
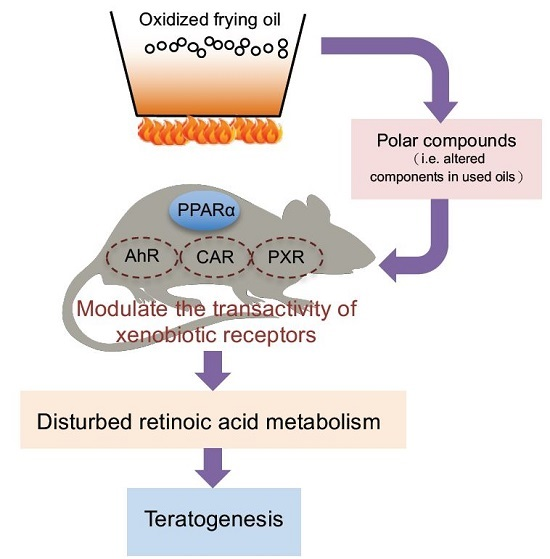
Peroxisome Proliferator-Activated Receptor α Activation Is Not the Main Contributor to Teratogenesis Elicited by Polar Compounds from Oxidized Frying Oil


Abstract
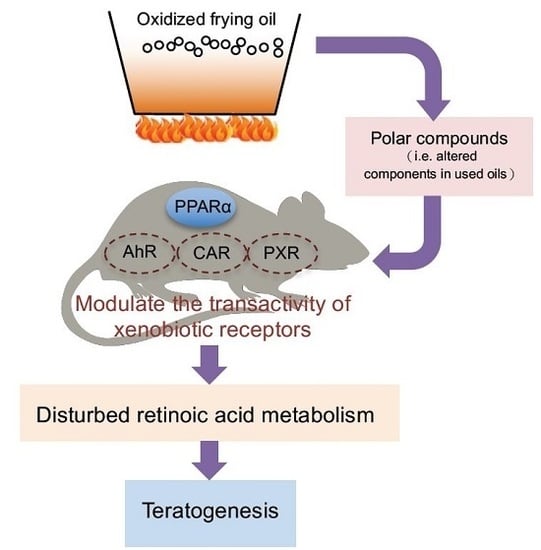
:
1. Introduction
2. Results
2.1. Effects of PO Diet or PPARα Deficiency on Embryotocixity and Reproductive Characteristics
2.2. Effects of PO Diet or PPARα Deficiency on Teratogenesis
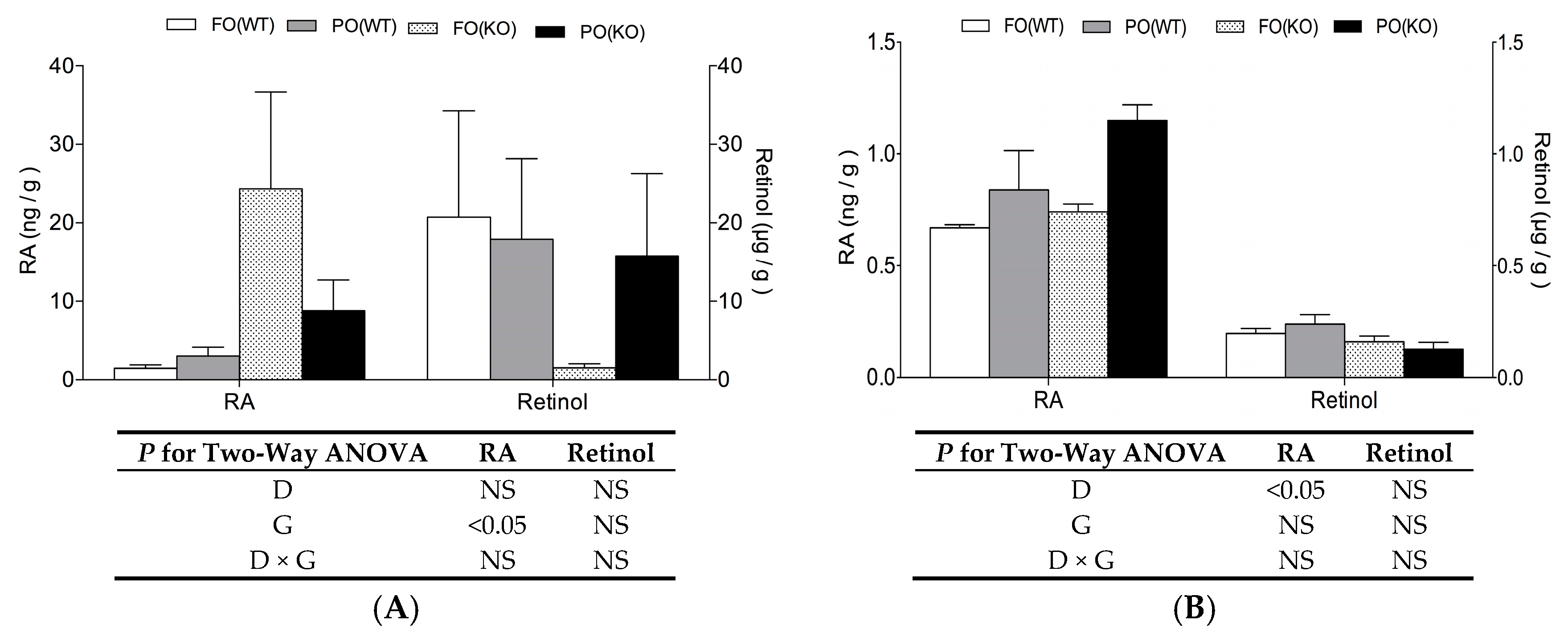
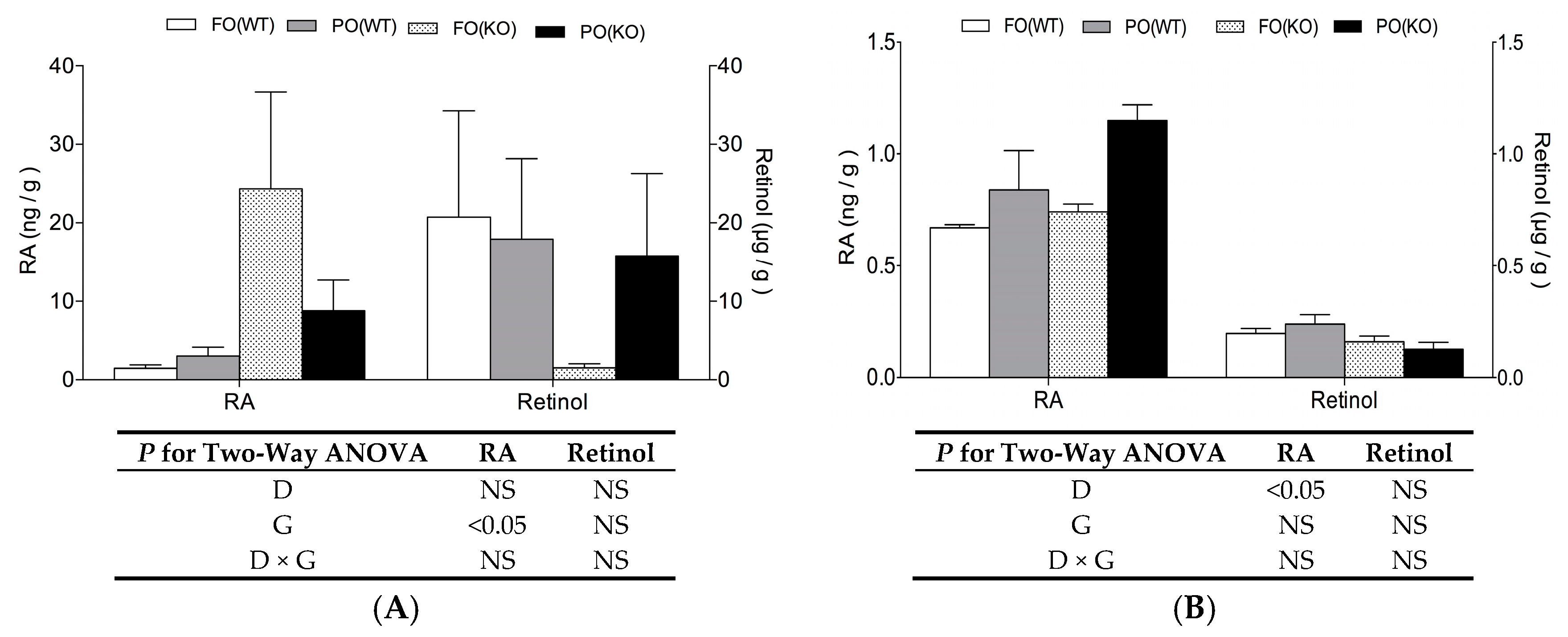
2.3. Effects of PO Diet or PPARα Deficiency on Vitamin A Status
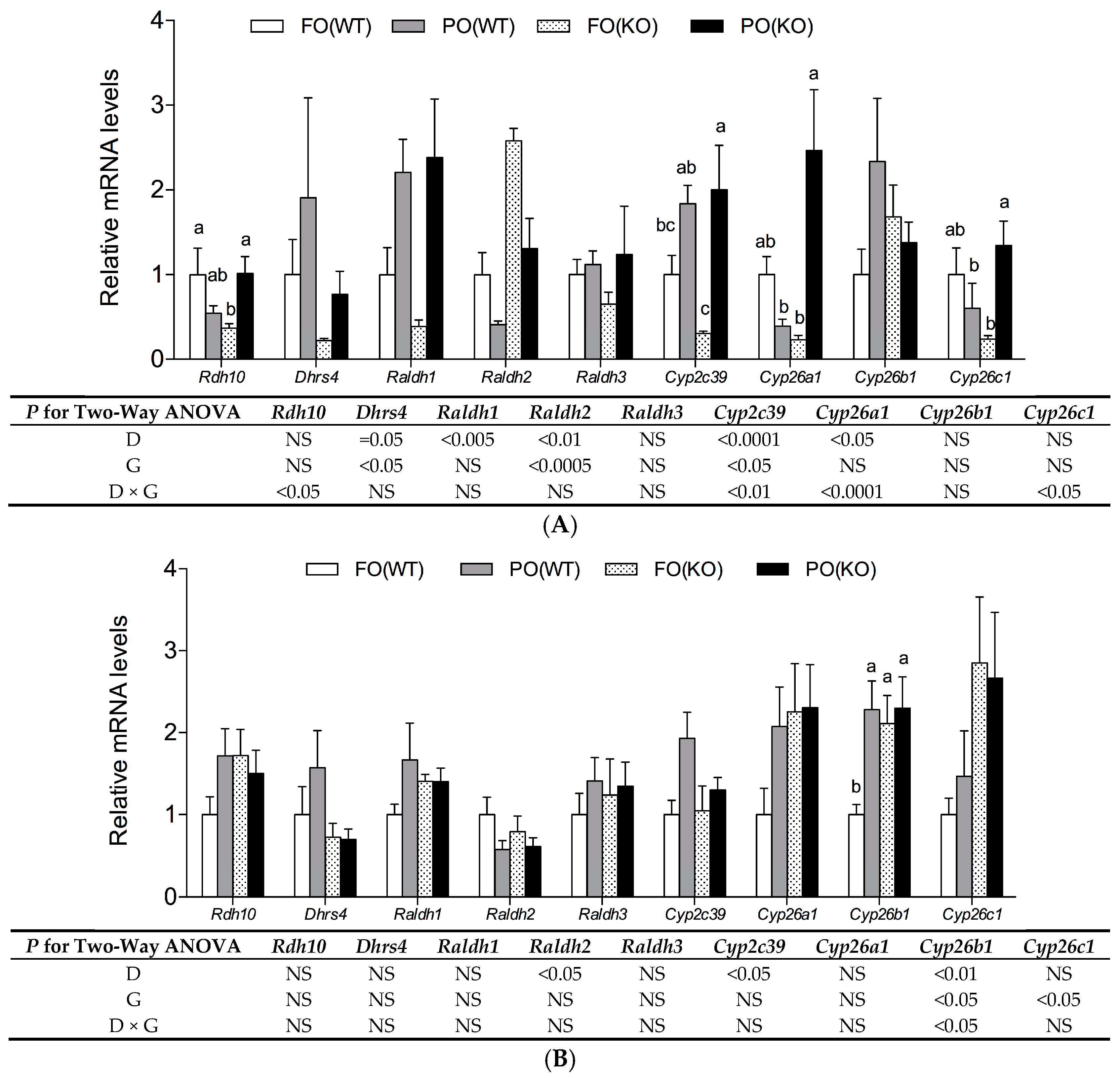
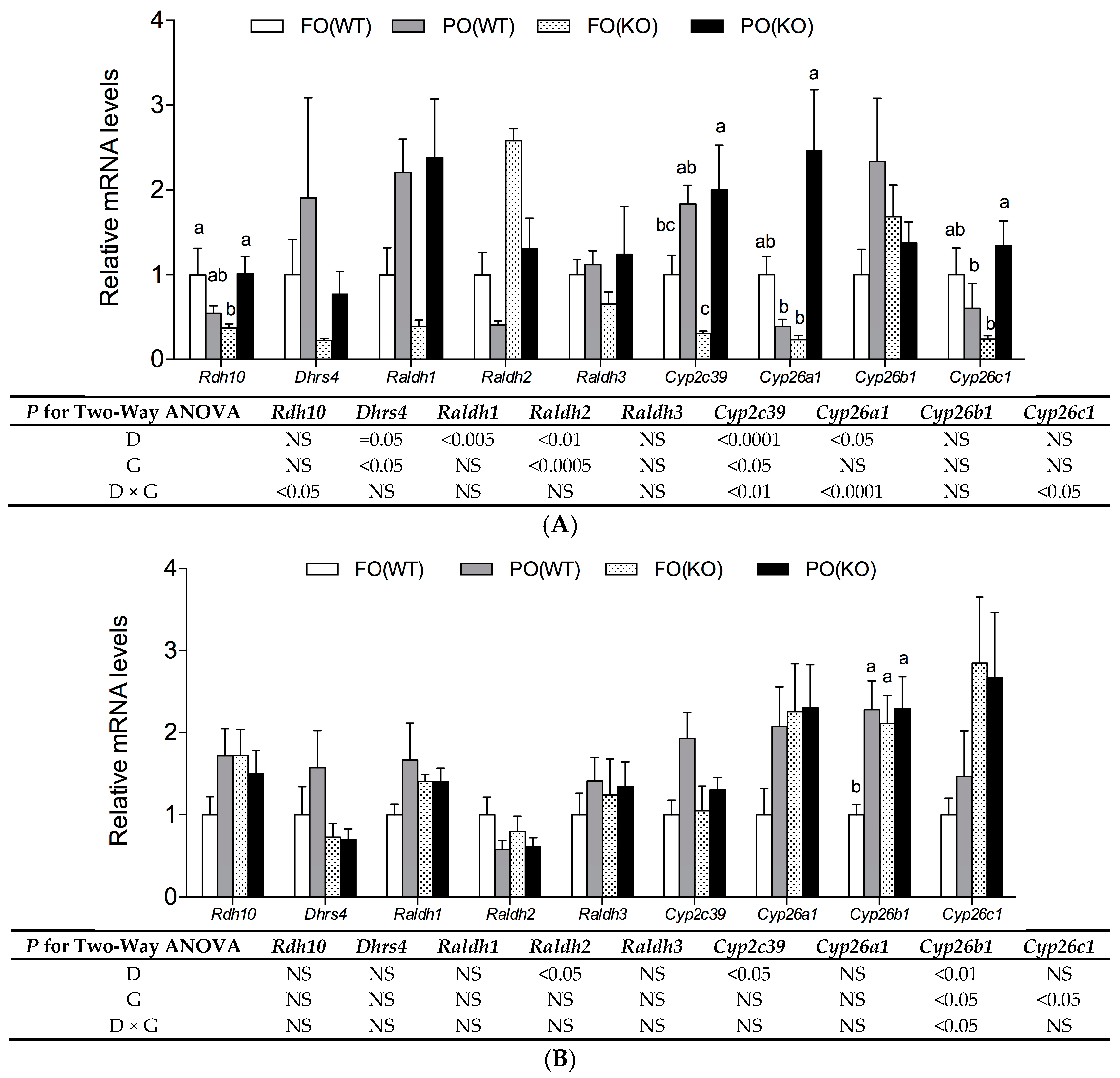
2.4. Effects of PO Diet or PPARα Deficiency on RA Metabolic Gene Expression
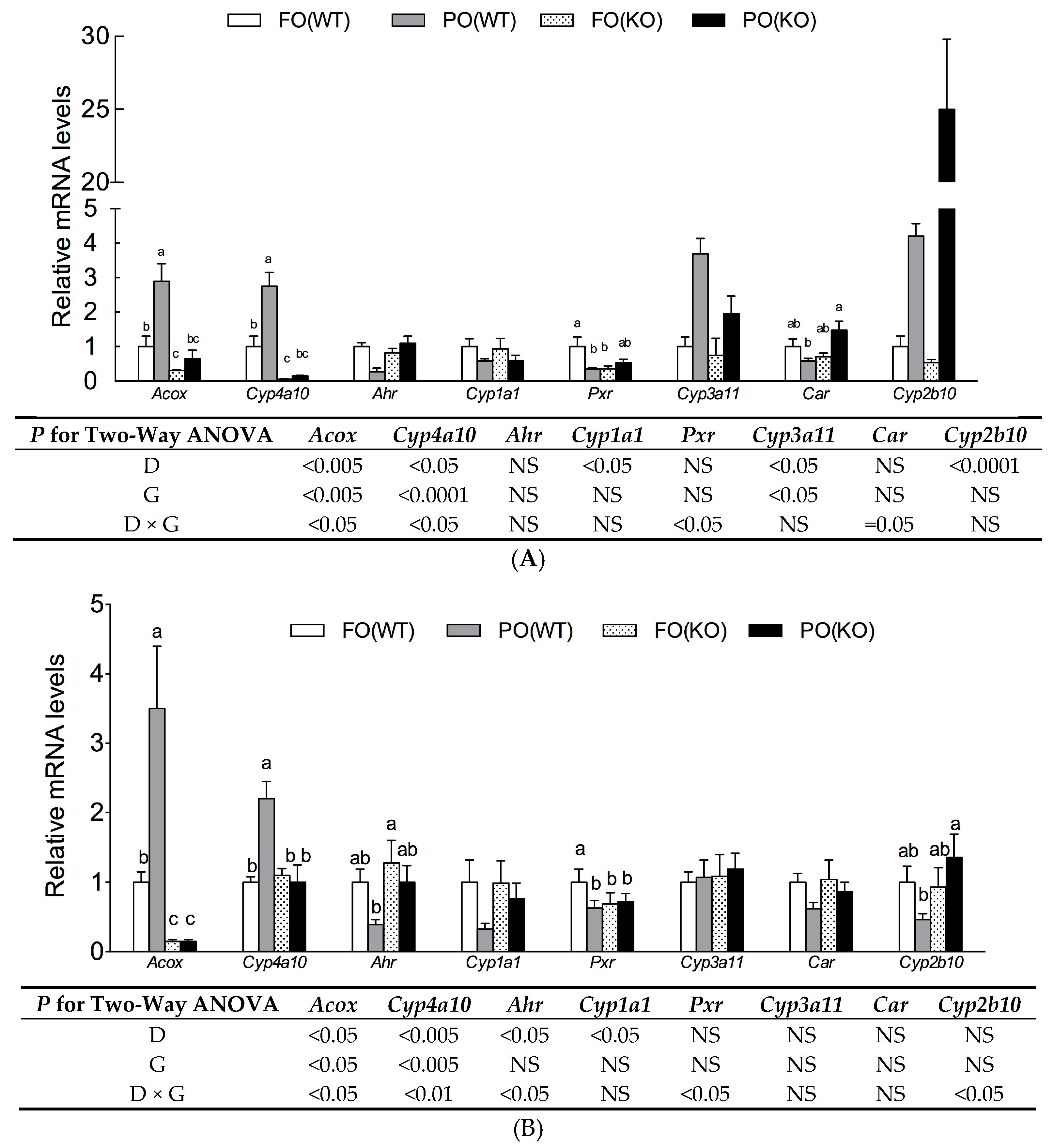
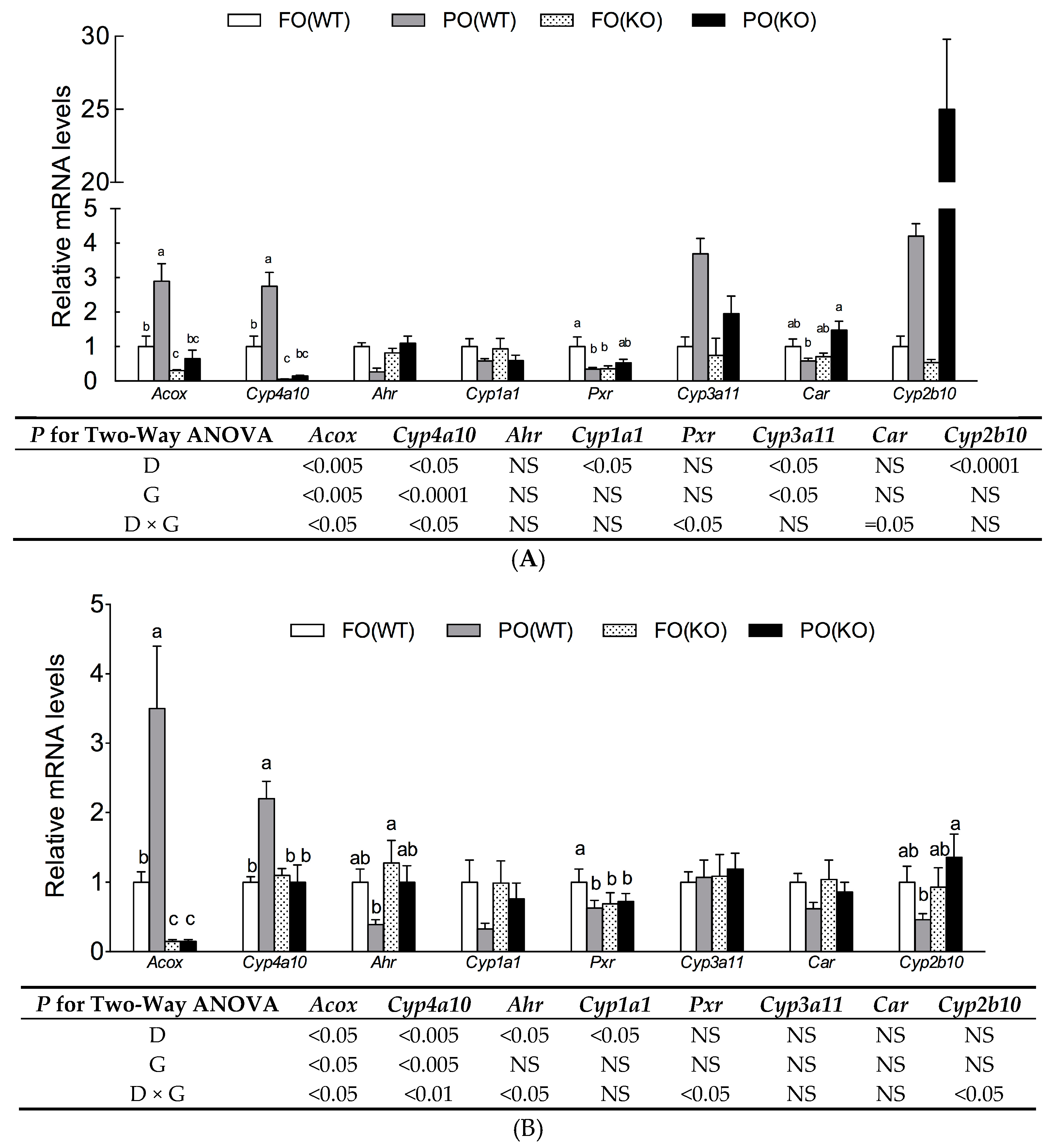
2.5. Effects of PO Diet or PPARα Deficiency on Transactivity of Xenobiotic Receptors
3. Discussion
4. Materials and Methods
4.1. Preparation of PO
4.2. Animals and Diets
4.3. Embryonic Toxicity and Morphogenesis
4.4. Quantification of Retinol and Retinoic Acid
4.5. RNA Isolation and mRNA Detection
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AhR | Aryl hydrocarbon receptor |
| CAR | Constitutive androstane receptor |
| DHRS | Dehydrogenase/reductase SDR family |
| FO | Fresh oil |
| KO | Knock out |
| OFO | Oxidized frying oil |
| PO | Polar compounds |
| PPs | Peroxisome proliferators |
| PXR | Pregnane-X receptor |
| RA | Retinoic acid |
| RALDH | Retinal dehydrogenase |
| RAR | Retinoic acid receptor |
| RARE | Retinoic acid response element |
| RDH | Retinol dehydrogenase |
| RXR | Retinoid X receptor |
| WT | Wild type |
References
