
The CpG Dinucleotide Adjacent to a κB Site Affects NF-κB Function through Its Methylation

Abstract
:
1. Introduction
2. Results
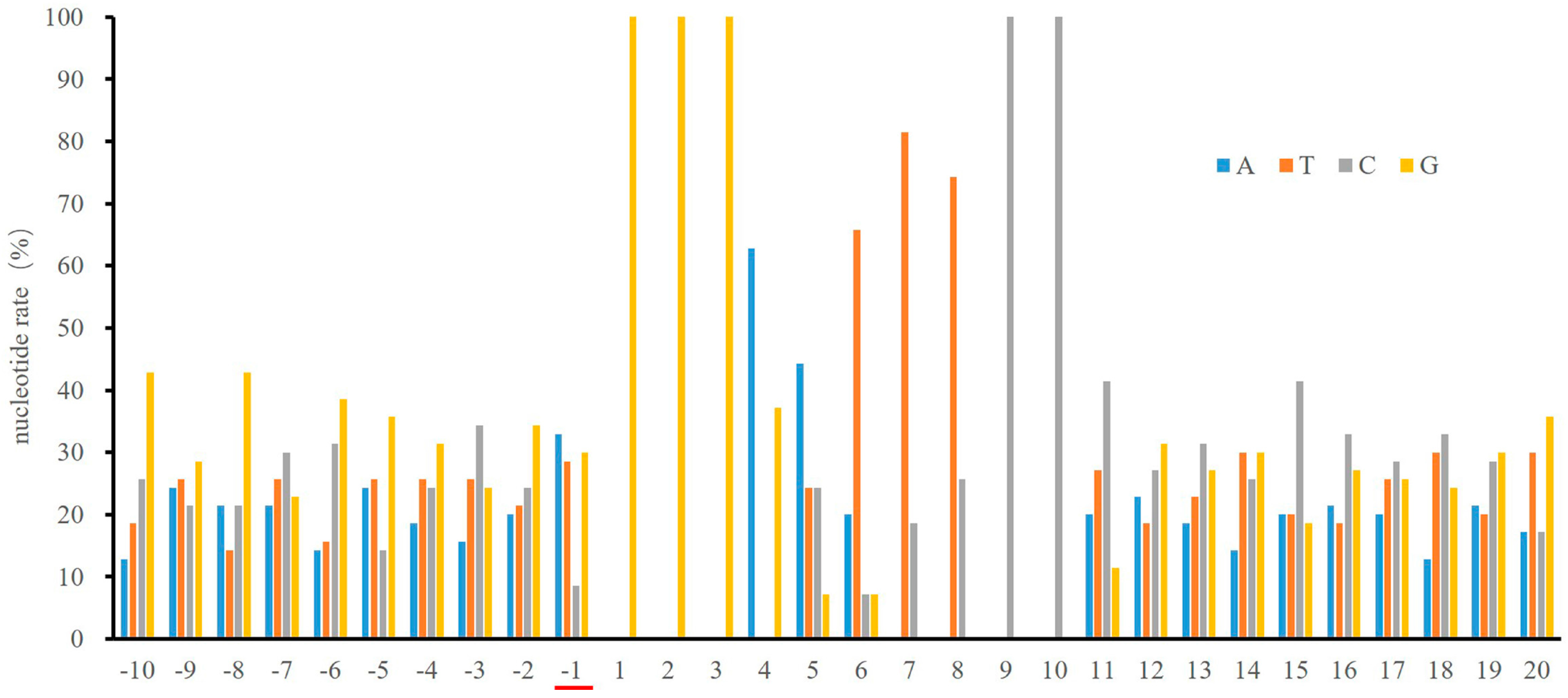
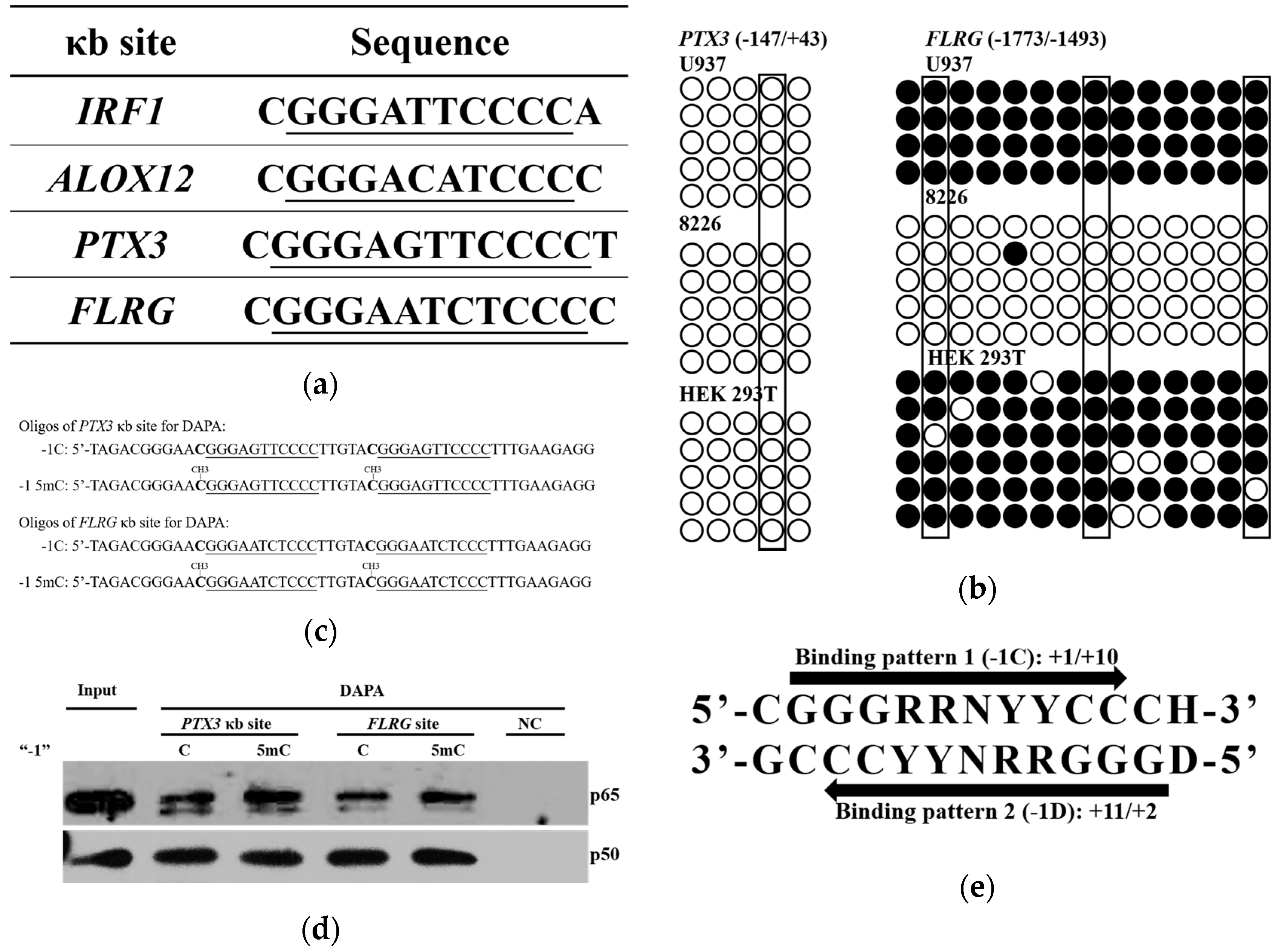
2.1. Low Frequency of a Cytosine at the -1 Position of the κB Sites
2.2. NF-κB DNA Binding Capacity is not Affected by the Nucleotide at the -1 Position
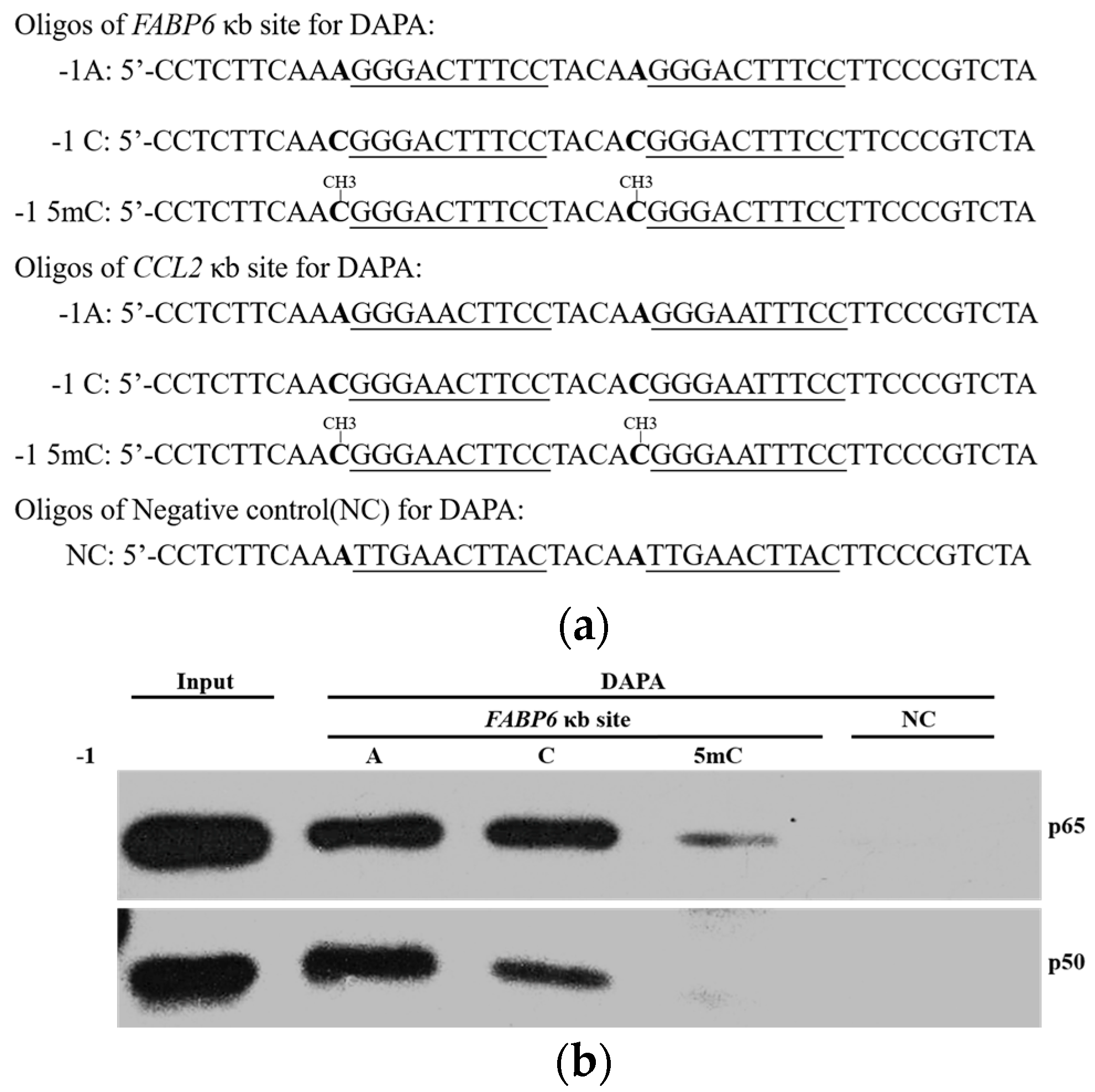
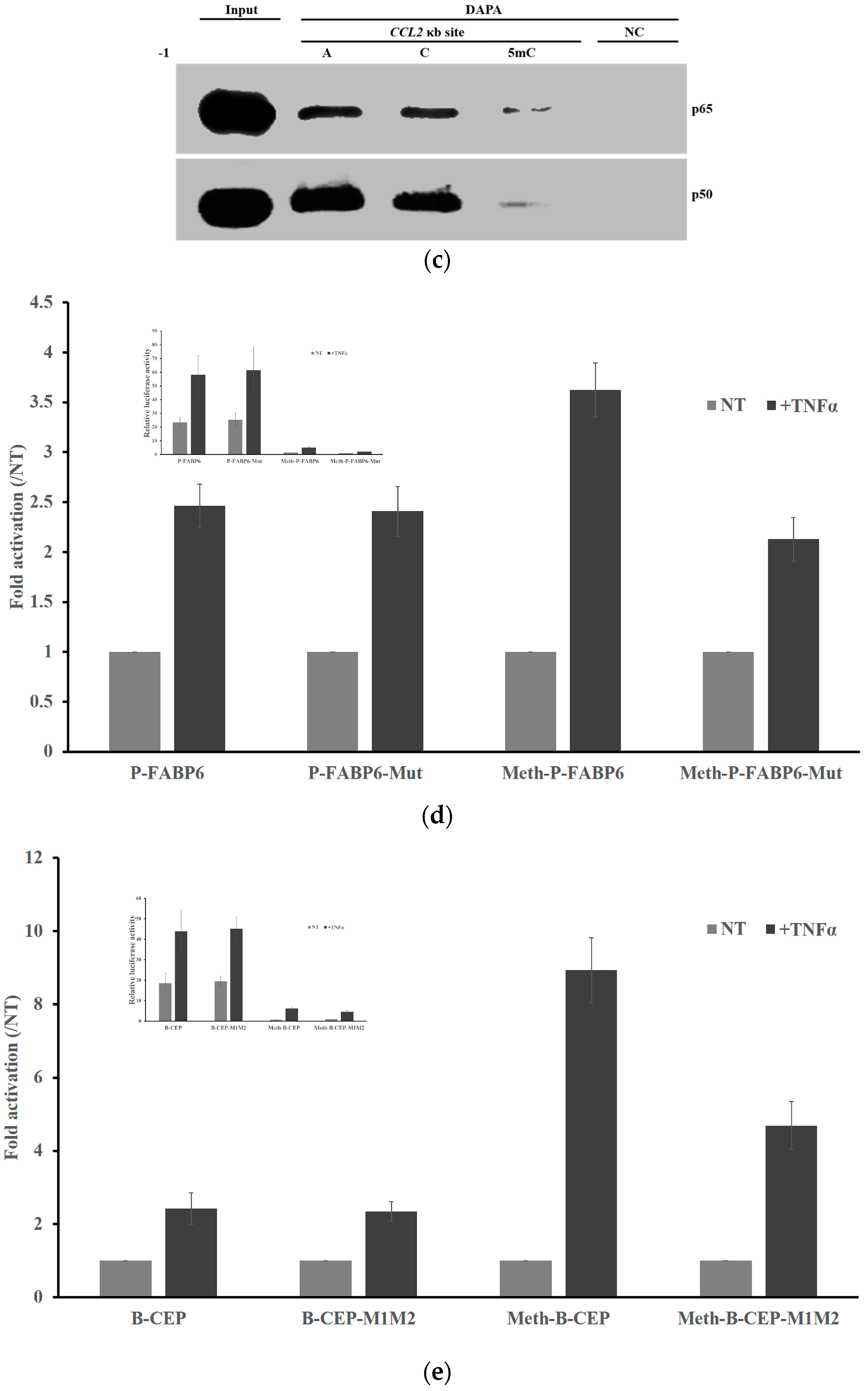
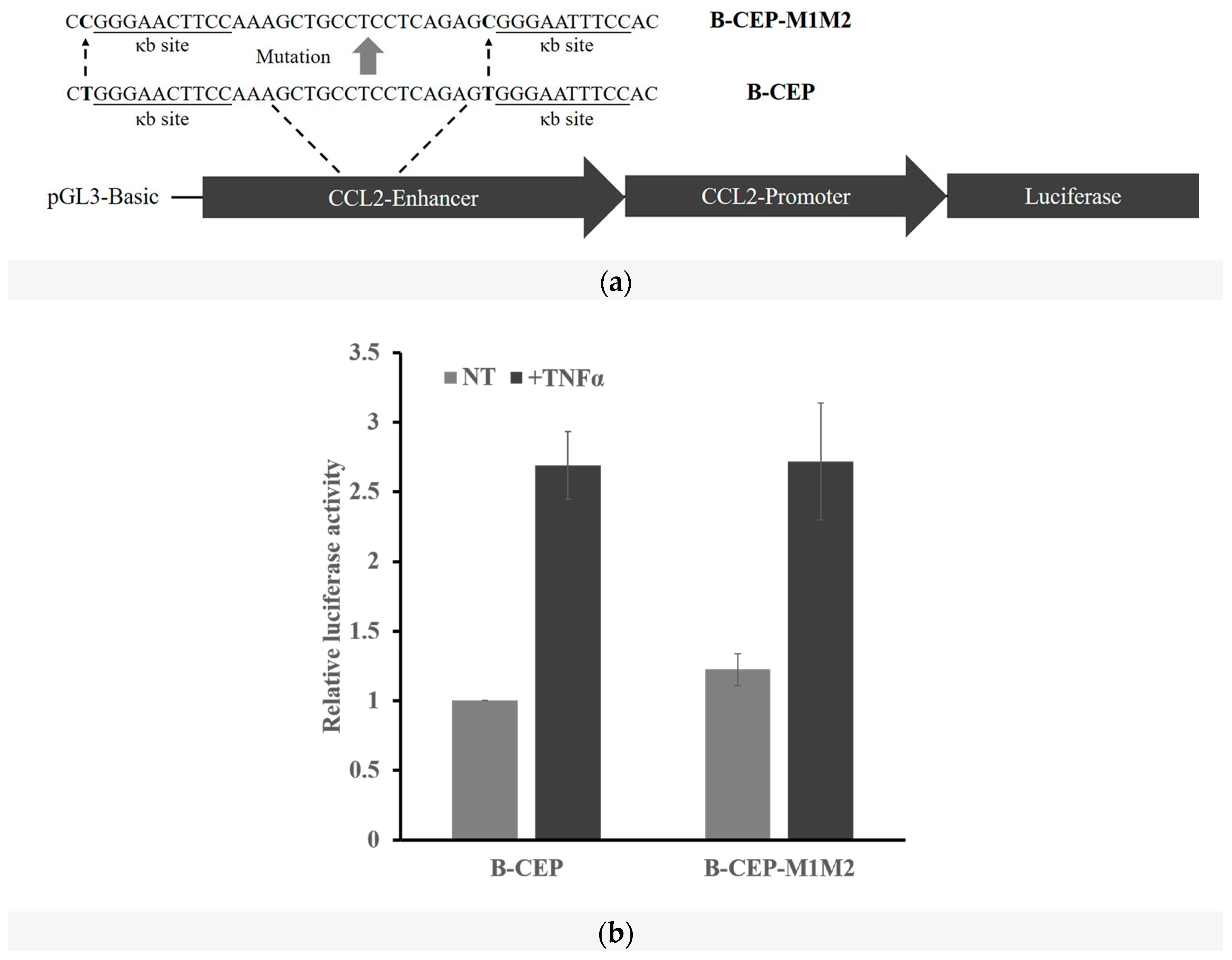
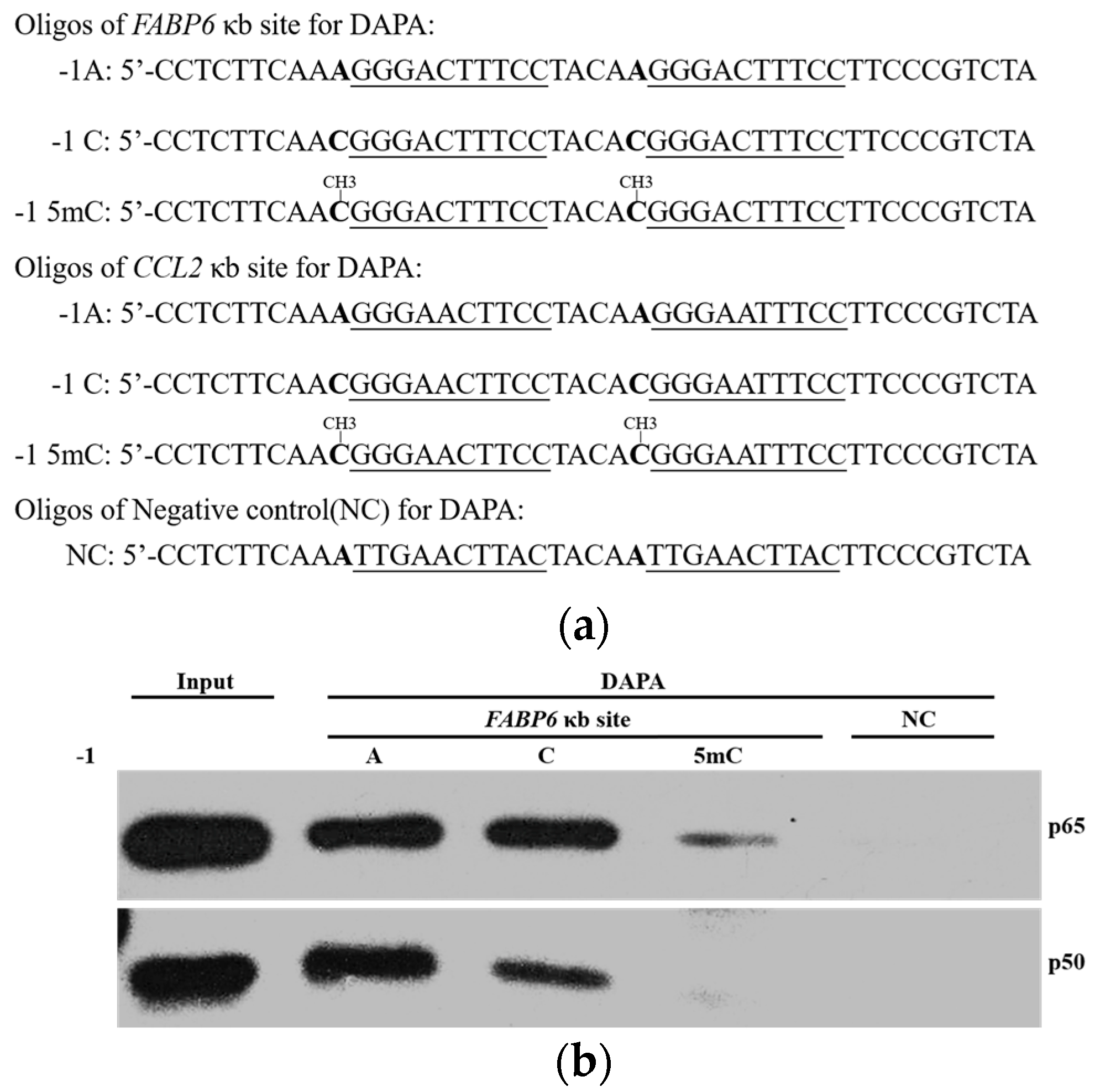
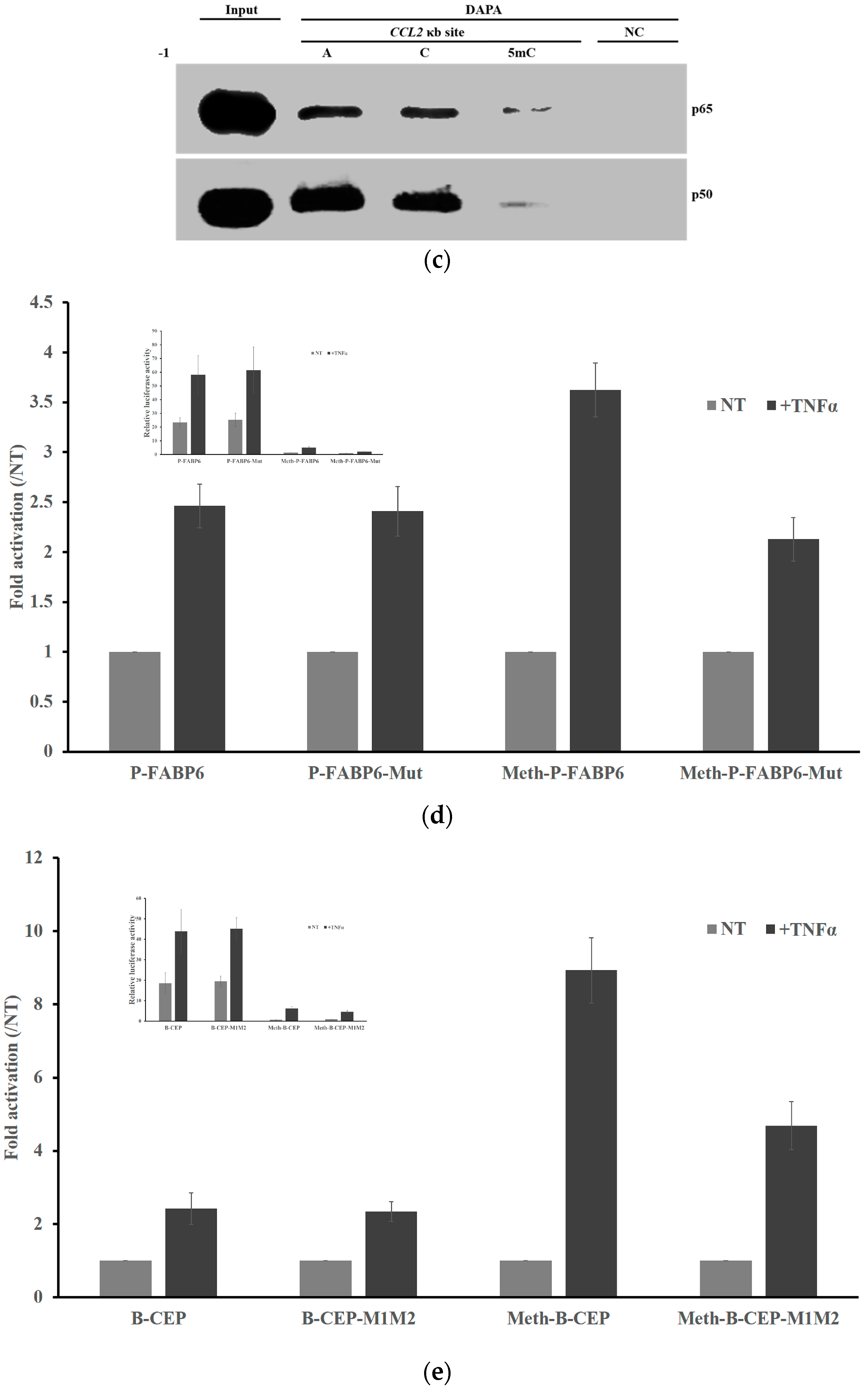
2.3. DNA Methylation at the -1C Inhibits DNA Binding and Thereafter the Function of NF-κB
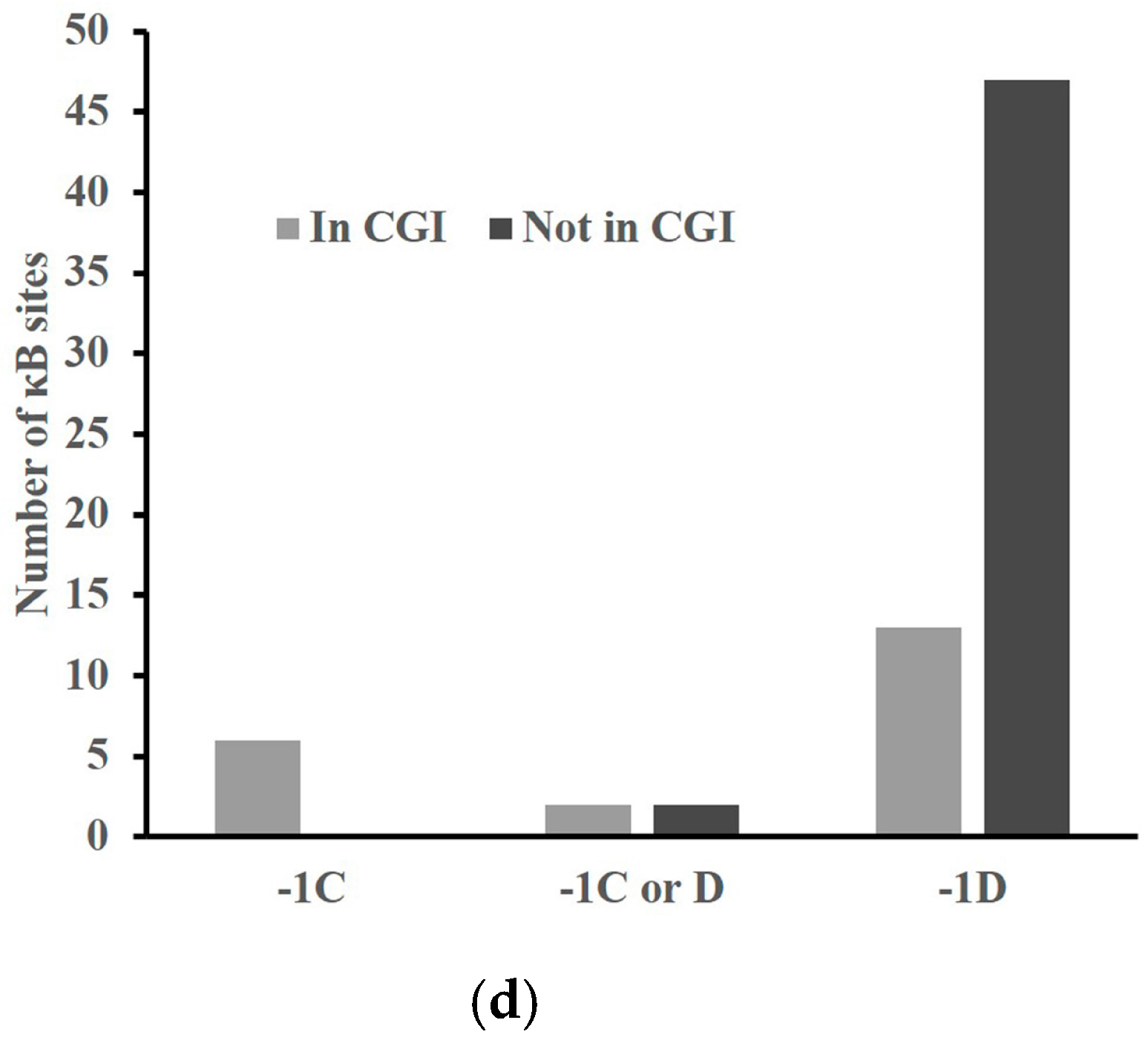
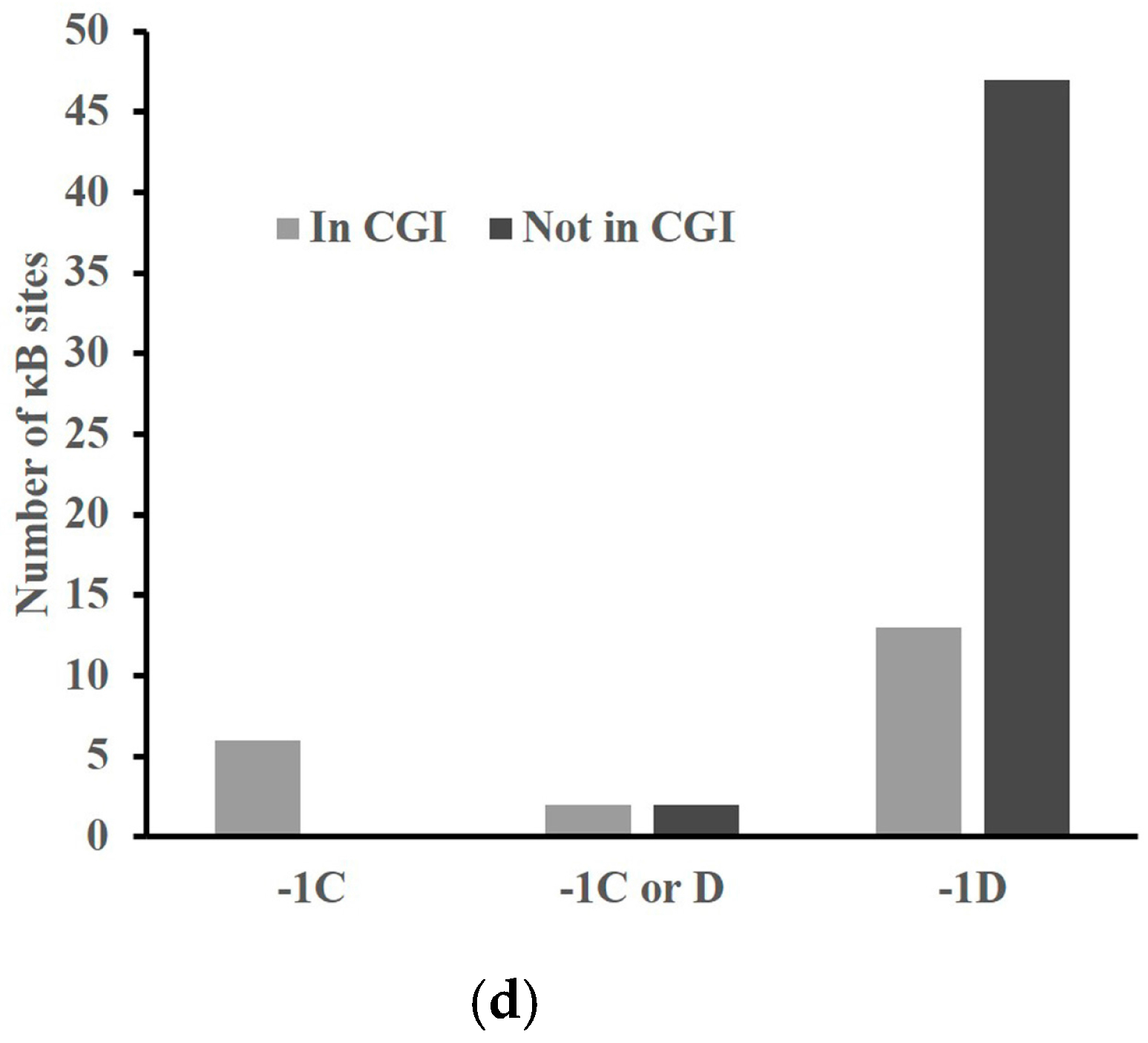
2.4. A -1C κB Site Is Preferably Located in a CGI to Avoid Methylation
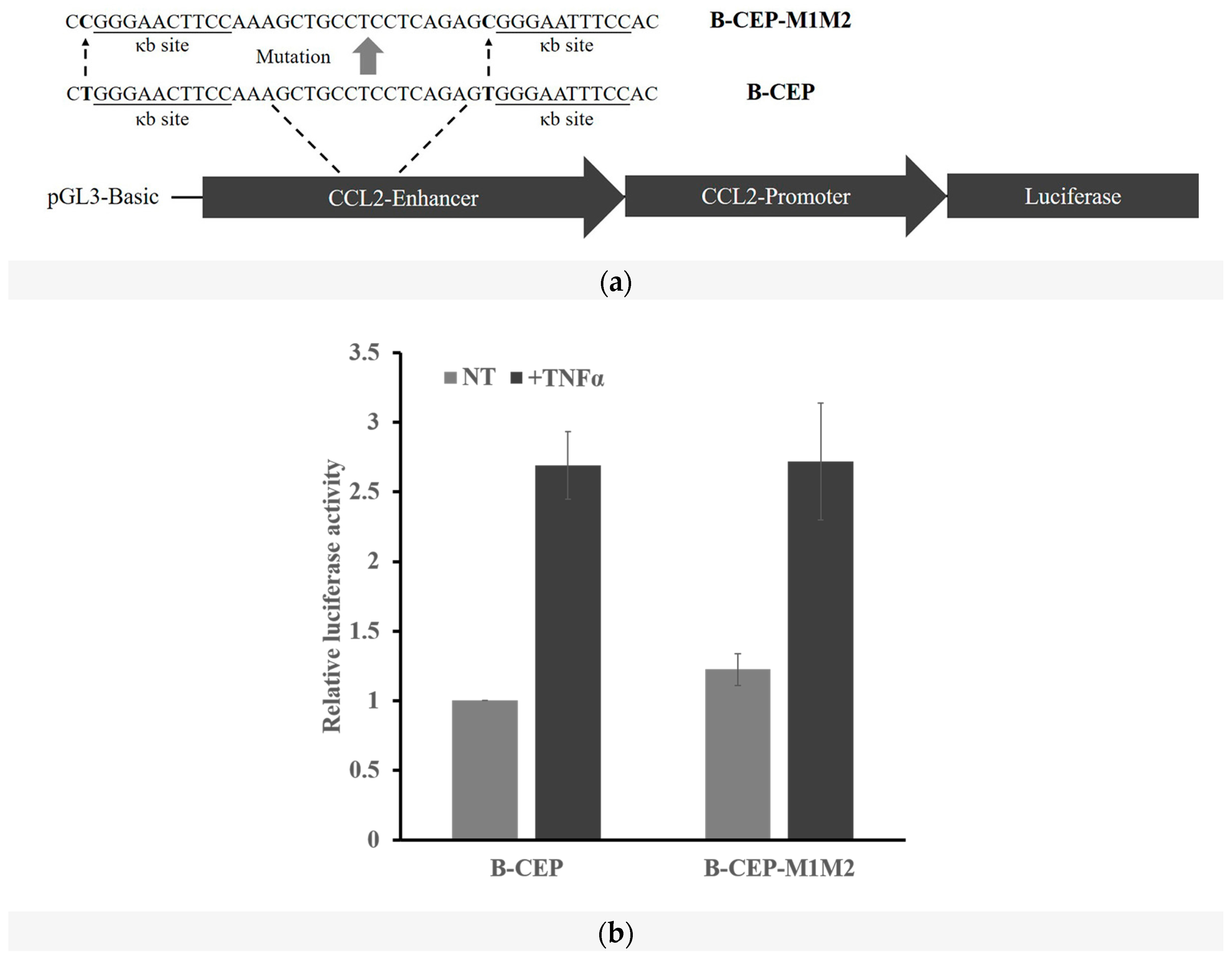
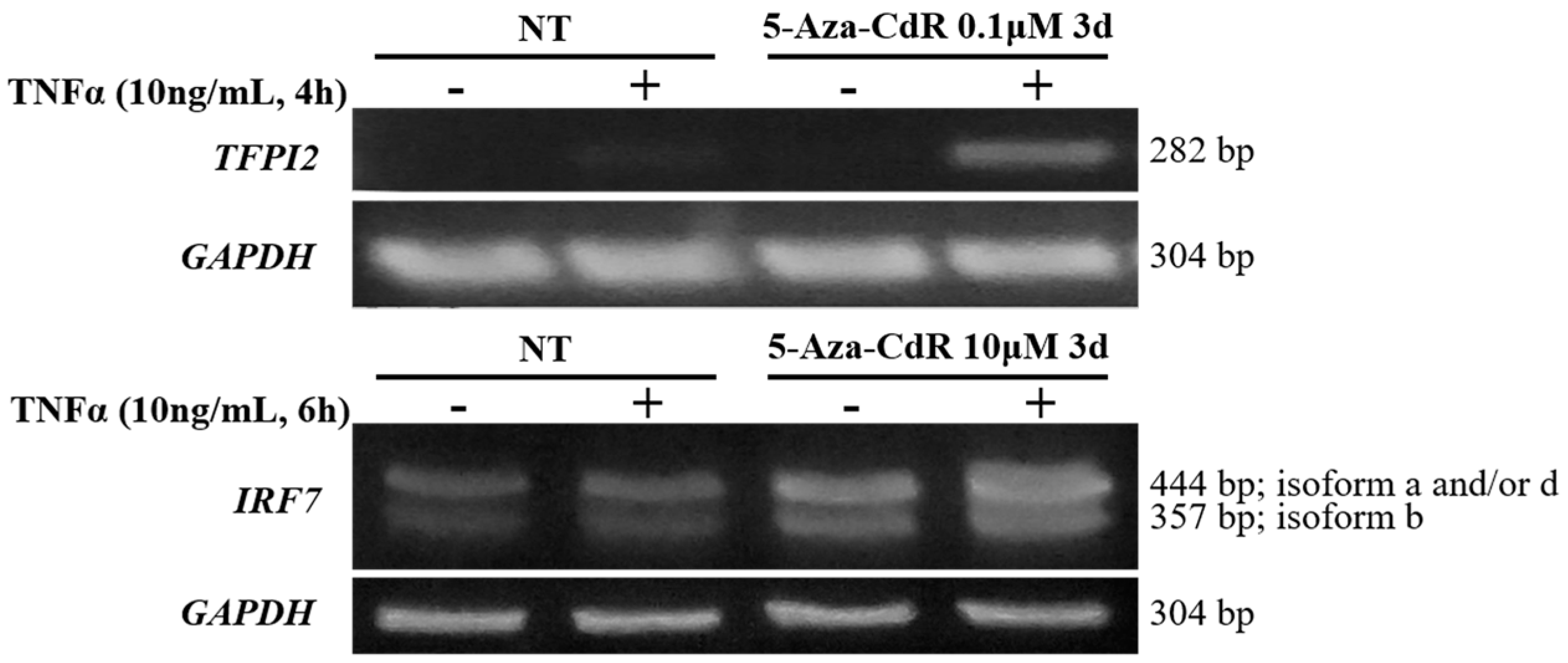
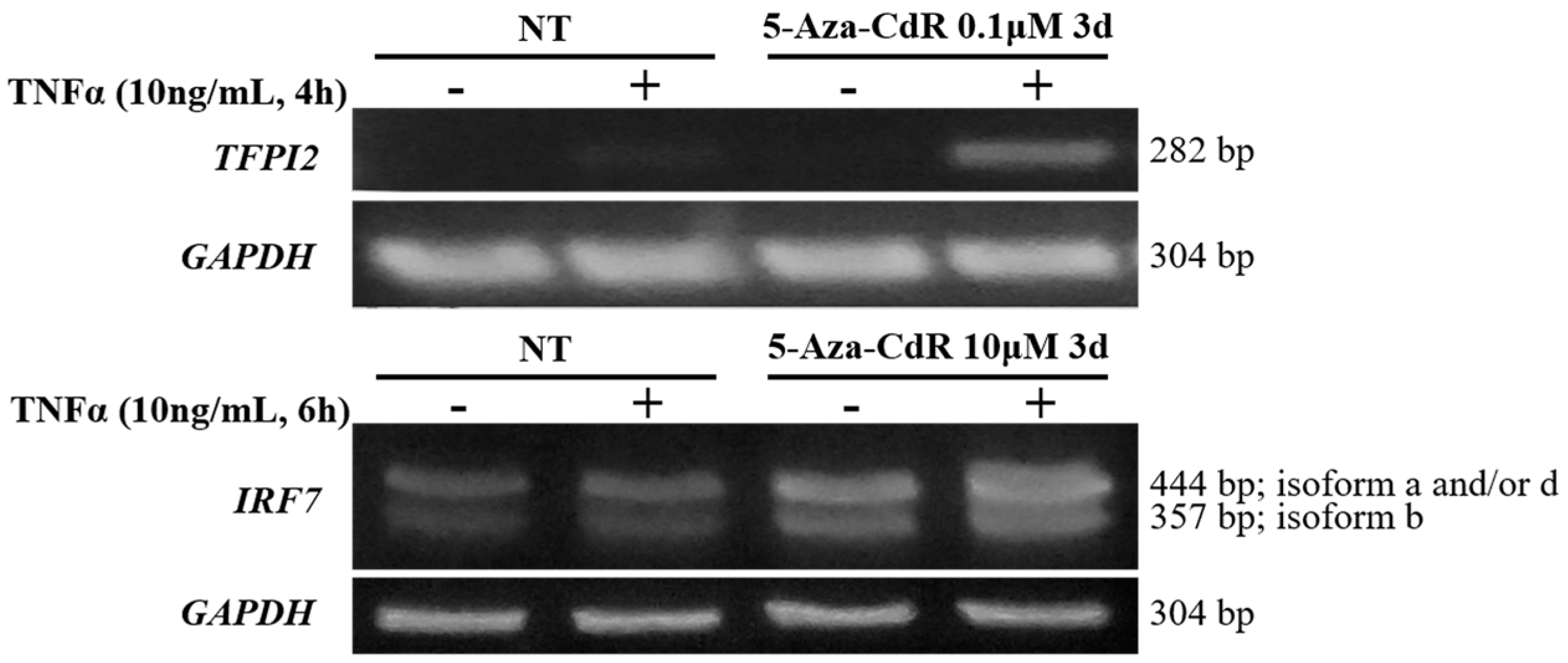
2.5. Methylation Dramatically Affects the Function of -1C Genes In Vivo
2.6. Methylation of -1C Would Not Affect Target Gene Expression When There is Another NF-κB Binding Possibility

2.7. The -1C κB Sites Are Evolutionarily Conserved Only When Located in CGIs
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. DNA Affinity Precipitation Assays (DAPA)
4.3. Plasmid Construction and Mutagenesis
4.4. Transfection and Luciferase Reporter System
