
Autophagy Induced by Areca Nut Extract Contributes to Decreasing Cisplatin Toxicity in Oral Squamous Cell Carcinoma Cells: Roles of Reactive Oxygen Species/AMPK Signaling

Abstract
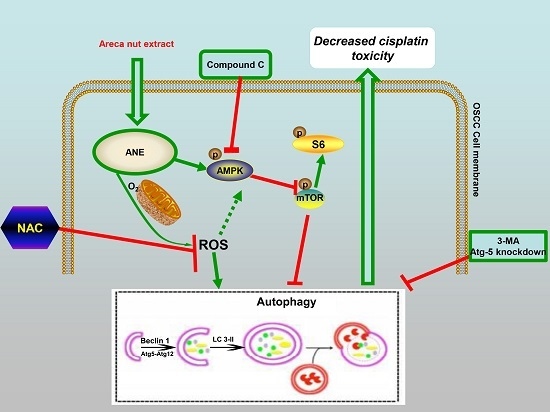
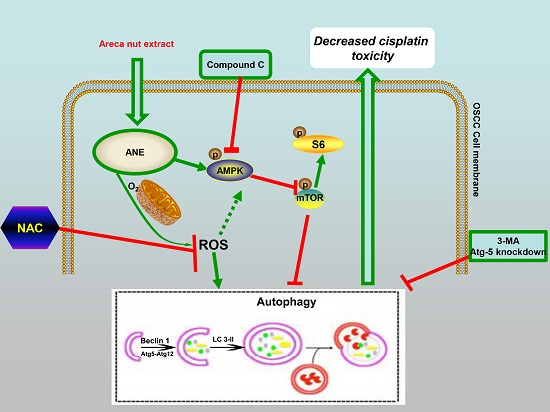
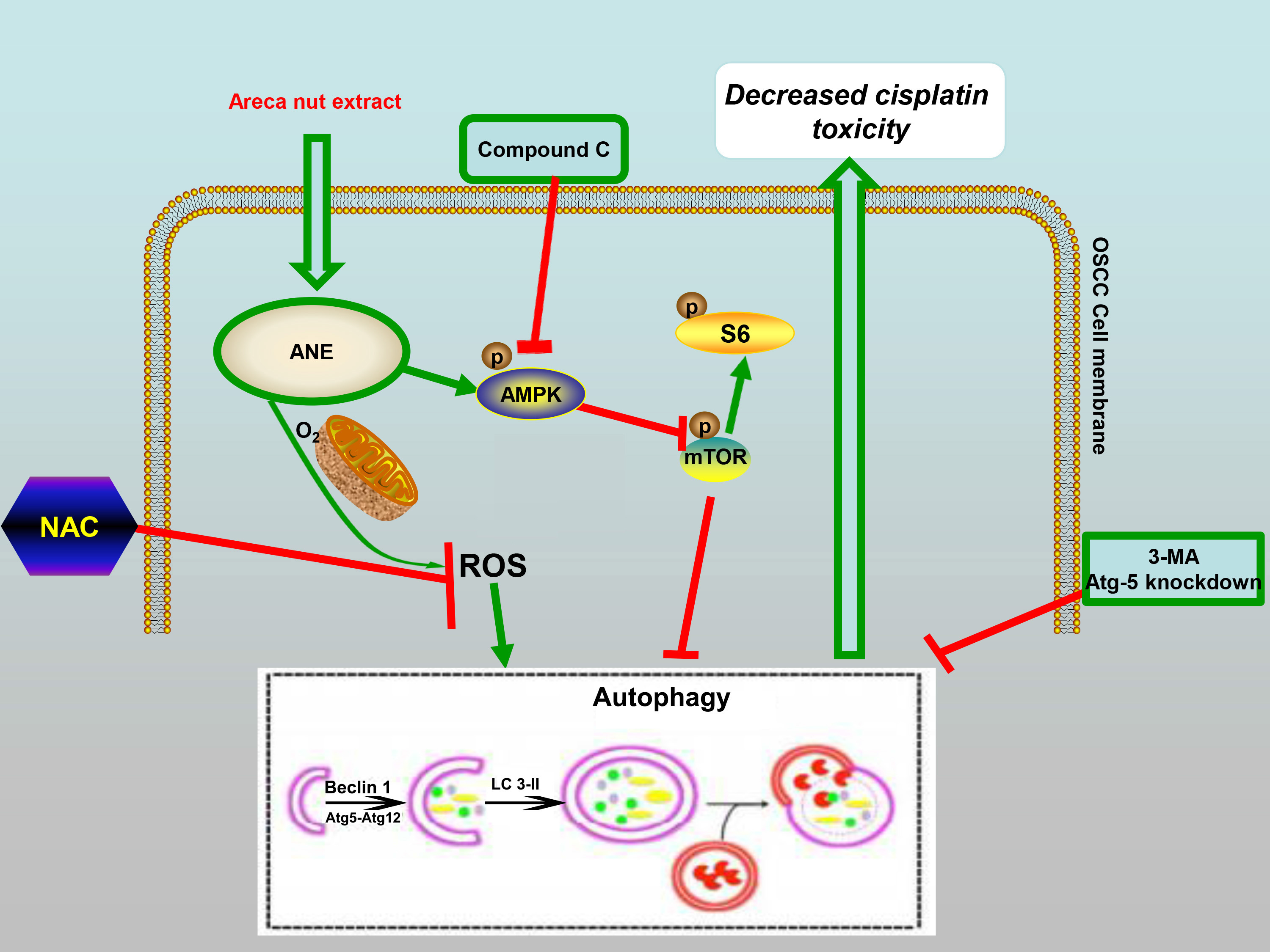
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
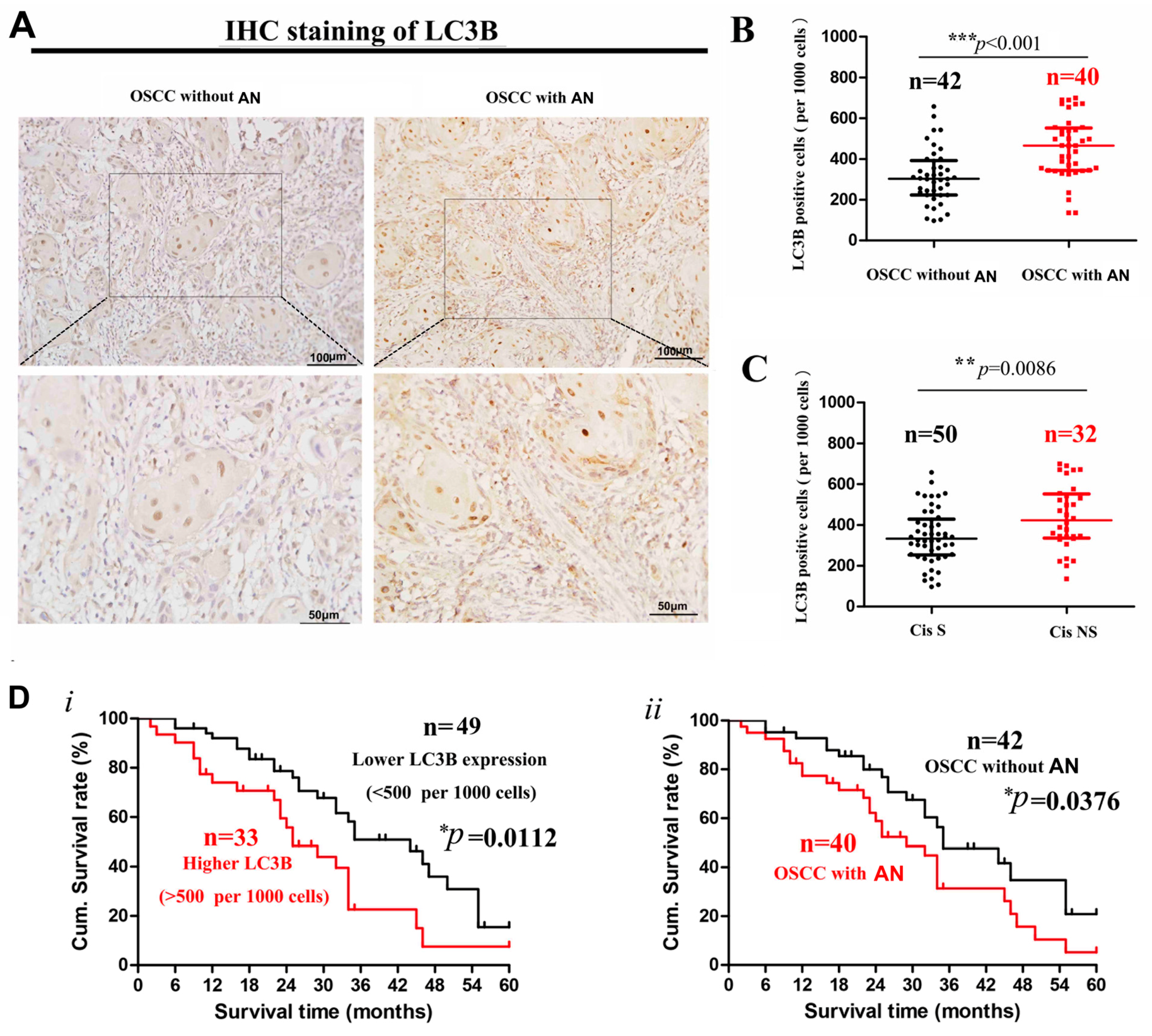
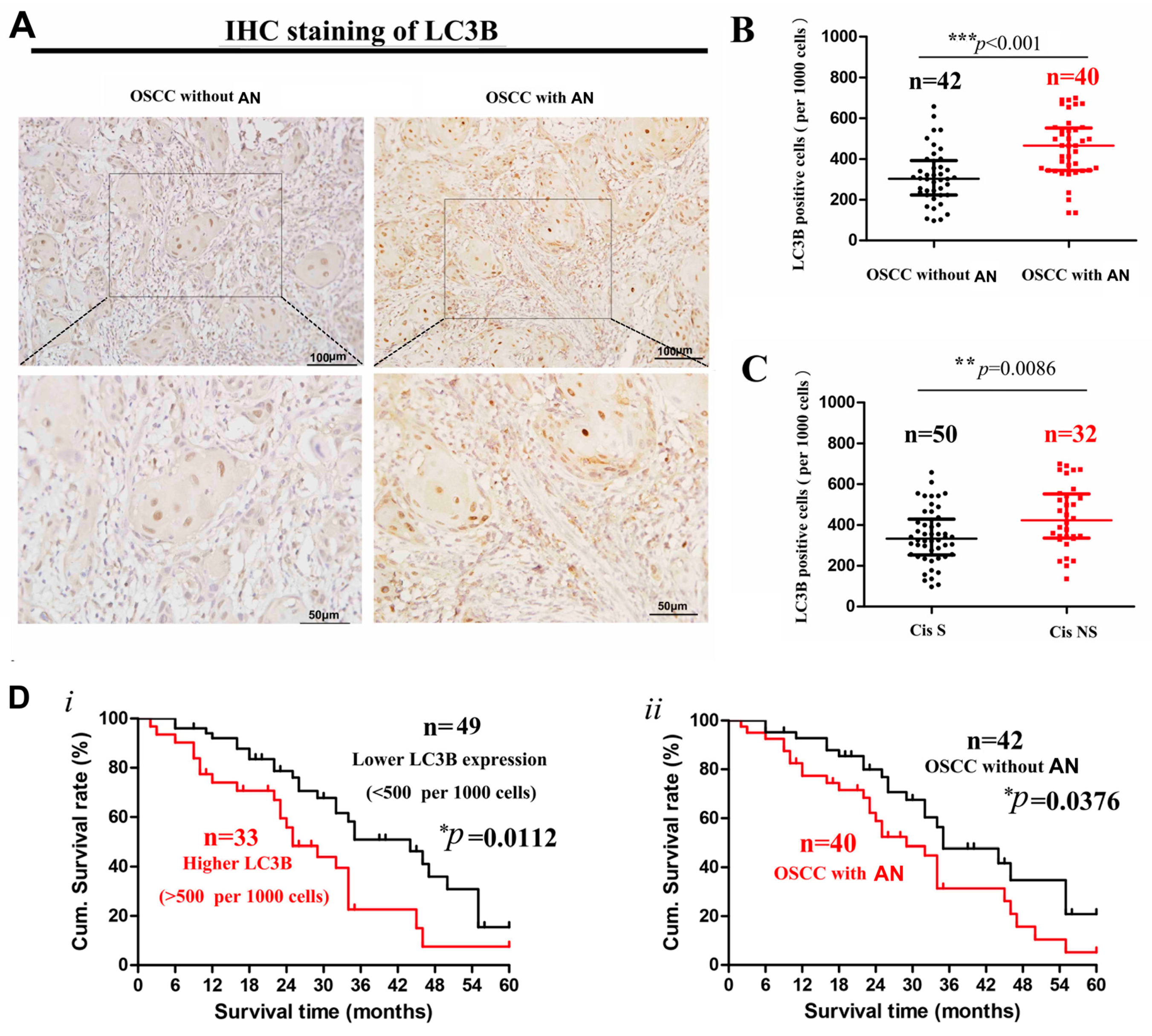
2.1. Decreased Cisplatin Sensitivity and Higher LC3 Expression in OSCC Patients with Areca Nut Chewing
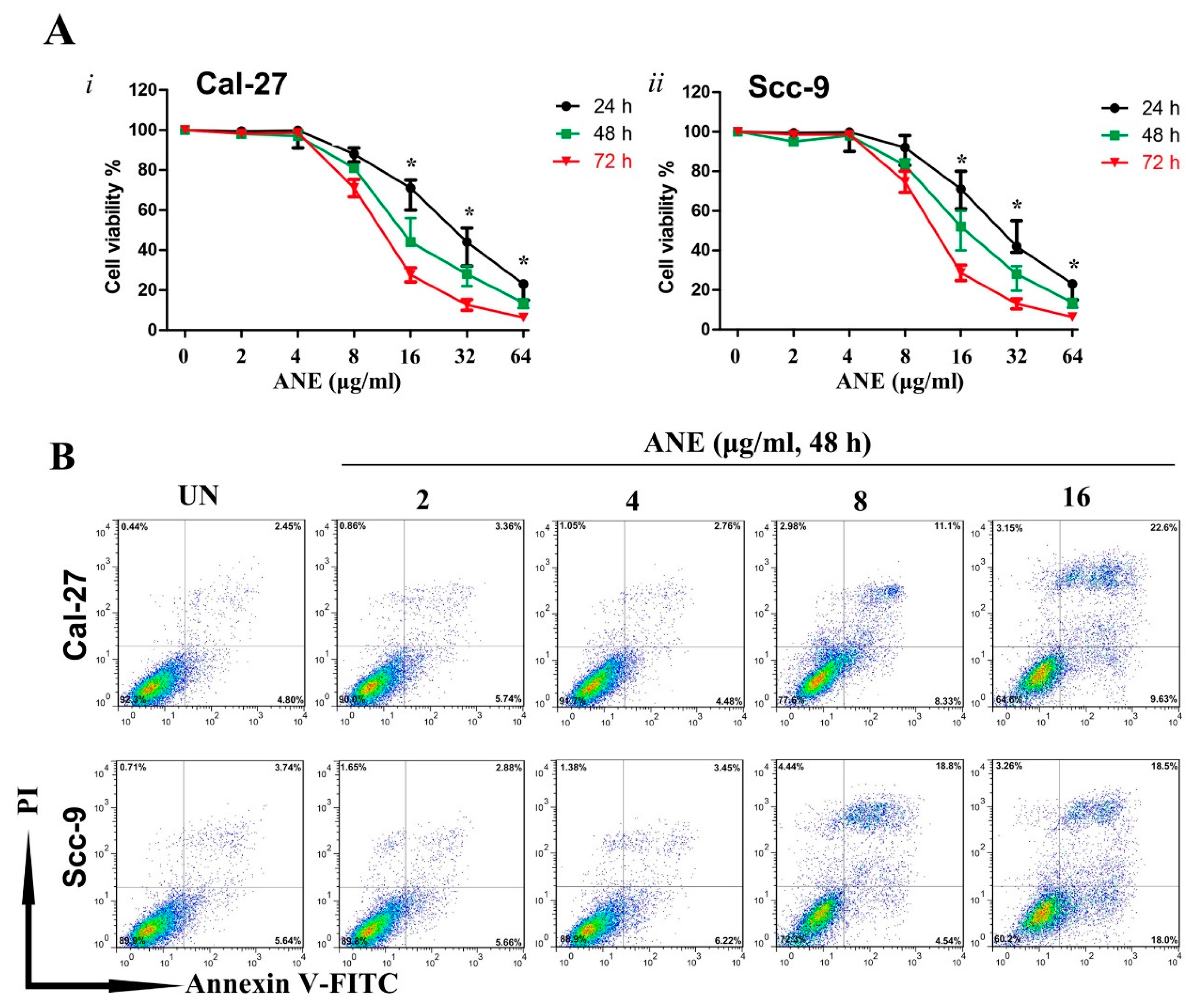
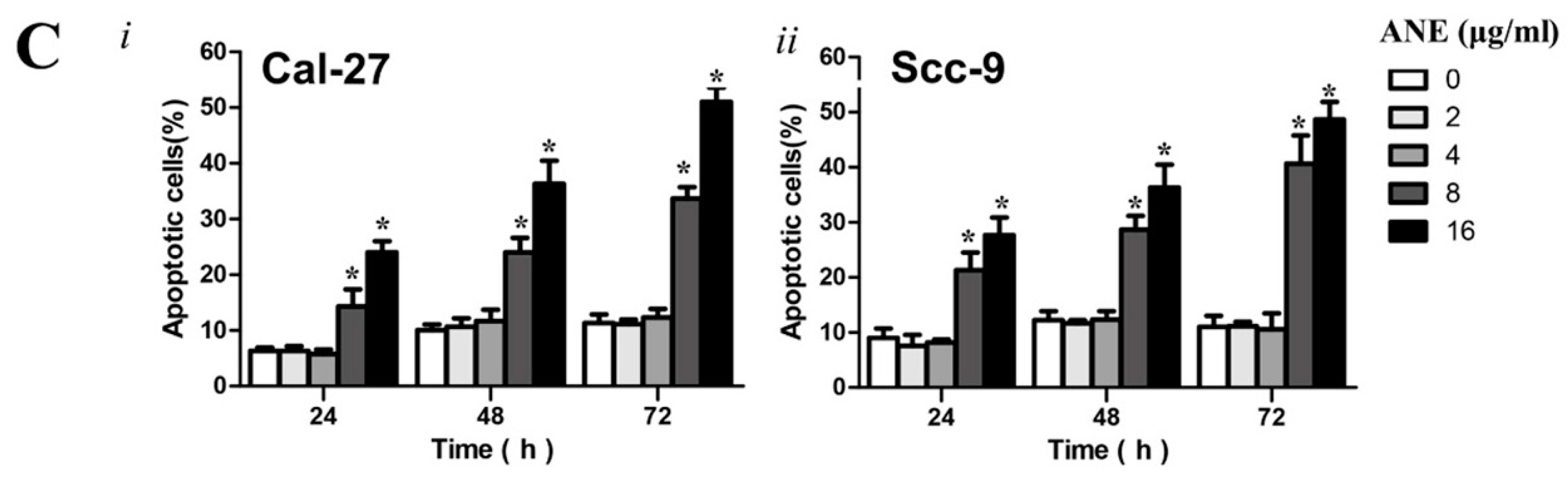
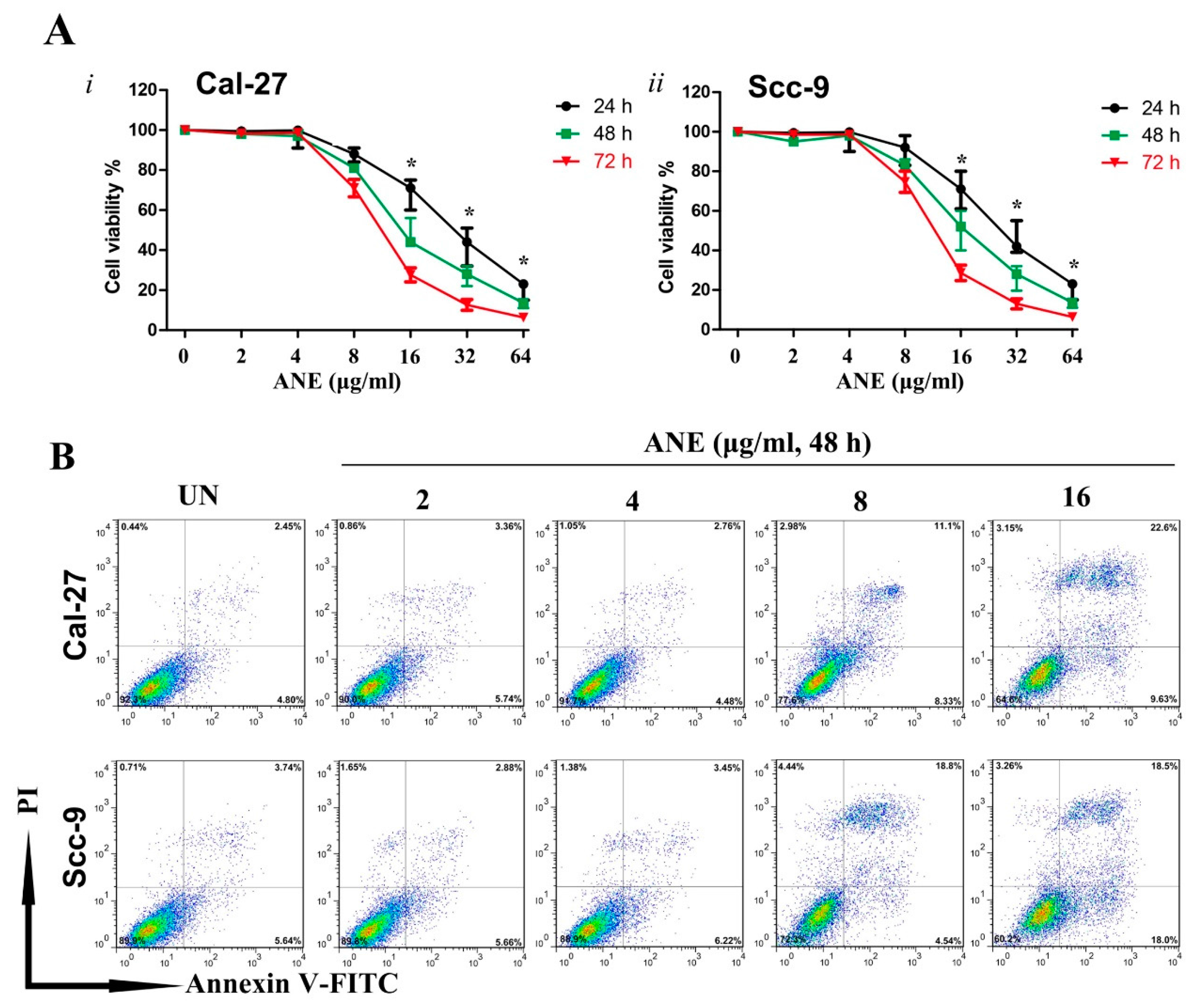
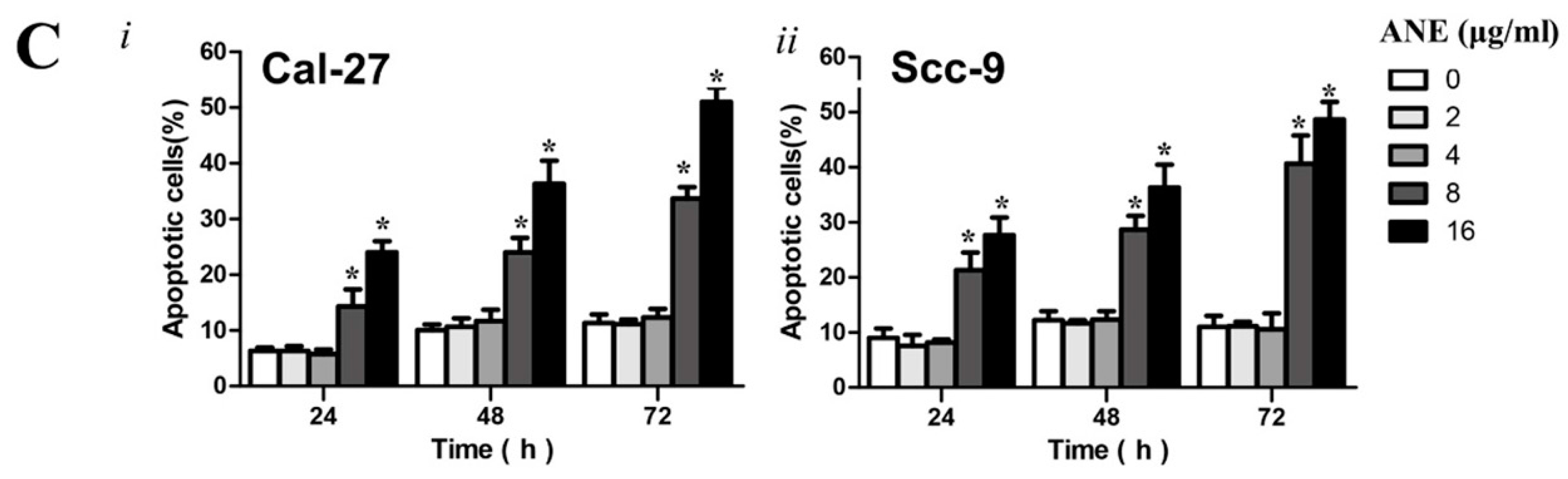
2.2. Low-Dose Usage of Areca Nut Extract (ANE) Showed No Significant Effects on Cell Viability and Apoptosis of Oral Squamous Cell Carcinoma (OSCC) Cells
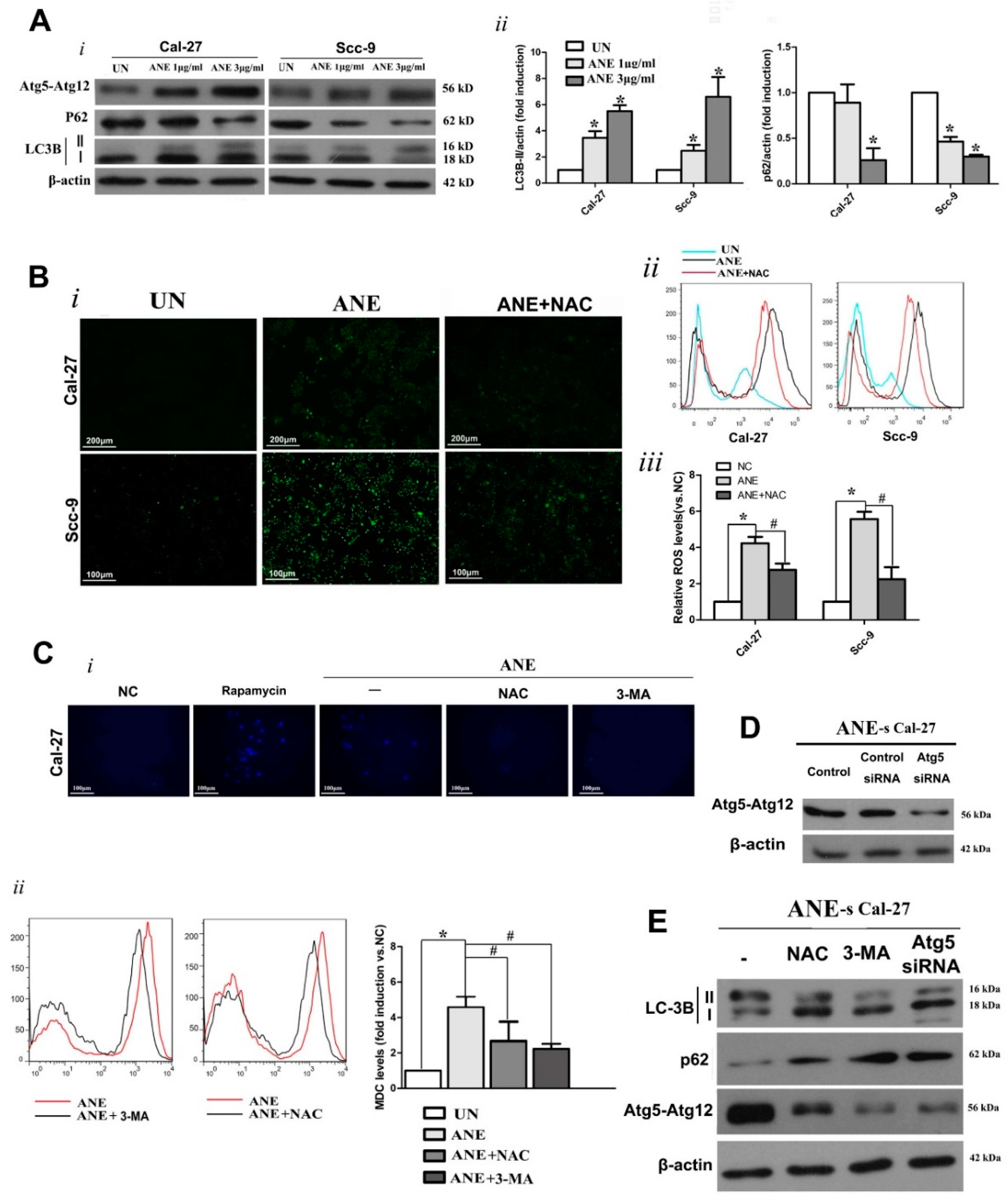
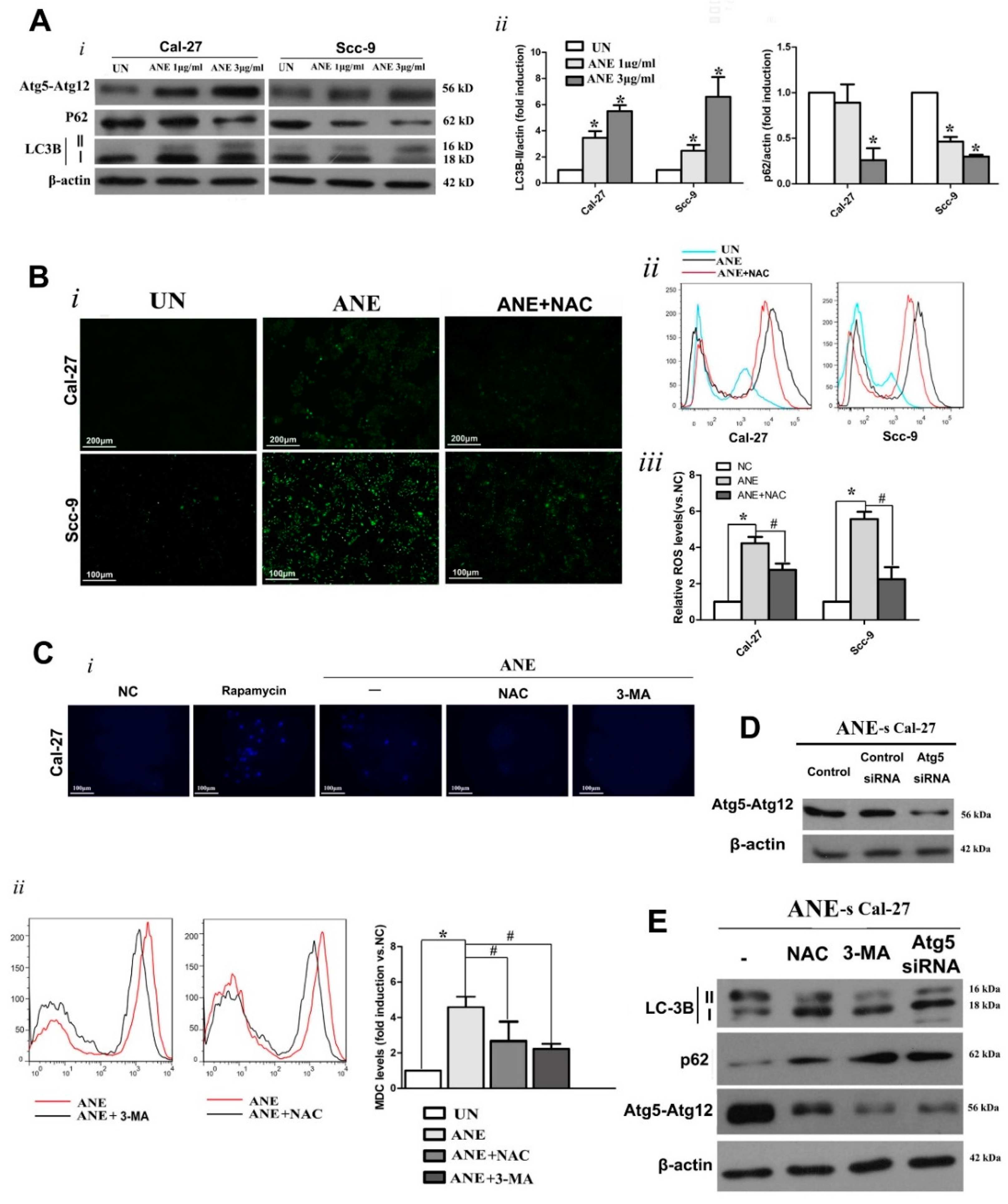
2.3. Autophagy Induced by ANE Was Mediated by Reactive Oxygen Species (ROS)
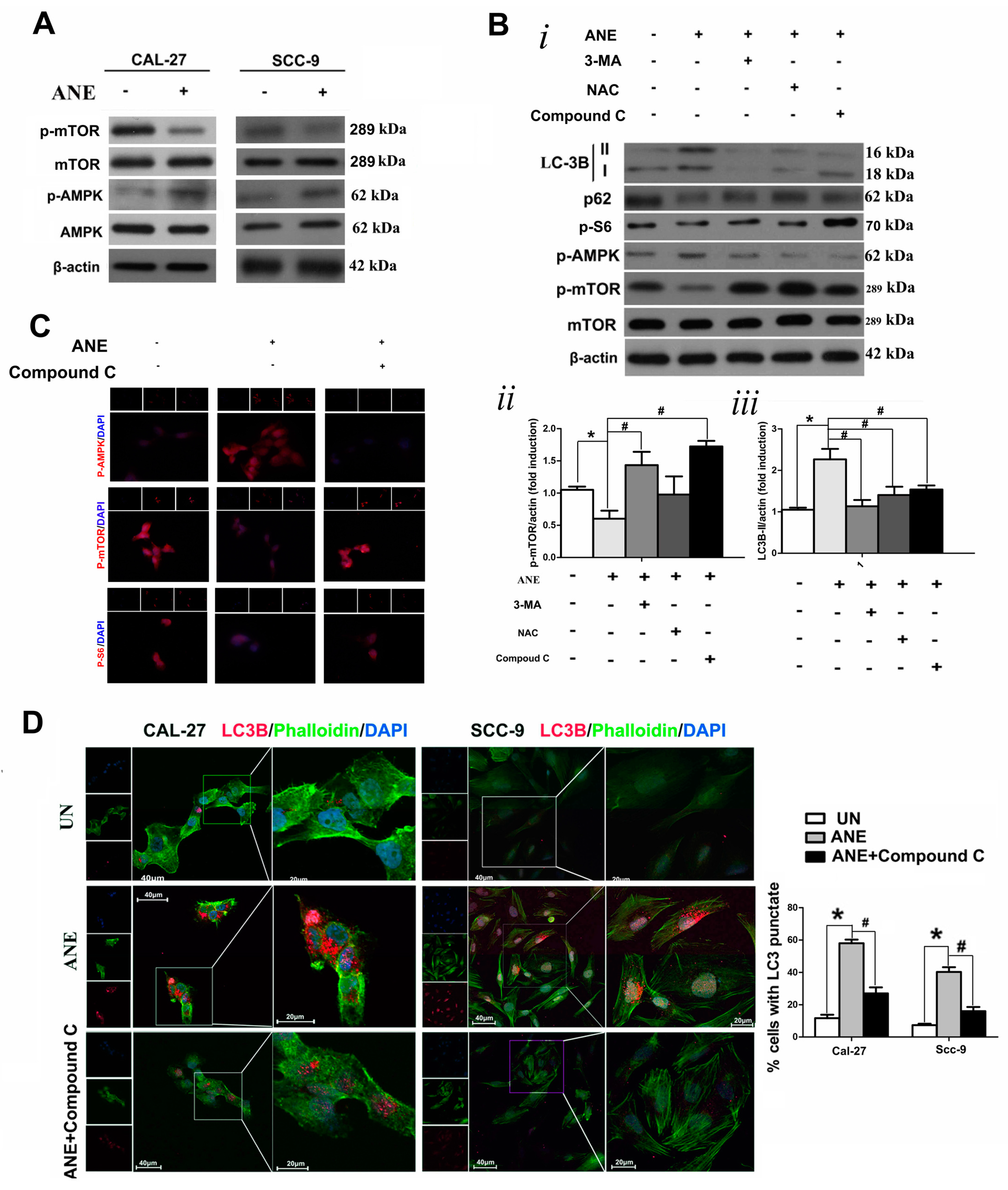
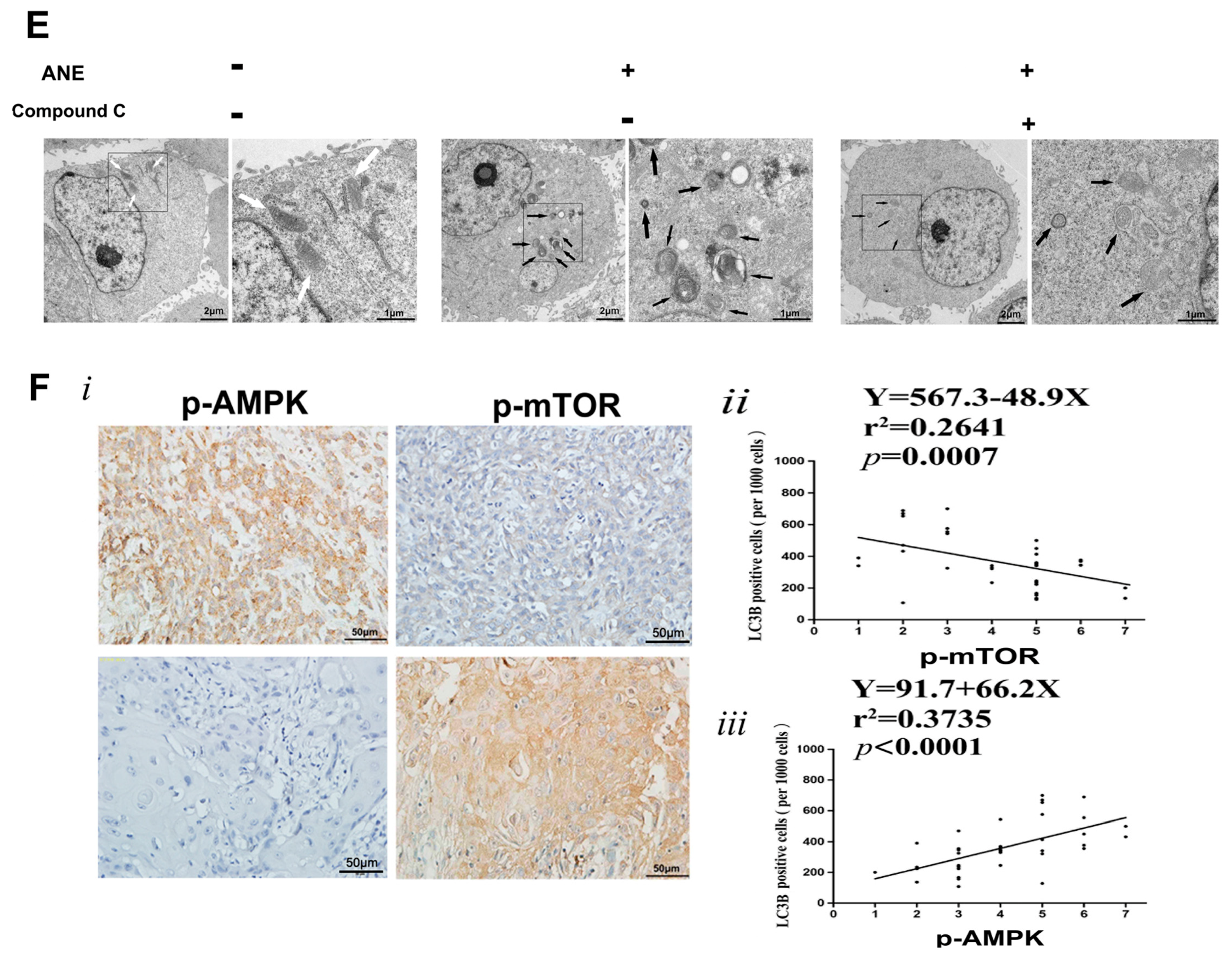
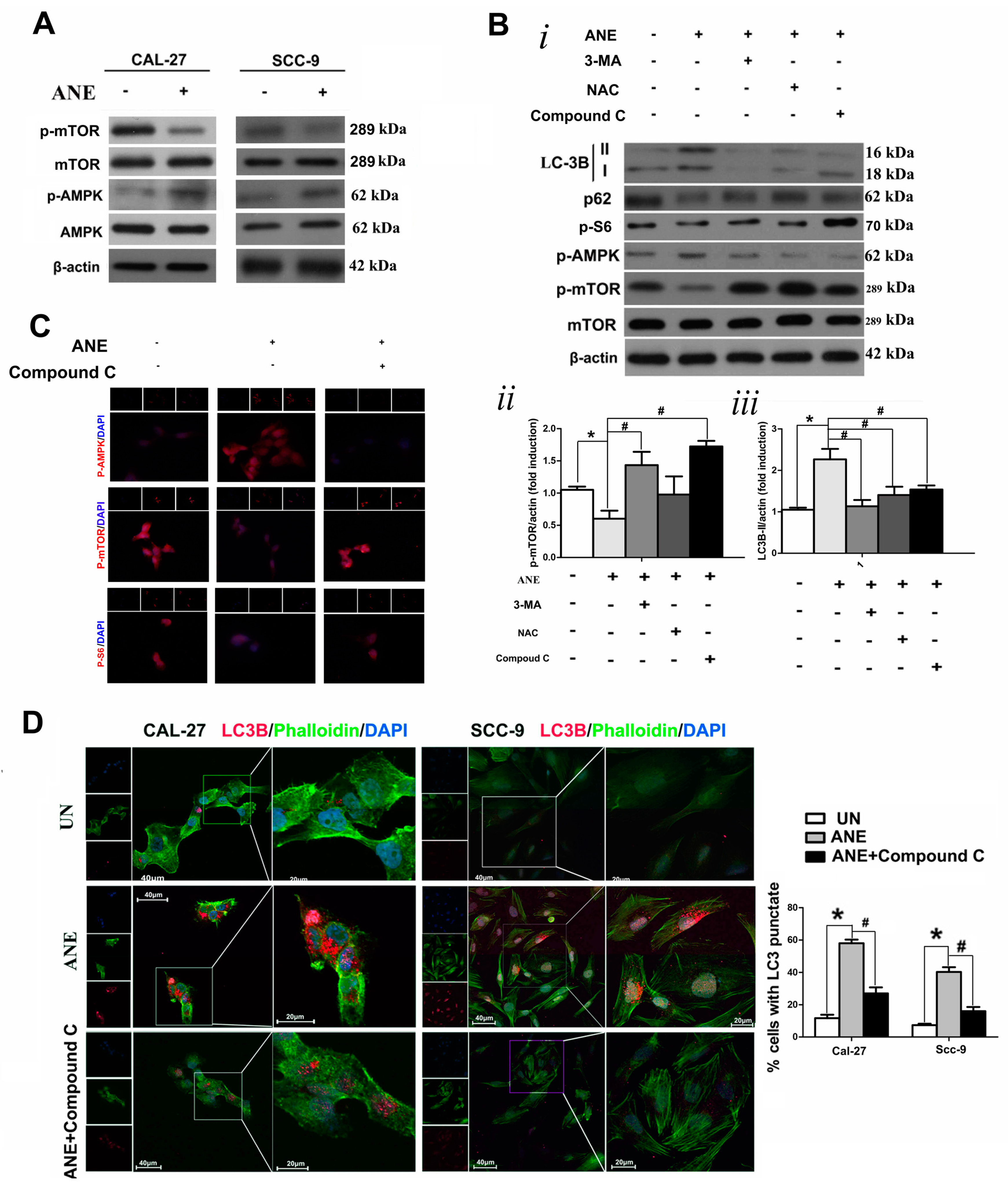
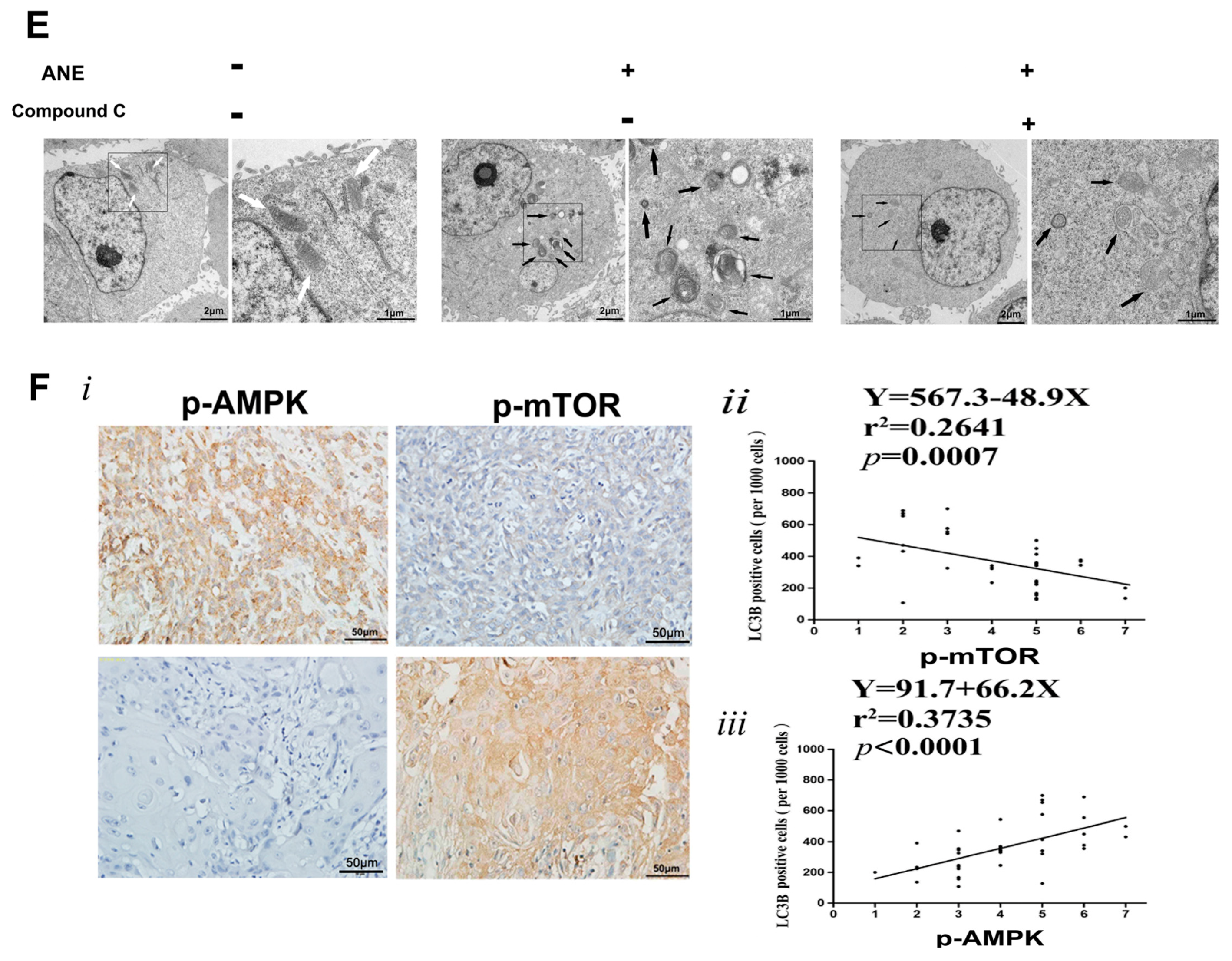
2.4. AMP-Activated Protein Kinase (AMPK)/mTOR Signaling Pathway Was Involved in ANE-Induced Autophagy
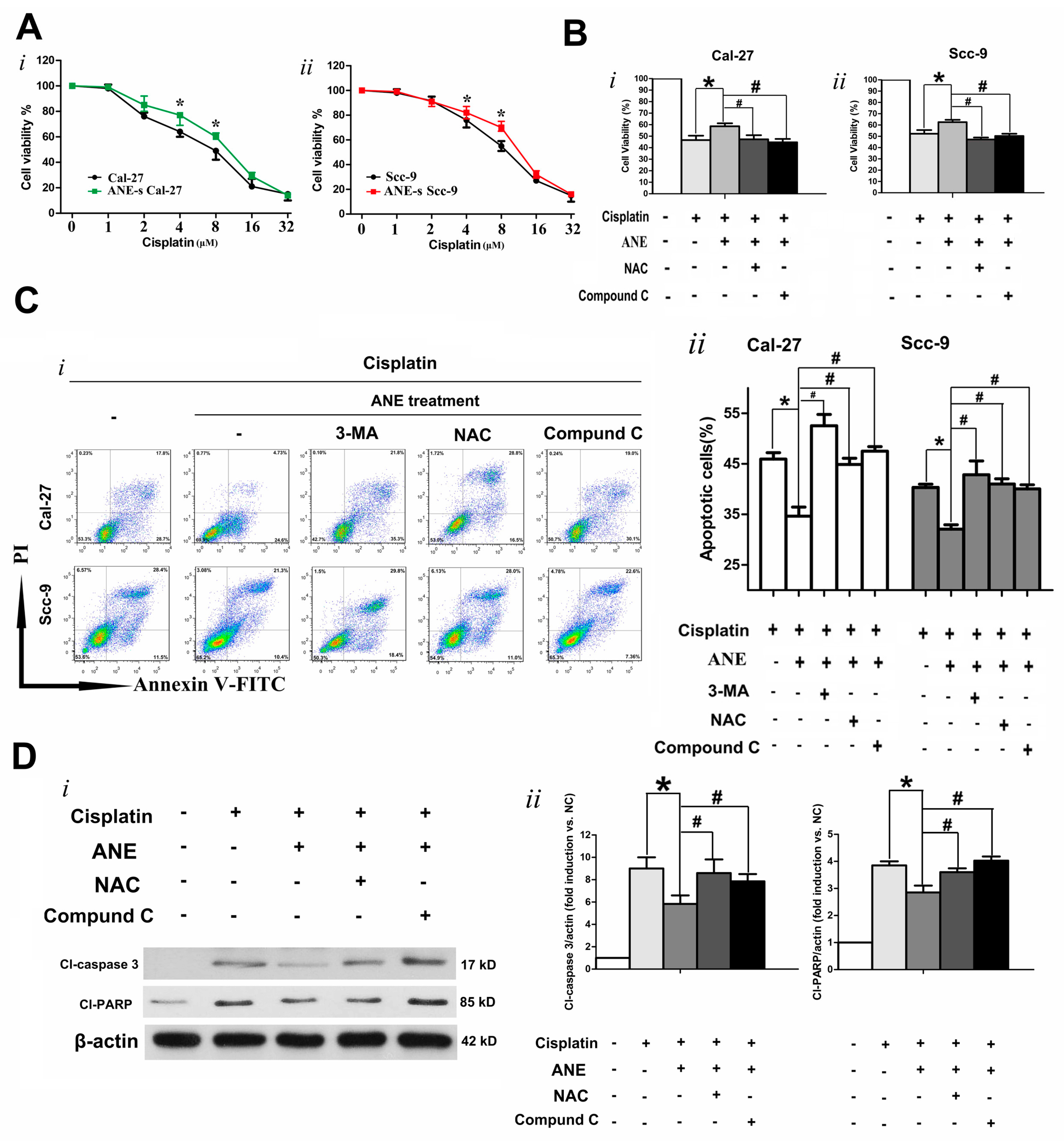
2.5. ROS/AMPK Mediated Autophagy Decreased Cisplatin Sensitivity in OSCC Cells
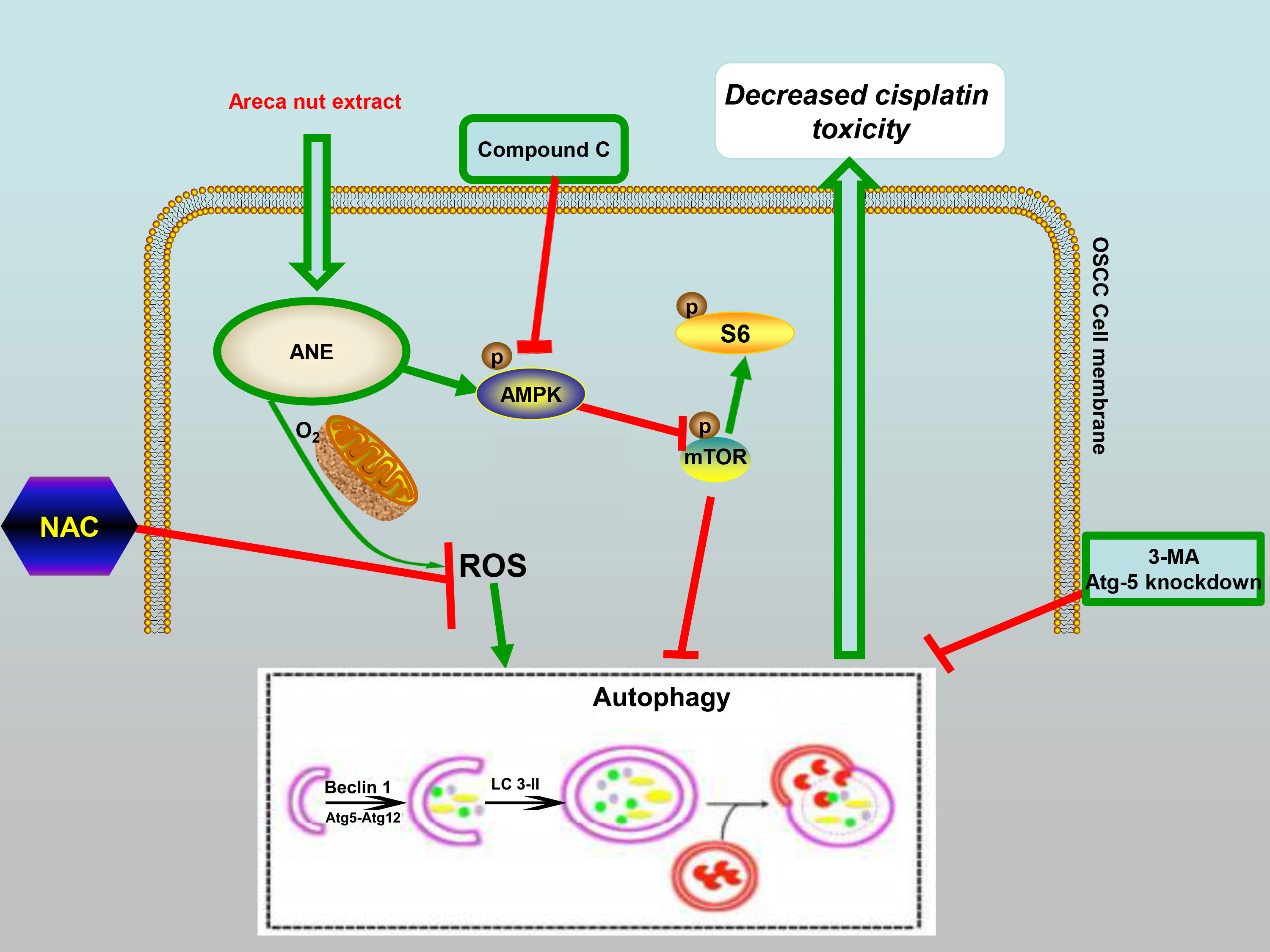