- Sogorb, M.A.; Pamies, D.; de Lapuente, J.; Estevan, C.; Estevez, J. An integrated approach for detecting embryotoxicity and developmental toxicity of environmental contaminants using in vitro alternative methods. Toxicol. Lett. 2014, 230, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Toft, G. Persistent organochlorine pollutants and human reproductive health. Dan. Med. J. 2014, 61, B4967. [Google Scholar] [PubMed]
- Huang, C.J.; Cheung, N.S.; Lu, V.R. Effects of deteriorated frying oil and dietary protein levels on liver microsomal enzymes in rats. J. Am. Oil Chem. Soc. 1988, 65, 1796–1803. [Google Scholar] [CrossRef]
- Huang, C.F.; Lin, Y.S.; Chiang, Z.C.; Lu, S.Y.; Kuo, Y.H. Oxidized frying oil and its polar fraction fed to pregnant mice are teratogenic and alter mRNA expressions of vitamin A metabolism genes in the liver of dams and their fetuses. J. Nutr. Biochem. 2014, 25, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.; Morrison, A.; Studer, M.; Popperl, H.; Krumlauf, R. Retinoids and Hox genes. FASEB J. 1996, 10, 969–978. [Google Scholar] [PubMed]
- Pares, X.; Farres, J.; Kedishvili, N.; Duester, G. Medium- and short-chain dehydrogenase/reductase gene and protein families: Medium-chain and short-chain dehydrogenases/reductases in retinoid metabolism. Cell. Mol. Life Sci. 2008, 65, 3936–3949. [Google Scholar] [CrossRef] [PubMed]
- Ashique, A.M.; May, S.R.; Kane, M.A.; Folias, A.E.; Phamluong, K. Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis 2012, 50, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Chen, W.; Zhang, M.; Napoli, J.L. Reduction of all-trans-retinal in the mouse liver peroxisome fraction by the short-chain dehydrogenase/reductase RRD: Induction by the PPAR α ligand clofibrate. Biochemistry 2003, 42, 4190–4196. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.K.; Belvaeva, O.V.; Wu, L.; Kedishvili, N.Y. The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 2014, 289, 14868–14880. [Google Scholar] [CrossRef] [PubMed]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar] [PubMed]