4.5. Bisulfite Sequencing PCR (BSP)
4.6. Reverse transcription PCR (RT-PCR)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Wang, V.Y.-F.; Huang, D.-B.; Fusco, A. NF-κB regulation: Lessons from structures. Immunol. Rev. 2012, 246, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Chiu, J.J.S.; Lenardo, M.J. IMF-κB and rel: Participants in a multiform transcriptional regulatory system. In International Review of Cytology; Kwang, W., Jeon, M.F., Jonathan, J., Eds.; Academic Press: Cambridge, MA, USA, 1993; Volume 143, pp. 1–62. [Google Scholar]
- Chen, F.; Huang, D.; Chen, Y.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Ghosh, G. Regulation of DNA binding by Rel/ NF-κB transcription factors: Structural views. Oncogene 1999, 18, 6845–6852. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sengchanthalangsy, L.; Hackett, A.; Ghosh, G. NF-κB p65 (rela) homodimer uses distinct mechanisms to recognize DNA targets. Structure 2000, 8, 419–428. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-κB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Kunsch, C.; Ruben, S.M.; Rosen, C.A. Selection of optimal κB/rel DNA-binding motifs: Interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 4412–4421. [Google Scholar] [CrossRef] [PubMed]
- Siggers, T.; Chang, A.B.; Teixeira, A.; Wong, D.; Williams, K.J.; Ahmed, B.; Ragoussis, J.; Udalova, I.A.; Smale, S.T.; Bulyk, M.L. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 2012, 13, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.H.; Hoffmann, A.; Baltimore, D. One nucleotide in a κB site can determine co-factor specificity for NF-κB dimers. Cell 2004, 118, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.Y.; Huang, W.; Asagiri, M.; Spann, N.; Hoffmann, A.; Glass, C.; Ghosh, G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Reports 2012, 2, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.; Taggart, M.; Frommer, M.; Miller, O.J.; Macleod, D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985, 40, 91–99. [Google Scholar] [CrossRef]
- Bird, A.P. CPG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Taggart, M.H.; Bird, A.P. Unmethlated domains in vertebrate DNA. Nucleic Acids Res. 1983, 11, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Antequera, F. Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 2003, 60, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.; Korner, M.; Rosen, H.; Baeuerle, P.A.; Citri, Y. Nuclear factor kappa B activates proenkephalin transcription in T lymphocytes. Mol. Cell. Biol. 1991, 11, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.; Halden, N.; Lenardo, M.; Leonard, W. Functionally distinct NF-κB binding sites in the immunoglobulin κ and IL-2 receptor α chain genes. Science 1989, 244, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Bednarik, D.P.; Duckett, C.; Kim, S.U.; Perez, V.L.; Griffis, K.; Guenthner, P.C.; Folks, T.M. Chromatin dynamics associated with HIV-1 TAT-activated transcription. New Biol. 1991, 3, 969–976. [Google Scholar] [PubMed]
- Xia, C.; Hu, J.; Ketterer, B.; Taylor, J.B. The organization of the human GSTP1–1 gene promoter and its response to retinoic acid and cellular redox status. Biochem. J. 1996, 313, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the RelB gene is regulated by NF-κB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Moore, P.A.; Pitha, P.M. Stimulation of IRF-7 gene expression by tumor necrosis factor α: Requirement for NF-κB transcription factor and gene accessibility. J. Biol. Chem. 2002, 277, 16592–16598. [Google Scholar] [CrossRef] [PubMed]
- Krikos, A.; Laherty, C.D.; Dixit, V.M. Transcriptional activation of the tumor necrosis factor α-inducible zinc finger protein, a20, is mediated by κB elements. J. Biol. Chem. 1992, 267, 17971–17976. [Google Scholar] [PubMed]
- Hinz, M.; Lemke, P.; Anagnostopoulos, I.; Hacker, C.; Krappmann, D.; Mathas, S.; Dörken, B.; Zenke, M.; Stein, H.; Scheidereit, C. Nuclear factor κB–dependent gene expression profiling of hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J. Exp. Med. 2002, 196, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-C.; Dahiya, R. Methprimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Tomizawa, S.