
The PRR11-SKA2 Bidirectional Transcription Unit Is Negatively Regulated by p53 through NF-Y in Lung Cancer Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
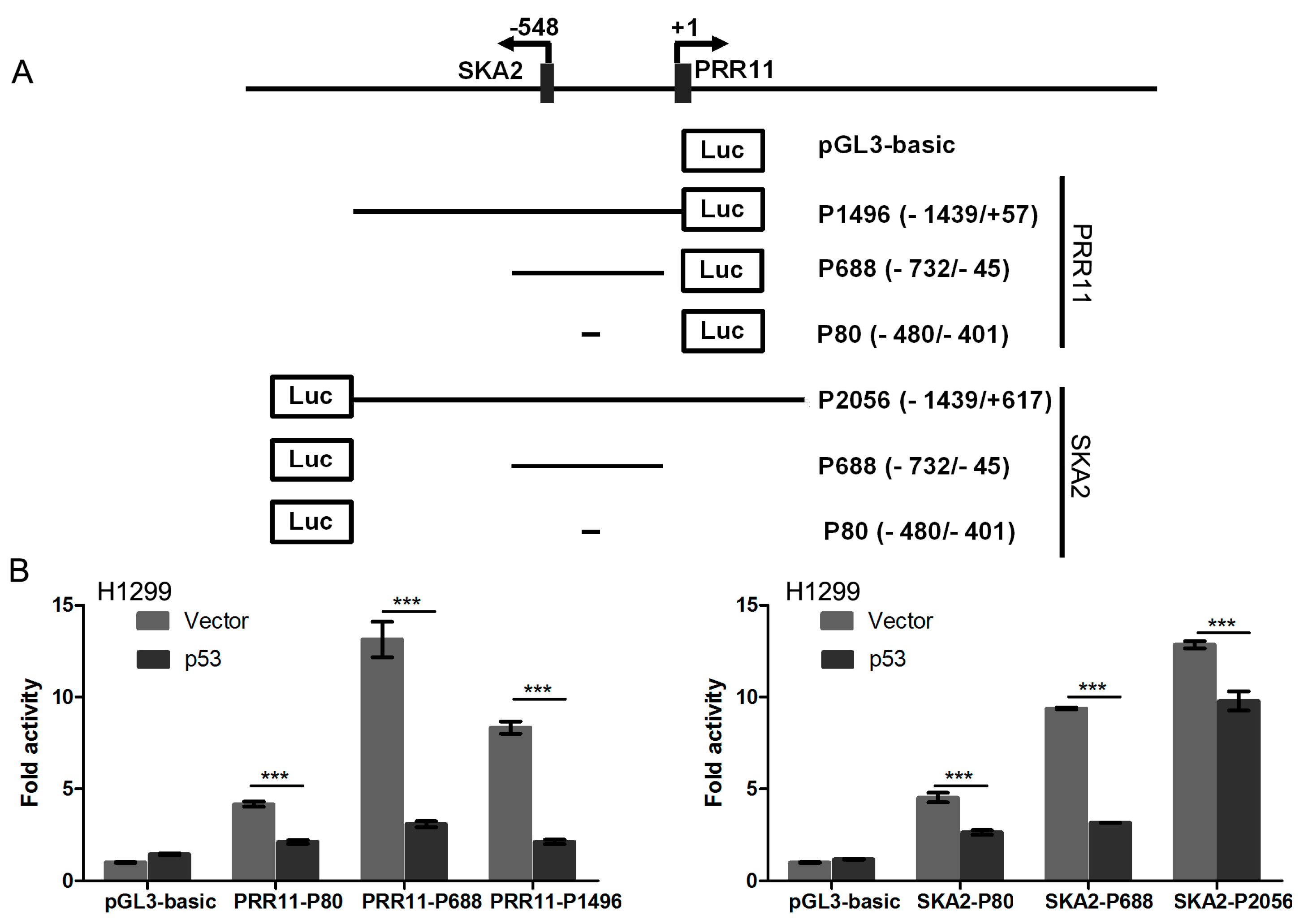
2.1. p53 Represses PRR11-SKA2 Bidirectional Promoter Activity
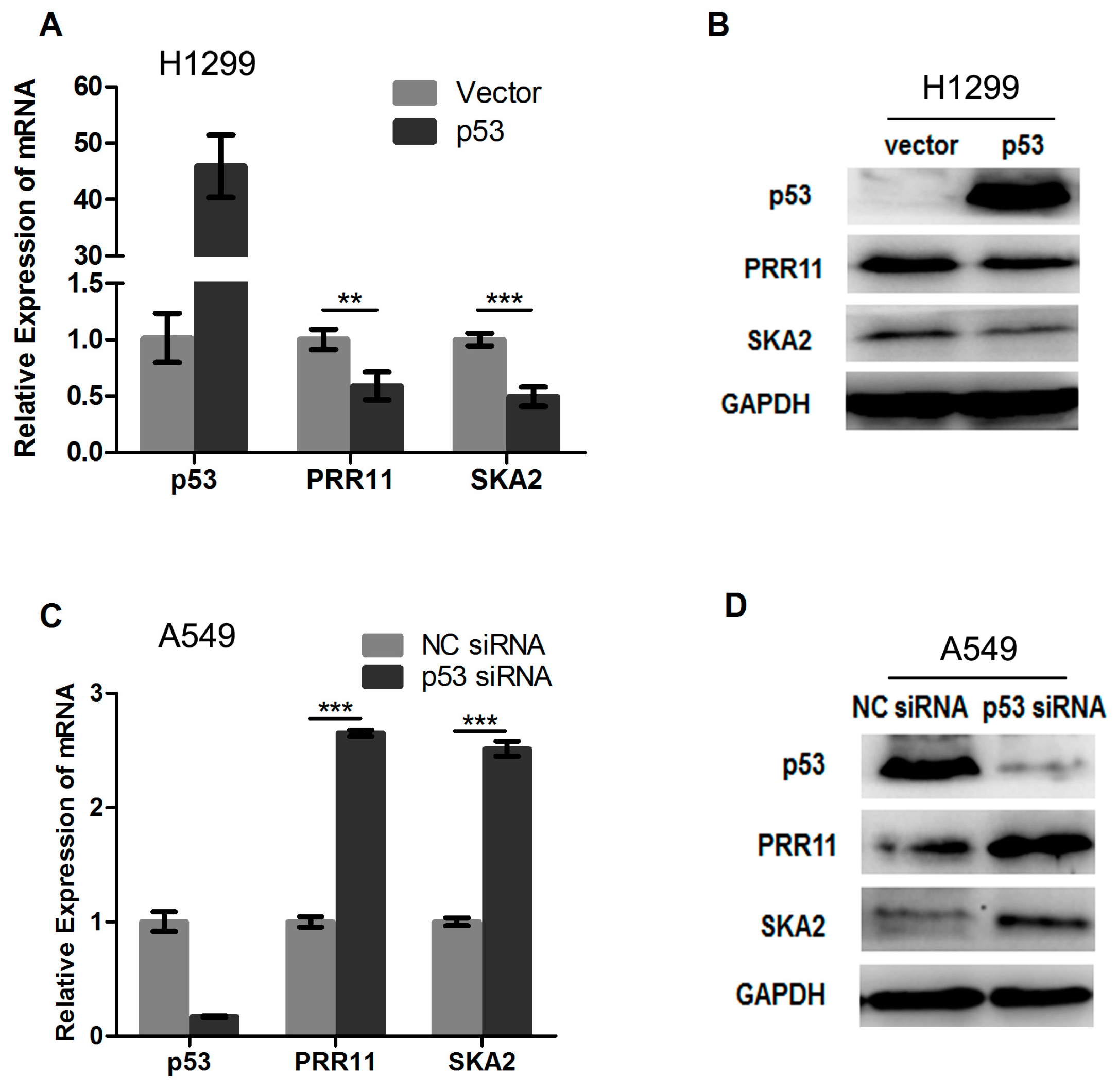
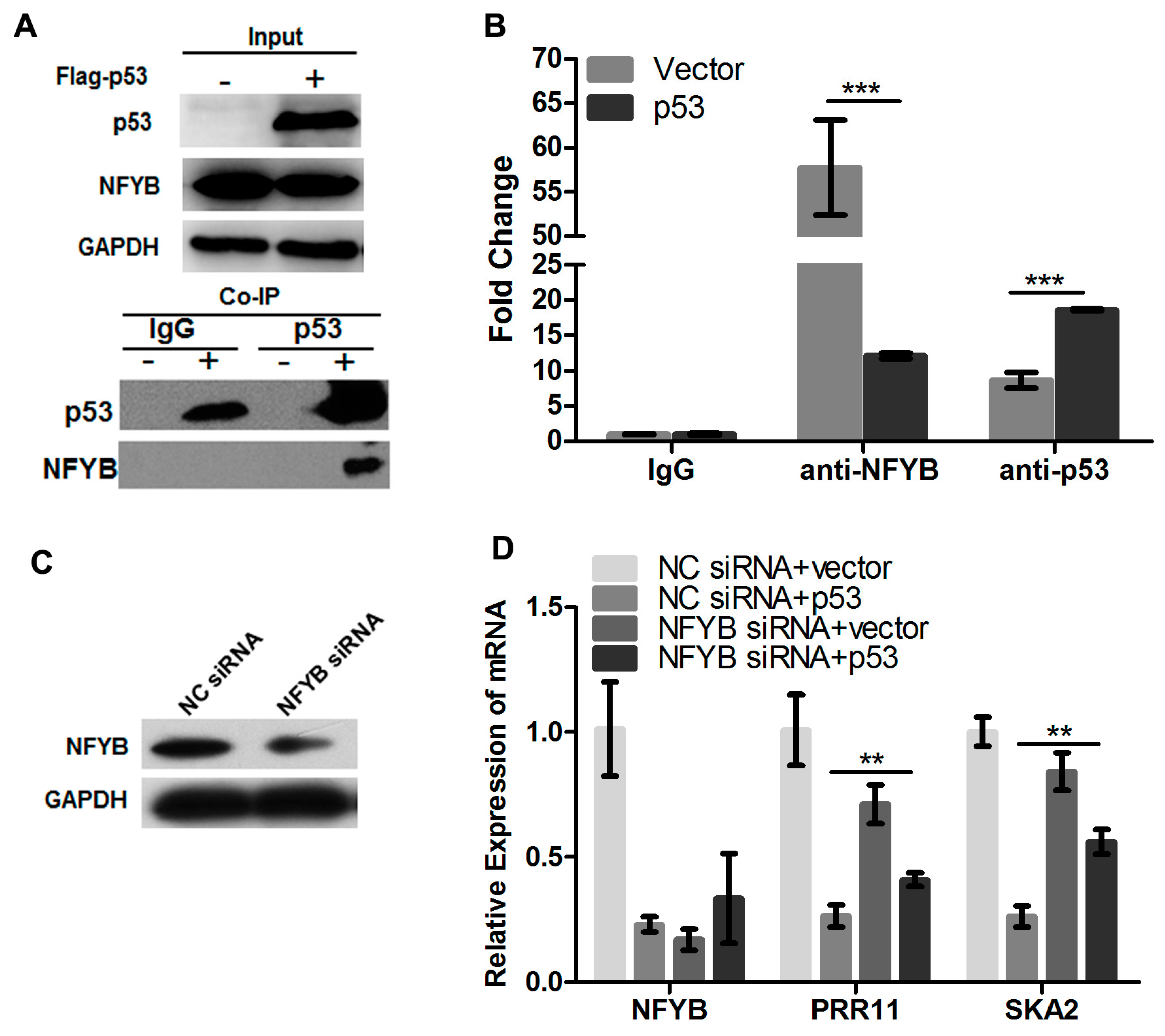
2.2. p53 Represses the Endogenous Transcription of the PRR11-SKA2 Bidirectional Unit
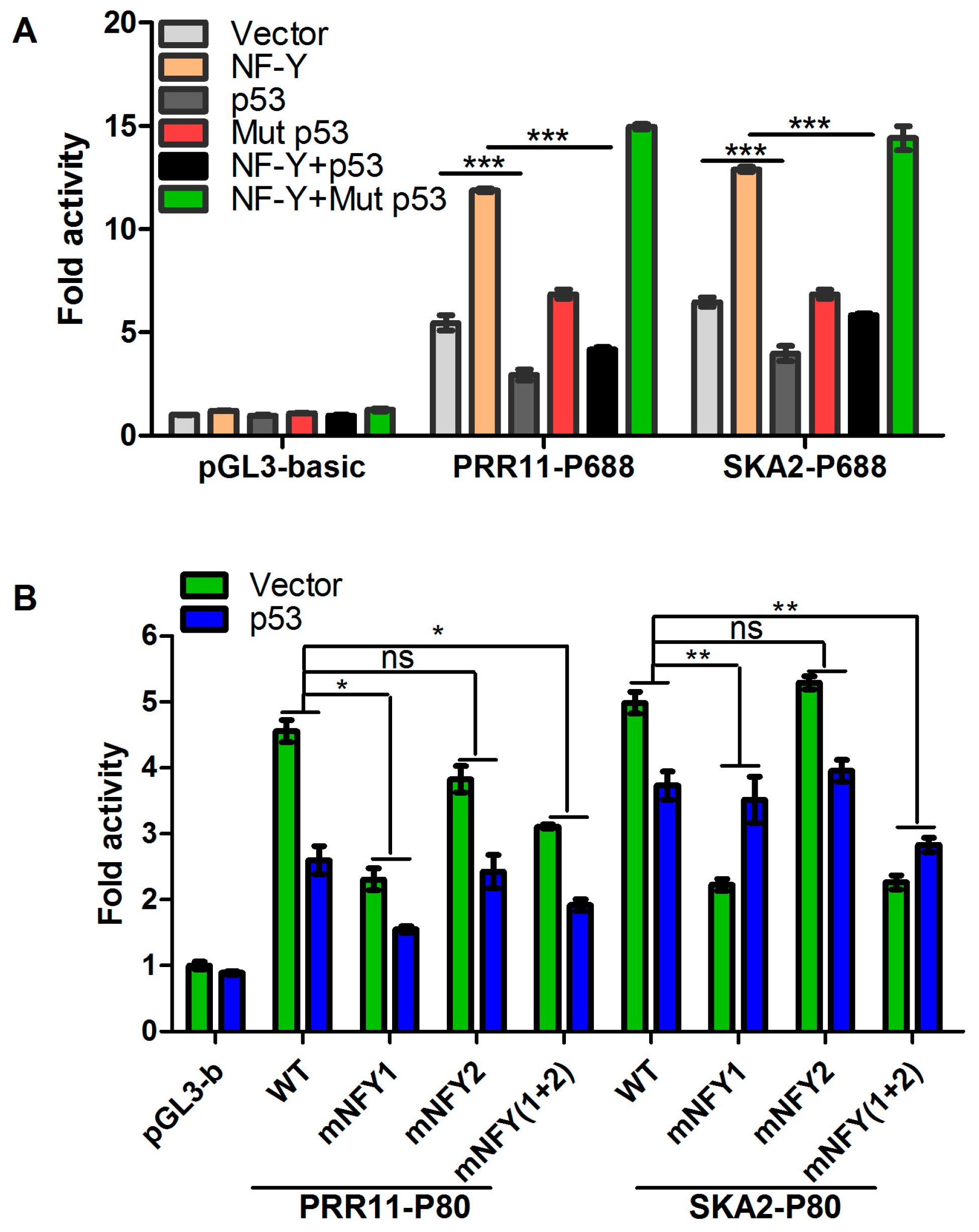
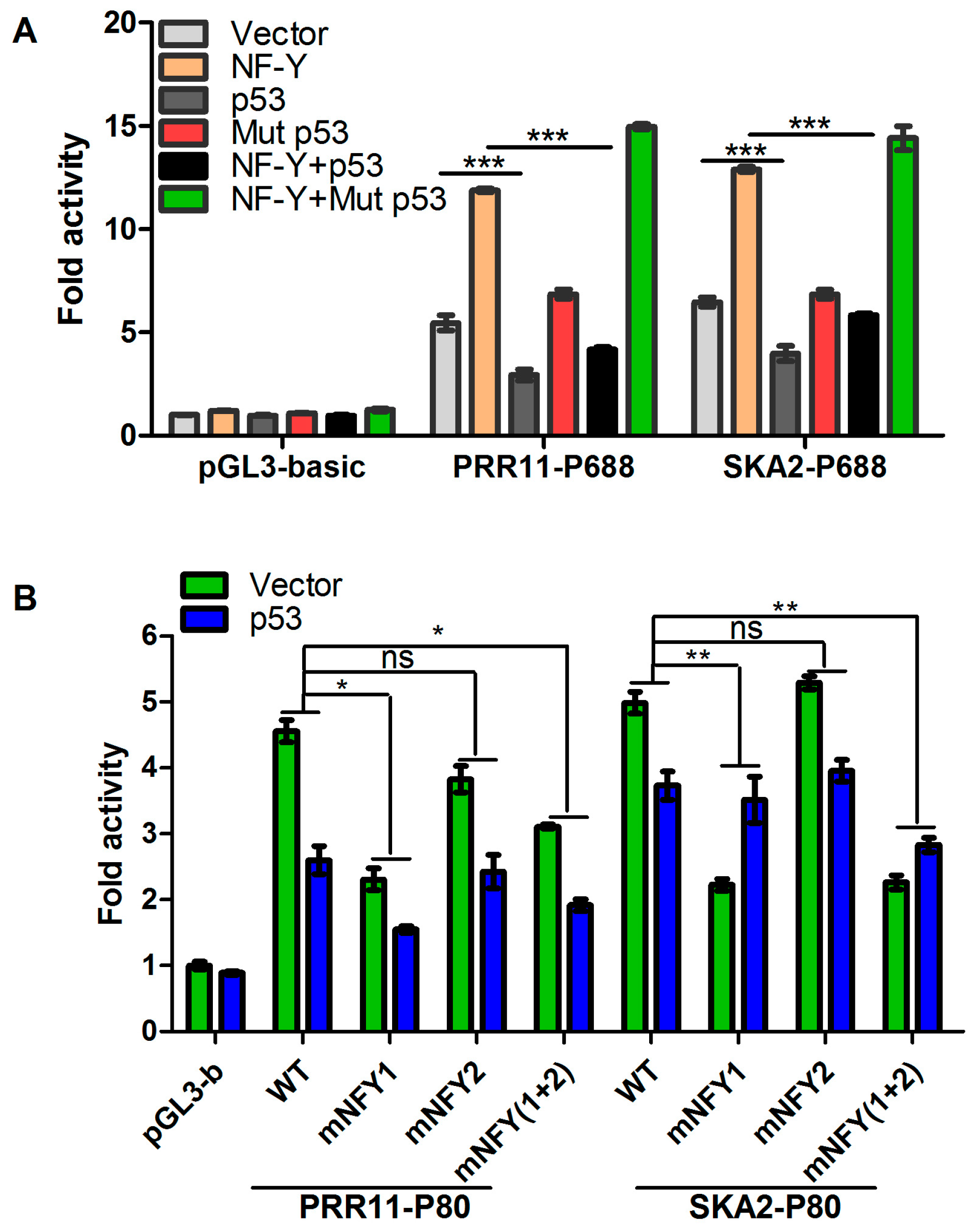
2.3. p53 Represses the Transcription of the PRR11-SKA2 Bidirectional Promoter through NF-Y
2.4. p53 Reduces the Recruitment of NF-Y to the PRR11-SKA2 Bidirectional Promoter
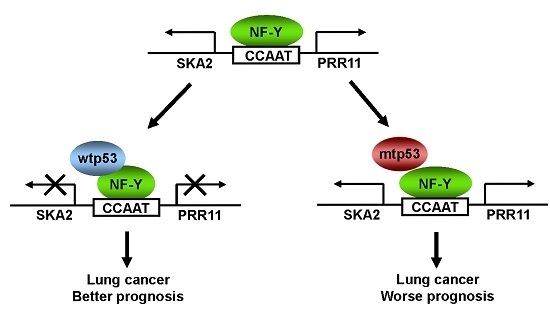
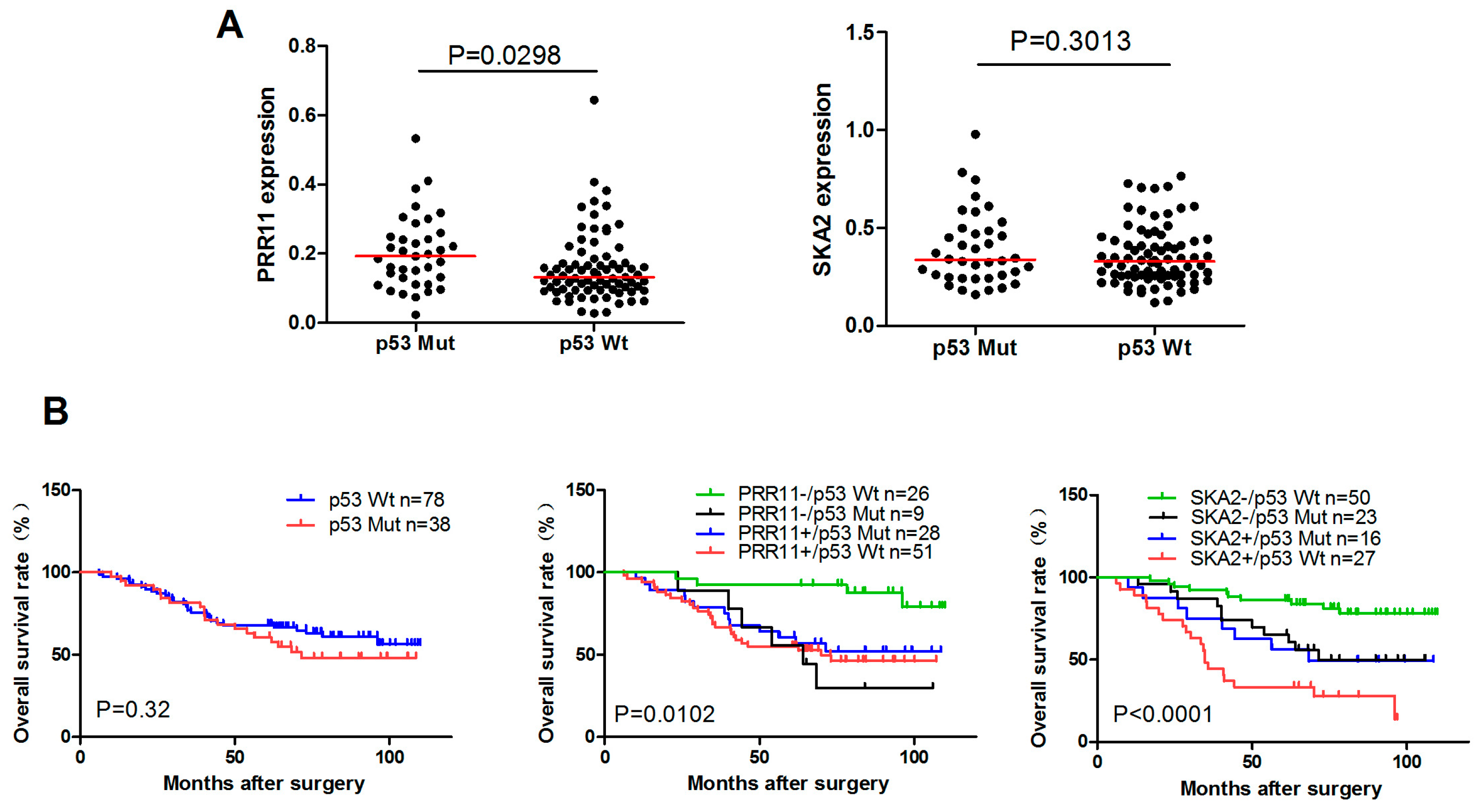
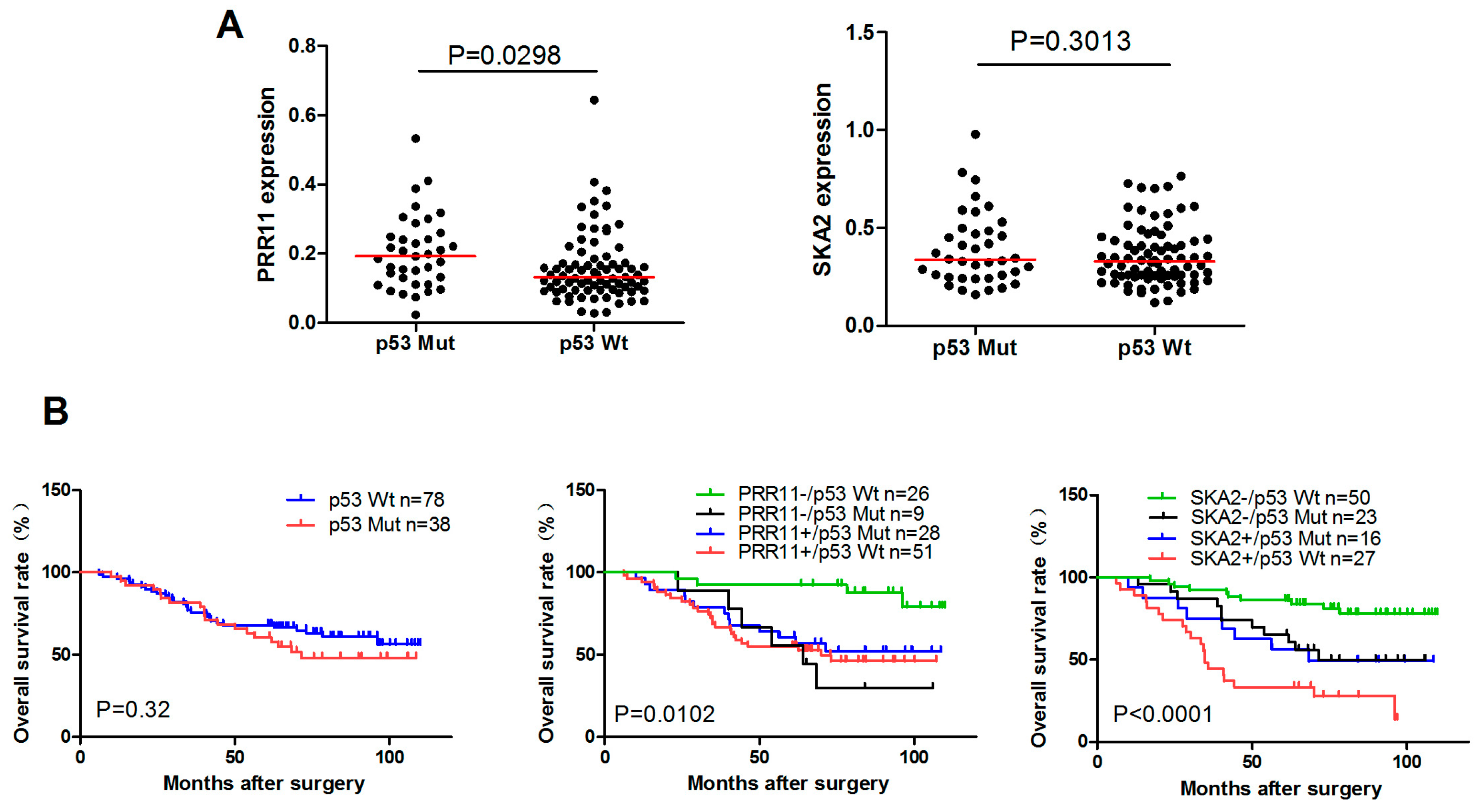
2.5. The Clinical Significance of p53-Mediated PRR11-SKA2 Repression in Lung Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Luciferase Reporter Constructs and Reporter Assays