- Ross, A.C.; Zolfaghari, R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu. Rev. Nutr. 2011, 31, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Andreola, F.; Hayhurst, G.P.; Luo, G.; Ferguson, S.S.; Gonzalez, F.J. Mouse liver CYP2C39 is a novel retinoic acid 4-hydroxylase. Its down-regulation offers a molecular basis for liver retinoid accumulation and fibrosis in aryl hydrocarbon receptor-null mice. J. Biol. Chem. 2004, 279, 3434–3438. [Google Scholar] [CrossRef] [PubMed]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Li, H.; Abu-Abed, S. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Reijntjes, S.; Blentic, A.; Gale, E.; Maden, M. The control of morphogen signalling: Regulation of the synthesis and catabolism of retinoic acid in the developing embryo. Dev. Biol. 2005, 285, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Choe, E.; Min, D.B. Chemistry of deep-fat frying oils. J. Food Sci. 2007, 72, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M. Regulation of frying fat and oil. In Deep Frying: Chemistry, Nutrition and Practical Applications; Erickson, M.D., Ed.; AOCS Press: Champaign, IL, USA, 2007; pp. 373–385. [Google Scholar]
- Issemann, I.; Green, I.S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.Q.; Brown-Borg, H.; Brown, S.; Westin, S.; Mode, A. PPARalpha activators down-regulate CYP2C7, a retinoic acid and testosterone hydroxylase. Toxicology 2004, 203, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.; Dickmann, L.; Dixit, V.; Isoherranen, N. A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation. Mol. Pharmacol. 