-I.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Creusot, F.; Acs, G.; Christman, J.K. Inhibition of DNA methyltransferase and induction of friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2‘-deoxycytidine. J. Biol. Chem. 1982, 257, 2041–2048. [Google Scholar] [PubMed]
- Christman, J.K.; Mendelsohn, N.; Herzog, D.; Schneiderman, N. Effect of 5-azacytidine on differentiation and DNA methylation in human promyelocyte leukemia cells (HL-60). Cancer Res. 1983, 43, 763–769. [Google Scholar] [PubMed]
- Christman, J.K. 5-azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, V.; Racanicchi, S.; Martelli, M.P.; Nocentini, G.; Fettucciari, K.; Riccardi, C.; Marconi, P.; Di Nardo, P.; Grignani, F.; Binaglia, L.; et al. Eicosapentaenoic acid demethylates a single CPG that mediates expression of tumor suppressor CCAAT/enhancer-binding protein δ in u937 leukemia cells. J. Biol. Chem. 2011, 286, 27092–27102. [Google Scholar] [CrossRef] [PubMed]
- Konduri, S.D.; Srivenugopal, K.S.; Yanamandra, N.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Foster, D.C.; Kisiel, W.; Ali-Osman, F.; Kondraganti, S.; et al. Promoter methylation and silencing of the tissue factor pathway inhibitor-2 (TFPI-2), in human glioma cells. Oncogene 2003, 22, 4509–4516. [Google Scholar] [CrossRef] [PubMed]