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Specimens
4.2. Chemicals and Reagents
4.3. Areca Nut Extract (ANE) Preparation
4.4. Cell Lines and Culture
4.5. Cell Viability Assay
4.6. Intracellular Reactive Oxygen Species (ROS) Level Detection
4.7. Cellular Immunofluorescence of OSCC
4.8. Monodansylcadaverine (MDC) Staining
4.9. Transmission Electron Microscopy (TEM) Analysis
4.10. Flow Cytometry Analysis of Cell Apoptosis
4.11. Small Interfering RNA (siRNA) Knockdown
4.12. Western Blot Analysis
4.13. Immunohistochemistry (IHC) Staining and Evaluation
4.14. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| OSCC | oral squamous cell carcinoma |
| ANE | areca nut extract |
| ROS | reactive oxygen species |
| CCK-8 | cell counting kit-8 |
| MDC | monodansylcadeverine |
| NAC | N-acetyl cysteine |
| DCF-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| 3-MA | 3-methyladenine |
| LC3 | microtubule-associated protein 1 light chain 3 |
| Atg5 | autophagy related 5 |
| Atg12 | autophagy related 12 |
| SQSTM1 | sequestosome 1 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| IC50 | 50% inhibitory concentration |
| IHC | immunohistochemical |
| Cl-PARP | cleaved-Poly-(ADP-ribose) polymerase |
| AMPK | adenosine monophosate-activated protein kinase |
| mTOR | mechanistic target of rapamycin |
| DMEM | Dulbecco’s modified Eagle’s medium |
| TEM | transmission electron microscopy |