2010, 77, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Thibodeaux, J.R.; Hanson, R.G.; Narotsky, M.G.; Rogers, J.M. Effects of perfluorooctanoic acid exposure during pregnancy in the mouse. Toxicol. Sci. 2006, 90, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Shiota, K.; Mima, S. Assessment of the teratogenicity of di(2-ethylhexyl)phthalate and mono(2-ethylhexyl)phthalate in mice. Arch. Toxicol. 1985, 56, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Tyl, R.W.; Price, C.J.; Marr, M.C.; Kimmel, C.A. Developmental toxicity evaluation of dietary di(2-ethylhexyl)phthalate in Fischer 344 rats and CD-1 mice. Fundam. Appl. Toxicol. 1988, 10, 395–412. [Google Scholar] [CrossRef]
- Peraza, M.A.; Burdick, A.D.; Marin, H.E.; Gonzalez, F.J.; Peters, J.M. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR). Toxicol. Sci. 2006, 90, 269–295. [Google Scholar] [CrossRef] [PubMed]
- Costet, P.; Legendre, C.; More, J.; Edgar, A.; Galtier, P.; Pineau, T. PPARα isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.; Lalwai, N.D. Carcinogenesis by hepatic peroxisome proliferators: Evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. Crit. Rev. Toxicol. 1983, 12, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Boily, M.H.; Berube, V.E.; Spear, P.A.; DeBlois, C.; Dassylva, N. Hepatic retinoids of bullfrogs in relation to agricultural pesticides. Environ. Toxicol. Chem. 2005, 24, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Benisek, M.; Hilscherova, K. Disruption of retinoid transport, metabolism and signaling by environmental pollutants. Environ. Int. 2008, 34, 898–913. [Google Scholar] [CrossRef] [PubMed]
- Kot-Leibovich, H.; Fainsod, A. Ethanol induces embryonic malformations by competing for retinaldehyde dehydrogenase activity during vertebrate gastrulation. Dis. Model. Mech. 2009, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Ulven, S.M.; Gundersen, T.E.; Weedon, M.S.; Landaas, V.O.; Sakhi, A.K. Identification of endogenous retinoids, enzymes, binding