- Hube, F.; Reverdiau, P.; Iochmann, S.; Rollin, J.; Cherpi-Antar, C.; Gruel, Y. Transcriptional silencing of the TFPI-2 gene by promoter hypermethylation in choriocarcinoma cells. Biol. Chem. 2003, 384, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Glöckner, S.C.; Dhir, M.; Yi, J.M.; McGarvey, K.E.; van Neste, L.; Louwagie, J.; Chan, T.A.; Kleeberger, W.; de Bruïne, A.P.; Smits, K.M.; et al. Methylation of TFPI2 in stool DNA: A potential novel biomarker for the detection of colorectal cancer. Cancer Res. 2009, 69, 4691–4699. [Google Scholar] [CrossRef] [PubMed]
- Ferraresso, S.; Bresolin, S.; Aricò, A.; Comazzi, S.; Gelain, M.E.; Riondato, F.; Bargelloni, L.; Marconato, L.; Kronnie, G.T.; Aresu, L. Epigenetic silencing of TFPI-2 in canine diffuse large B-cell lymphoma. PLoS ONE 2014, 9, e92707. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Hsiao, Y.-H.; Lai, Y.-H.; Chen, Y.-C.; Chen, Y.-J.; Chou, J.-L.; Chan, M.W.Y.; Lin, Y.-H.; Tsou, Y.-A.; Tsai, M.-H.; et al. DNA methylation profiles and biomarkers of oral squamous cell carcinoma. Epigenetics 2015, 10, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tan, Q.; Tao, L.; Pan, X.; Pang, L.; Liang, W.; Liu, W.; Zhang, W.; Li, F.; Jia, W. Hypermethylation of TFPI2 correlates with cervical cancer incidence in the Uygur and Han populations of Xinjiang, china. Int. J. Clin. Exp. Pathol. 2015, 8, 1844–1854. [Google Scholar] [PubMed]