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.I.; Shin, D.M. Recent advances in head and neck cancer. N. Engl. J. Med. 2008, 359, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Guneri, P.; Epstein, J.B. Late stage diagnosis of oral cancer: Components and possible solutions. Oral Oncol. 2014, 50, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.K.; Li, Y.; Murphy, B.; Hussain, M.H.; DeConti, R.C.; Ensley, J.; Forastiere, A.A.; Eastern Cooperative Oncology Group. Randomized phase III evaluation of cisplatin plus fluorouracil versus cisplatin plus paclitaxel in advanced head and neck cancer (E1395): An intergroup trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2005, 23, 3562–3567. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chang, J.T.; Liao, C.T.; Wang, H.M.; Yen, T.C.; Chiu, C.C.; Lu, Y.C.; Li, H.F.; Cheng, A.J. Head and neck cancer in the betel quid chewing area: Recent advances in molecular carcinogenesis. Cancer Sci. 2008, 99, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Petti, S. Lifestyle risk factors for oral cancer. Oral Oncol. 2009, 45, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Pant, I.; Narra, S.; Radhesh, R.; Ranganathan, K.; Rao, S.G.; Kondaiah, P. Epithelial atrophy in oral submucous fibrosis is mediated by copper(II) and arecoline of areca nut. J. Cell. Mol. Med. 2015, 19, 2397–2412. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, K.; Liu, Y.; Nair, J.; Bartsch, H.; Arvidson, K.; Grafstrom, R.C. Cytotoxic and genotoxic effects of areca nut-related compounds in cultured human buccal epithelial cells. Cancer Res. 1989, 49, 5294–5298. [Google Scholar] [PubMed]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; Wang, X.; He, C.; Pan, H. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Sannigrahi, M.K.; Singh, V.; Sharma, R.; Panda, N.K.; Khullar, M. Role of autophagy in head and neck cancer and therapeutic resistance. Oral Dis. 2015, 21, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yu, H.; Qin, H.; Kang, J.; Yu, C.; Zhong, J.; Su, J.; Li, H.; Sun, L. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012, 314, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.M.; Bourachot, B.; Zago, G.; Bieche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy 2014, 10, 2122–2142. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Lu, R.; Ye, X.J.; Zhang, J.; Tan, Y.Q.; Zhou, G. Icaritin Reduces Oral Squamous Cell Carcinoma Progression via the Inhibition of STAT3 Signaling. Int. J. Mol. Sci. 2017, 18, 132. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, L.; Ju, Y.; Li, W.; Zhang, M.; Jiao, Y.; Zhang, J.; Wang, S.; Wang, Y.; Zhao, M.; et al. A novel androstenedione derivative induces ROS-mediated autophagy and attenuates drug resistance in osteosarcoma by inhibiting macrophage migration inhibitory factor (MIF). Cell Death Dis. 2014, 5, e1361. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Teo, A.E.; McCarty, N. ROS-Induced CXCR4 Signaling Regulates Mantle Cell Lymphoma (MCL) Cell Survival and Drug Resistance in the Bone Marrow Microenvironment via Autophagy. Clin. Cancer Res. 2015, 22, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Chen, Y.S.; Chou, S.H.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Huang, C.Y.; Lo, J.F. Distinct Subpopulations of Head and Neck Cancer Cells with Different Levels of Intracellular Reactive Oxygen Species Exhibit Diverse Stemness, Proliferation, and Chemosensitivity. Cancer Res. 2014, 74, 6291–6305. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Hsieh, W.-F.; Chiang, W.-F.; Hong, W.-Z.; Hsu, Y.-R.; Cheng, Y.-C.; Chen, T.-C.; Hsu, K.-C.; Lina, P.-Y.; Liu, S.-Y.; et al. Autophagy induction by the 30–100 kDa fraction of areca nut in both normal and malignant cells through reactive oxygen species. Oral Oncol. 2010, 46, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Kemp, B.E.; Stapleton, D.; Campbell, D.J.; Chen, Z.P.; Murthy, S.; Walter, M.; Gupta, A.; Adams, J.J.; Katsis, F.; van Denderen, B.; et al. AMP-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 2003, 31 Pt 1, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Vaishampayan, S.S.; Nair, S.; Nair, D.; Agarwal, J.P.; Kane, S.V.; Pawar, P.; Datta, S. Oral squamous cell carcinoma arising in background of oral submucous