proteins, and receptors during early postimplantation development in mouse: Important role of retinal dehydrogenase type 2 in synthesis of all-trans-retinoic acid. Dev. Biol. 2000, 220, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Tzimas, G.; Collins, M.D.; Burgin, H.; Hummler, H.; Nau, H. Embryotoxic doses of vitamin A to rabbits result in low plasma but high embryonic concentrations of all-trans-retinoic acid: Risk of vitamin A exposure in humans. J. Nutr. 1996, 126, 2159–2171. [Google Scholar] [PubMed]
- Tickle, C.; Alberts, B.; Wolpert, L.; Lee, J. Local application of retinoic acid to the limb bond mimics the action of the polarizing region. Nature 1982, 296, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Goldbeter, A.; Gonze, D.; Pourquié, O. Sharp developmental thresholds defined through bistability by antagonistic gradients of retinoic acid and FGF signaling. Dev. Dyn. 2007, 236, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Dobbs-McAuliffe, B.; Zhao, Q.; Linney, E. Feedback mechanisms regulate retinoic acid production and degradation in the zebrafish embryo. Mech. Dev. 2004, 121, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Loudig, O.; Maclean, G.A.; Dore, N.L.; Luu, L.; Petkovich, M. Transcriptional co-operativity between distant retinoic acid response elements in regulation of Cyp26A1 inducibility. Biochem. J. 2005, 392, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Ihunnah, C.A.; Jiang, M.; Xie, W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. Biophys. Acta 2011, 1812, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Indart, A.; Viana, M.; Grootveld, M.C.; Silwood, C.J.; Sanchez-Vera, I.; Bonet, B. Teratogenic actions of thermally-stressed culinary oils in rats. Free Radic. Res. 2002, 36, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Saguy, S.; Dana, D. Integrated approach to deep fat frying: Engineering, nutrition, health and consumer aspects. J. Food Eng. 2003, 56, 143–152. [Google Scholar] [CrossRef]