- Sun, F.-K.; Fan, Y.-C.; Zhao, J.; Zhang, F.; Gao, S.; Zhao, Z.-H.; Sun, Q.; Wang, K. Detection of TFPI2 methylation in the serum of hepatocellular carcinoma patients. Dig. Dis. Sci. 2013, 58, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Long, H.K.; Sims, D.; Heger, A.; Blackledge, N.P.; Kutter, C.; Wright, M.L.; Grützner, F.; Odom, D.T.; Patient, R.; Ponting, C.P.; et al. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. eLife 2013, 2, e00348. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. DNA methylation and the frequency of CPG in animal DNA. Nucleic. Acids Res. 1980, 8, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Species | VCAM1 | CCL2 | RelB | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mammal | Homo sapiens | D | D | D | D | D | D | D | C | |||||||||||
| Pan troglodytes | D | D | D | D | D | D | C | |||||||||||||
| Mus musculus | D | D | D | D | D | D | D | C | ||||||||||||
| Bos taurus | D | D | D | D | D | D | D | D | C | |||||||||||
| Canis lupus familiaris | D | D | D | C | D | D | D | D | D | D | C | |||||||||
| Lipotes vexillifer | D | D | D | D | D | D | D | D | D | D | C | D | ||||||||
| Eptesicus fuscus | D | D | C | D | D | D | D | D | ||||||||||||
| Pteropus alecto | D | D | D | C/D | D | D | D | D | ||||||||||||
| Aves | Pseudopodoces humilis | D | C/D | C/D | D | C | D | D | D | D | D | C | C/D | |||||||
| Aquila chrysaetos canadensis | C/D | C/D | D | C | ||||||||||||||||
