, J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front. Cell. Neurosci. 2014, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Todd, K.G. Microglia in cerebral ischemia: Molecular actions and interactions. Can. J. Physiol. Pharmacol. 2006, 84, 49–59. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A.; Rivest, S. Microglia in Alzheimer's disease: A multifaceted relationship. Brain Behav. Immun. 2016, 55, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Cuadros, M.A.; Navascues, J. The origin and differentiation of microglial cells during development. Prog. Neurobiol. 1998, 56, 173–189. [Google Scholar] [CrossRef]
- Alliot, F.; Lecain, E.; Grima, B.; Pessac, B. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc. Natl. Acad. Sci. USA 1991, 88, 1541–1545. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.Y.; Kohsaka, S.; Rezaie, P. The origin and cell lineage of microglia: New concepts. Brain Res. Rev. 2007, 53, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Hume, D.A.; Gordon, S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 1985, 15, 313–326. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFα. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, P.; Dean, A.; Male, D.; Ulfig, N. Microglia in the cerebral wall of the human telencephalon at second trimester. Cereb. Cortex 2005, 15, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, E.; Miki, A.; Mizoguti, H. Histochemical study of the differentiation of microglial cells in the developing human cerebral hemispheres. J. Anat. 1989, 166, 253–264. [Google Scholar] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Review. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The role of microglia in the healthy brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef] [PubMed]
- Soulet, D.; Rivest, S. Bone-marrow-derived microglia: Myth or reality? Curr. Opin. Pharmacol. 2008, 8, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Sievers, J.; Parwaresch, R.; Wottge, H.U. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: Morphology. Glia 1994, 12, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Elali, A.; Rivest, S. Innate immunity in the CNS: Redefining the relationship between the CNS and its environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, X.F.; Liu, C.S.; Wen, Z.L.; Du, J.L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.J.; Lee, J.E.; Wang, Y.D.; Ma, W.; Fontainhas, A.M.; Fariss, R.N.; Wong, W.T. Regulation of dynamic behavior of retinal microglia by CX3CR1 signaling. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4444–4451. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Miyauchi, J.; Tsirka, S.E. Microglia: An active player in the regulation of synaptic activity. Neural Plast. 2013, 2013, 627325. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M.E. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 2014, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Pavan, P.R. Arcuate retinal folds after intraocular gas injection. Arch. Ophthalmol. 1988, 106, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Rosas, M.; Liddiard, K.; Kimberg, M.; Faro-Trindade, I.; McDonald, J.U.; Williams, D.L.; Brown, G.D.; Taylor, P.R. The induction of inflammation by dectin-1 in vivo is dependent on myeloid cell programming and the progression of phagocytosis. J. Immunol. 2008, 181, 3549–3557. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A.; Rivest, S. Microglia ontology and signaling. Front. Cell Dev. Biol. 2016, 4, 72. [Google Scholar] [CrossRef] [PubMed]
- Dopp, J.M.; Mackenzie-Graham, A.; Otero, G.C.; Merrill, J.E. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J. Neuroimmunol. 1997, 75, 104–112. [Google Scholar] [CrossRef]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Arico, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-β signaling through smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Stockinger, B. TGFβ1, a “jack of all trades”: The link with pro-inflammatory il-17-producing t cells. Trends Immunol. 2006, 27, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. CCR2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. Trems in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Stefano, L.; Racchetti, G.; Bianco, F.; Passini, N.; Gupta, R.S.; Panina Bordignon, P.; Meldolesi, J. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J. Neurochem. 2009, 110, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Reutelingsperger, C.P.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.; LaFace, D.M.; Green, D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Mattson, M.P.; Arumugam, T.V. Involvement of Fc receptors in disorders of the central nervous system. Neuromol. Med. 2010, 12, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Lyapina, L.A.; Ul’yanov, A.M. Formation of complexes of heparin with proteins and its physiological role. II. Hum. Physiol. 1979, 4, 239–248. [Google Scholar] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 73–90. [Google Scholar] [PubMed]
- Ito, D.; Tanaka, K.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Enhanced expression of IBA1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 2001, 32, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.L.; Goodsall, A.L.; Hickey, W.F.; Sedgwick, J.D. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 1995, 154, 4309–4321. [Google Scholar] [PubMed]
- Mattiace, L.A.; Davies, P.; Dickson, D.W. Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am. J. Pathol. 1990, 136, 1101–1114. [Google Scholar] [PubMed]