- Dobarganes, C.; Márquez-Ruiz, G. Possible adverse effects of frying with vegetable oils. Br. J. Nutr. 2015, 113, S49–S57. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Huang, C.F.; Chang, Y.C.; Lin, Y.S.; Chao, P.M. Gestational ingestion of oxidized frying oil by C57BL/6J mice differentially affects the susceptibility of the male and female offspring to diet-induced obesity in adulthood. J. Nutr. 2013, 143, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Dieffenbacher, A.; Pocklington, W. UPAC Standard Method 2.507: Determination of polar compounds in frying fats. In Standard Methods for the Analysis of Oils, Fats and Derivatives, 7th ed.; International Union of Pure and Applied Chemistry, Blackwell Scientific: Oxford, UK, 1987. [Google Scholar]
- Sallee, E.M. Official and Tentative Methods of the American Oil Chemists’ Society, 3rd ed.; American Oil Chemists’ Society: Champaign, IL, USA, 1971. [Google Scholar]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [PubMed]
- Chao, P.M.; Department of Nutrition, China Medical University, Taichung, Taiwan. Teratogenesis Was Reproduced by PO Diet in Wild Type and PPARα KO Mice. Unpublished work. 2016. [Google Scholar]
- Liu, C.; Russell, R.M.; Wang, X.D. Exposing ferrets to cigarette smoke and a pharmacological dose of β-carotene supplementation enhance in vitro retinoic acid catabolism in lungs via induction of cytochrome P450 enzymes. J. Nutr. 2003, 133, 173–179. [Google Scholar] [PubMed]
- Liu, C.; Chung, J.; Seitz, H.K.; Russell, R.M.; Wang, X.D. Chlormethiazole treatment prevents reduced hepatic vitamin A levels in ethanol-fed rats. Alcohol. Clin. Exp. Res. 2002, 26, 1703–1709. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quality Index | FO | PO |