fibrosis: A clinicopathologically distinct disease. Head Neck 2013, 35, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, K.; Takahashi, H.; Kaira, K.; Toyoda, M.; Oyama, T.; Chikamatsu, K. Immunological significance of the accumulation of autophagy components in oral squamous cell carcinoma. Cancer Sci. 2015, 106, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Agarwal, C.; Wadhwa, R.; Serkova, N.J.; Agarwal, R. Energy deprivation by silibinin in colorectal cancer cells: A double-edged sword targeting both apoptotic and autophagic machineries. Autophagy 2013, 9, 697–713. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Liu, S.Y.; Liu, Y.C. Autophagy induction by a natural ingredient of areca nut. Autophagy 2008, 4, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.-T.; Yang, S.-R.; Chen, J.Y.-F.; Cheng, Y.-P.; Lee, Y.-R.; Chiang, M.-K.; Chen, H.-R. Arecoline downregulates levels of p21 and p27 through the reactive oxygen species/mTOR complex 1 pathway and may contribute to oral squamous cell carcinoma. Cancer Sci. 2012, 103, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Kim, Y.; Cho, D.H.; Jeong, S.Y.; Kim, S.B.; Suh, N.; Lee, J.S.; Choi, E.K.; Koh, J.Y.; Hwang, J.J.; et al. Raloxifene induces autophagy-dependent cell death in breast cancer cells via the activation of AMP-activated protein kinase. Mol. Cells 2015, 38, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Qiang, L.; Han, W.; Ming, M.; Viollet, B.; He, Y.Y. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene 2013, 32, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, G.D.; Ropelle, E.R.; Rocha, G.Z.; Carvalheira, J.B. The role of neuronal AMPK as a mediator of nutritional regulation of food intake and energy homeostasis. Metab. Clin. Exp. 2013, 62, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gollavilli, P.N.; Kanugula, A.K.; Koyyada, R.; Karnewar, S.; Neeli, P.K.; Kotamraju, S. AMPK inhibits MTDH expression via GSK3β and SIRT1 activation: Potential role in triple negative breast cancer cell proliferation. FEBS J. 2015, 282, 3971–3985. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK β1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 α protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-Y.; Lin, M.-H.; Liu, S.-Y.; Chiang, W.-F.; Hsieh, W.-F.; Cheng, Y.-C.; Hsu, K.-C.; Liu, Y.-C. Arecoline-mediated inhibition of AMP-activated protein kinase through reactive oxygen species is required for apoptosis induction. Oral Oncol. 2011, 47, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, X.-P.; Song, K.; Shang, Z.-J. Ephrin-A1 is up-regulated by hypoxia in cancer cells and promotes angiogenesis of HUVECs through a coordinated cross-talk with eNOS. PLoS ONE 2013, 8, e74464. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, X.P.; Xu, Z.; Yan, T.L.; Song, Y.; Song, K.; Huang, C.M.; Wang, L.; Zhou, X.C.; Jiang, E.H.; Shao, Z.; Shang, Z.J. EphA2 silencing promotes growth, migration, and metastasis in salivary adenoid cystic carcinoma: In vitro and in vivo study. Am. J. Transl. Res. 2016, 8, 1518–1529. [Google Scholar] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Huang, C.-M.; Shao, Z.; Zhao, X.-P.; Wang, M.; Yan, T.-L.; Zhou, X.-C.; Jiang, E.-H.; Liu, K.; Shang, Z.-J. Autophagy Induced by Areca Nut Extract Contributes to Decreasing Cisplatin Toxicity in Oral Squamous Cell Carcinoma Cells: Roles of Reactive Oxygen Species/AMPK Signaling. Int. J. Mol. Sci. 2017, 18, 524. https://doi.org/10.3390/ijms18030524
Xu Z, Huang C-M, Shao Z, Zhao X-P, Wang M, Yan T-L, Zhou X-C, Jiang E-H, Liu K, Shang Z-J. Autophagy Induced by Areca Nut Extract Contributes to Decreasing Cisplatin Toxicity in Oral Squamous Cell Carcinoma Cells: Roles of Reactive Oxygen Species/AMPK Signaling. International Journal of Molecular Sciences. 2017; 18(3):524. https://doi.org/10.3390/ijms18030524
Chicago/Turabian StyleXu, Zhi, Chun-Ming Huang, Zhe Shao, Xiao-Ping Zhao, Meng Wang, Ting-Lin Yan, Xiao-Cheng Zhou, Er-Hui Jiang, Ke Liu, and Zheng-Jun Shang. 2017. "Autophagy Induced by Areca Nut Extract Contributes to Decreasing Cisplatin Toxicity in Oral Squamous Cell Carcinoma Cells: Roles of Reactive Oxygen Species/AMPK Signaling" International Journal of Molecular Sciences 18, no. 3: 524. https://doi.org/10.3390/ijms18030524