| Reptilia | Chrysemys picta | N | C | |||||||||||||||||
| Alligator mississippiensis | C | |||||||||||||||||||
| Python bivittatus | D | D | D | |||||||||||||||||
| Alligator sinensis | D | N | ||||||||||||||||||
| Amphhibia | Xenopus (Silurana) tropicalis | D | D | D | D | D | D | D | ||||||||||||
| Pisce | Notothenia coriiceps | N | D | |||||||||||||||||
| Stegastes partitus | N | C | ||||||||||||||||||
| Poecilia reticulata | D | D | C | C/D | D | |||||||||||||||
| Zebra fish | D | D | ||||||||||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Li, J.; Ding, K.; Zhang, L.; Che, Q.; Sun, X.; Dai, Y.; Sun, W.; Bao, M.; Wang, X.; et al. The CpG Dinucleotide Adjacent to a κB Site Affects NF-κB Function through Its Methylation. Int. J. Mol. Sci. 2017, 18, 528. https://doi.org/10.3390/ijms18030528
Wang T, Li J, Ding K, Zhang L, Che Q, Sun X, Dai Y, Sun W, Bao M, Wang X, et al. The CpG Dinucleotide Adjacent to a κB Site Affects NF-κB Function through Its Methylation. International Journal of Molecular Sciences. 2017; 18(3):528. https://doi.org/10.3390/ijms18030528
Chicago/Turabian StyleWang, Tao, Jinge Li, Ke Ding, Li Zhang, Qiuru Che, Xiuming Sun, Yumeng Dai, Wei Sun, Meiying Bao, Xiaochun Wang, and et al. 2017. "The CpG Dinucleotide Adjacent to a κB Site Affects NF-κB Function through Its Methylation" International Journal of Molecular Sciences 18, no. 3: 528. https://doi.org/10.3390/ijms18030528