|---|---|---|
| Acid value, mg KOH/g | 0.056 ± 0.002 | 2.9 ± 0.1 |
| Conjugated diene, OD233/g | 392 ± 4.23 | 4933 ± 35.52 |
| Variables | FO(WT) | PO(WT) | FO(KO) | PO(KO) | p Values (Two-Way ANOVA) | ||
|---|---|---|---|---|---|---|---|
| D | G | D × G | |||||
| No. of dams | 5 | 5 | 5 | 5 | |||
| Maternal weight gain, g | 4.71 ± 0.52 | 2.97 ± 0.52 | 4.94 ± 0.86 | 1.98 ± 0.75 | <0.005 | NS | NS |
| Relative liver weight, % | 4.58 ± 0.15 c | 7.35 ± 0.12 a | 5.11 ± 0.23 bc | 5.90 ± 0.28 b | <0.0001 | NS | <0.005 |
| No. of corpora lutea/litter | 7.00 ± 0.32 | 8.60 ± 0.68 | 9.40 ± 0.51 | 9.20 ± 0.73 | NS | NS | NS |
| No. of implantations/litter | 7.00 ± 0.51 | 8.60 ± 0.68 | 9.40 ± 0.51 | 9.20 ± 0.73 | NS | NS | NS |
| No. of fetuses/litter | 5.40 ± 0.68 | 6.60 ± 0.75 | 8.60 ± 0.75 | 6.80 ± 1.69 | NS | NS | NS |
| No. of live fetuses/litter | 5.40 ± 0.68 | 6.40 ± 0.75 | 8.40 ± 0.93 | 6.60 ± 1.57 | NS | NS | NS |
| No. of dead fetuses/litter | 0.00 ± 0.00 | 0.20 ± 0.20 | 0.20 ± 0.20 | 0.20 ± 0.20 | NS | NS | NS |
| Pre-implantation loss/litter | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | NS | NS | NS |
| Post-implantation loss/litter | 1.00 ± 0.32 | 2.00 ± 0.45 | 0.80 ± 0.58 | 2.40 ± 1.21 | NS | NS | NS |
| Litters with resorptions ≥3, % | 0 (0/5) | 40 (2/5) | 20 (1/5) | 20 (1/5) | |||
| Full-litter resorptions, % | 0 (0/5) | 0 (0/5) | 0 (0/5) | 0 (0/5) | |||
| Variables | FO(WT) | PO(WT) | FO(KO) | PO(KO) | p Values (Two-Way ANOVA) | ||
|---|---|---|---|---|---|---|---|
| D | G | D × G | |||||
| No. of litter 2 | 5 (27) | 5 (33) | 5 (43) | 5 (34) | |||
| Body weight, g | 0.81 ± 0.03 | 0.88 ± 0.05 | 0.80 ± 0.36 | 0.76 ± 0.07 | NS | NS | NS |
| Placenta, g | 0.07 ± 0.00 | 0.09 ± 0.00 | 0.09 ± 0.04 | 0.10 ± 0.01 | <0.005 | NS | NS |