- Anttila, J.E.; Whitaker, K.W.; Wires, E.S.; Harvey, B.K.; Airavaara, M. Role of microglia in ischemic focal stroke and recovery: Focus on toll-like receptors. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Sansing, L.H. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol. 2013, 2013, 746068. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating resident microglia after focal cerebral ischaemia in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Murugan, M.; Ling, E.A.; Kaur, C. Glutamate receptors in microglia. CNS Neurol. Disord. Drug Targets 2013, 12, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Thery, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Slager-Earnest, S.E.; Hoffman, S.J.; Beckmann, C.J. Effects of a specialized prenatal adolescent program on maternal and infant outcomes. J. Obstet. Gynecol. Neonat. Nurs. JOGNN 1987, 16, 422–429. [Google Scholar] [CrossRef]
- Estevez, A.G.; Sahawneh, M.A.; Lange, P.S.; Bae, N.; Egea, M.; Ratan, R.R. Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neurons. J. Neurosci. 2006, 26, 8512–8516. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Moon, P.G.; Baek, M.C.; Kim, H.S. Comparison of the effects of matrix metalloproteinase inhibitors on TNF-α release from activated microglia and TNF-α converting enzyme activity. Biomol. Ther. 2014, 22, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.R.; Koerner, I.P.; Moller, T. Microglia in ischemic brain injury. Future Neurol. 2010, 5, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Schlichter, L.C. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008, 28, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 2007, 115, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Ma, J.; Ha, T.; Xia, Y.; Kelley, J.; Williams, D.L.; Kao, R.L.; Browder, I.W.; Schweitzer, J.B.; Kalbfleisch, J.H.; et al. Activation of toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J. Neuroimmunol. 2007, 190, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Pradillo, J.M.; Fernandez-Lopez, D.; Garcia-Yebenes, I.; Sobrado, M.; Hurtado, O.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J. Neurochem. 2009, 109, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lathia, J.D.; Selvaraj, P.K.; Jo, D.G.; Mughal, M.R.; Cheng, A.; Siler, D.A.; Markesbery, W.R.; Arumugam, T.V.; Mattson, M.P. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid β-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 2008, 213, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hebert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Takata, K.; Inden, M.; Tsuchiya, D.; Yanagisawa, D.; Nakata, J.; Taniguchi, T. Intracerebroventricular injection of microglia protects against focal brain ischemia. J. Pharmacol. Sci. 2004, 94, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Thored, P.; Heldmann, U.; Gomes-Leal, W.; Gisler, R.; Darsalia, V.; Taneera, J.; Nygren, J.M.; Jacobsen, S.E.; Ekdahl, C.T.; Kokaia, Z.; et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 2009, 57, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Szalay, G.; Martinecz, B.; Lenart, N.; Kornyei, Z.; Orsolits, B.; Judak, L.; Csaszar, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [PubMed]
- Breckwoldt, M.O.; Chen, J.W.; Stangenberg, L.; Aikawa, E.; Rodriguez, E.; Qiu, S.; Moskowitz, M.A.; Weissleder, R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. USA 2008, 105, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Malm, T.; Koistinaho, M.; Muona, A.; Magga, J.; Koistinaho, J. The role and therapeutic potential of monocytic cells in Alzheimer’s disease. Glia 2010, 58, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Mausberg, A.K.; Lee, J.I.; Simiantonakis, I.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 2012, 71, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.X.; Broughton, B.R.; Kim, H.A.; Lee, S.; Drummond, G.R.; Sobey, C.G. Evidence that Ly6C(hi) monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke 2015, 46, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.P.; Bellavance, M.A.; Prefontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid β. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Yang, J.; Beltran, C.D.; Cho, S. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Song, W.; Ji, Y.; Liu, X.; Tian, L.; Wang, C.; Chen, D.; Zhang, X.; Zhang, M. Microglia and microglia-like cell differentiated from DC inhibit CD4 T cell proliferation. PLoS ONE 2009, 4, e7869. [Google Scholar] [CrossRef] [PubMed]
- Ozen, I.; Deierborg, T.; Miharada, K.; Padel, T.; Englund, E.; Genove, G.; Paul, G. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol. 2014, 128, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Kawahara, M.; Nakano-Doi, A.; Takahashi, A.; Tanaka, Y.; Narita, A.; Kuwahara-Otani, S.; Hayakawa, T.; Yagi, H.; Matsuyama, T.; et al. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J. Neuroinflamm. 2016, 13, 57. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guruswamy, R.; ElAli, A. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. Int. J. Mol. Sci. 2017, 18, 496. https://doi.org/10.3390/ijms18030496
Guruswamy R, ElAli A. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. International Journal of Molecular Sciences. 2017; 18(3):496. https://doi.org/10.3390/ijms18030496
Chicago/Turabian StyleGuruswamy, Revathy, and Ayman ElAli. 2017. "Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions" International Journal of Molecular Sciences 18, no. 3: 496. https://doi.org/10.3390/ijms18030496