| Liver, g | 0.04 ± 0.00 | 0.04 ± 0.00 | 0.04 ± 0.02 | 0.04 ± 0.00 | NS | NS | NS |
| Mortality rate, % | 0 ± 0 | 3.4 ± 3.3 | 0 ± 0 | 2.8 ± 2.7 | NS | NS | NS |
| Incidence of externally visible abnormalities, % | |||||||
| Eye defect | 0 ± 0 | 3.4 ± 3.3 | 0 ± 0 | 2.8 ± 2.7 | NS | NS | NS |
| Edema | 0 ± 0 | 3.4 ± 3.3 | 0 ± 0 | 3.4 ± 3.3 | NS | NS | NS |
| Brain defect | 0 ± 0 | 2.8 ± 2.7 | 0 ± 0 | 2.8 ± 2.7 | NS | NS | NS |
| Haematoma | 0 ± 0 | 8.8 ± 3.6 | 0 ± 0 | 20 ± 12 | <0.05 | NS | NS |
| Shrivelling | 0 ± 0 | 12.4 ± 5.3 | 0 ± 0 | 11.2 ± 4.8 | <0.05 | NS | NS |
| Spina bifida | 0 ± 0 | 3.4 ± 3.3 | 0 ± 0 | 6.2 ± 3.7 | NS | NS | NS |
| Limb defect | 0 ± 0 | 6.8 ± 4.0 | 0 ± 0 | 2.8 ± 2.7 | NS | NS | NS |
| Total 3 | 0 ± 0 | 30.2 ± 8.4 | 0 ± 0 | 32.5 ± 9.7 | <0.05 | NS | NS |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-S.; Lin, T.-Y.; Wu, J.-J.; Yao, H.-T.; Chang, S.L.-Y.; Chao, P.-M. Peroxisome Proliferator-Activated Receptor α Activation Is Not the Main Contributor to Teratogenesis Elicited by Polar Compounds from Oxidized Frying Oil. Int. J. Mol. Sci. 2017, 18, 510. https://doi.org/10.3390/ijms18030510
Lin Y-S, Lin T-Y, Wu J-J, Yao H-T, Chang SL-Y, Chao P-M. Peroxisome Proliferator-Activated Receptor α Activation Is Not the Main Contributor to Teratogenesis Elicited by Polar Compounds from Oxidized Frying Oil. International Journal of Molecular Sciences. 2017; 18(3):510. https://doi.org/10.3390/ijms18030510
Chicago/Turabian StyleLin, Yu-Shun, Ting-Yi Lin, Jia-Jiuan Wu, Hsien-Tsung Yao, Sunny Li-Yun Chang, and Pei-Min Chao. 2017. "Peroxisome Proliferator-Activated Receptor α Activation Is Not the Main Contributor to Teratogenesis Elicited by Polar Compounds from Oxidized Frying Oil" International Journal of Molecular Sciences 18, no. 3: 510. https://doi.org/10.3390/ijms18030510