4.3. siRNA and Plasmid Transfection
4.4. RNA Isolation and RT-PCR
4.5. Western Blotting
4.6. Co-Immunoprecipitation (Co-IP)
4.7. Chromatin Immunoprecipitation (ChIP)
4.8. Prognostic Analysis of PRR11 and SKA2 in Lung Cancer Patients
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PRR11 | proline-rich protein 11 |
| SKA2 | spindle and kinetochore associated complex subunit 2 |
| NF-Y | nuclear transcription factor Y |
| ChIP | chromatin immunoprecipitation |
References
- Ji, Y.; Xie, M.; Lan, H.; Zhang, Y.; Long, Y.; Weng, H.; Li, D.; Cai, W.; Zhu, H.; Niu, Y.; et al. PRR11 is a novel gene implicated in cell cycle progression and lung cancer. Int. J. Biochem. Cell Biol. 2013, 45, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Y.; Li, Y.; Zhu, H.; Wang, Y.; Cai, W.; Zhu, J.; Ozaki, T.; Bu, Y. PRR11 regulates late-S to G2/M phase progression and induces premature chromatin condensation (PCC). Biochem. Biophys. Res. Commun. 2015, 458, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, W.; Xiao, Y.; Zhang, M.; Luo, Y.; Yuan, W.; Xu, Y.; Yu, G.; Hu, Y. PRR11 is a prognostic marker and potential oncogene in patients with gastric cancer. PLoS ONE 2015, 10, e0128943. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cha, Z.; Fang, W.; Qian, B.; Yu, W.; Li, W.; Yu, G.; Gao, Y. The prognostic potential and oncogenic effects of PRR11 expression in hilar cholangiocarcinoma. Oncotarget 2015, 6, 20419–20433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Zhang, C.; Weng, H.; Li, Y.; Cai, W.; Xie, M.; Long, Y.; Ai, Q.; Liu, Z.; et al. The gene pair PRR11 and SKA2 shares a NF-Y-regulated bidirectional promoter and contributes to lung cancer development. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2015, 1849, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Lieber, M.R. Bidirectional gene organization: A common architectural feature of the human genome. Cell 2002, 109, 807–809. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Aldred, S.F.; Hartman, S.J.; Schroeder, D.I.; Otillar, R.P.; Myers, R.M. An abundance of bidirectional promoters in the human genome. Genome Res. 2004, 14, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Pelechano, V.; Järvelin, A.I.; Steinmetz, L.M. Functional consequences of bidirectional promoters. Trends Genet. TIG 2011, 27, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wakano, C.; Byun, J.S.; Di, L.-J.; Gardner, K. The dual lives of bidirectional promoters. Biochim. Biophys. Acta 2012, 1819, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Orekhova, A.S.; Rubtsov, P.M. Bidirectional promoters in the transcription of mammalian genomes. Biochem. Biokhimiia 2013, 78, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Dolfini, D.; Gatta, R.; Mantovani, R. NF-Y and the transcriptional activation of CCAAT promoters. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Van der Watt, P.J.; Leaner, V.D. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2011, 1809, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Salsi, V. Interactions between p300 and multiple NF-Y trimers govern cyclin B2 promoter function. J. Biol. Chem. 2003, 278, 6642–6650. [Google Scholar] [CrossRef] [PubMed]
- Imbriano, C.; Gnesutta, N.; Mantovani, R. The NF-Y/p53 liaison: Well beyond repression. Biochim. Biophys. Acta BBA Rev. Cancer 2012, 1825, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Imbriano, C.; Gurtner, A.; Cocchiarella, F.; di Agostino, S.; Basile, V.; Gostissa, M.; Dobbelstein, M.; del Sal, G.; Piaggio, G.; Mantovani, R. Direct p53 transcriptional repression: In vivo analysis of CCAAT-containing G2/M promoters. Mol. Cell. Biol. 2005, 25, 3737–3751. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, A.; Fuschi, P.; Martelli, F.; Manni, I.; Artuso, S.; Simonte, G.; Ambrosino, V.; Antonini, A.; Folgiero, V.; Falcioni, R.; et al. Transcription factor NF-Y induces apoptosis in cells expressing wild-type p53 through E2F1 upregulation and p53 activation. Cancer Res. 2010, 70, 9711–9720. [Google Scholar] [CrossRef] [PubMed]
- Benatti, P.; Basile, V.; Merico, D.; Fantoni, L.I.; Tagliafico, E.; Imbriano, C. A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res. 2008, 36, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Soussi, T.; Shomer, B.; Hollstein, M.; Greenblatt, M.; Hovig, E.; Harris, C.C.; Montesano, R. Database of p53 gene somatic mutations in human tumors and cell lines: Updated compilation and future prospects. Nucleic Acids Res. 1997, 25, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Liebermann, D.A.; Hoffman, B.; Vesely, D. p53 induced growth arrest versus apoptosis and its modulation by survival cytokines. Cell Cycle Georget. Tex. 2007, 6, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Ceribelli, M.; Dolfini, D.; Merico, D.; Gatta, R.; Viganò, A.M.; Pavesi, G.; Mantovani, R. The histone-like NF-Y is a bifunctional transcription factor. Mol. Cell. Biol. 2008, 28, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Dalvai, M.; Mondesert, O.; Bourdon, J.-C.; Ducommun, B.; Dozier, C. Cdc25B is negatively regulated by p53 through Sp1 and NF-Y transcription factors. Oncogene 2011, 30, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Tomida, S.; Yatabe, Y.; Kosaka, T.; Osada, H.; Yanagisawa, K.; Mitsudomi, T.; Takahashi, T. Expression profile-defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Niu, Y.; Li, C.; Yang, Y.; Gao, C. Integrating multiple microarray datasets on oral squamous cell carcinoma to reveal dysregulated networks. Head Neck 2012, 34, 1789–1797. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Weng, H.; Zhang, Y.; Long, Y.; Li, Y.; Niu, Y.; Song, F.; Bu, Y. The PRR11-SKA2 Bidirectional Transcription Unit Is Negatively Regulated by p53 through NF-Y in Lung Cancer Cells. Int. J. Mol. Sci. 2017, 18, 534. https://doi.org/10.3390/ijms18030534
Wang Y, Weng H, Zhang Y, Long Y, Li Y, Niu Y, Song F, Bu Y. The PRR11-SKA2 Bidirectional Transcription Unit Is Negatively Regulated by p53 through NF-Y in Lung Cancer Cells. International Journal of Molecular Sciences. 2017; 18(3):534. https://doi.org/10.3390/ijms18030534
Chicago/Turabian StyleWang, Yitao, Huali Weng, Ying Zhang, Yinjiang Long, Yi Li, Yulong Niu, Fangzhou Song, and Youquan Bu. 2017. "The PRR11-SKA2 Bidirectional Transcription Unit Is Negatively Regulated by p53 through NF-Y in Lung Cancer Cells" International Journal of Molecular Sciences 18, no. 3: 534. https://doi.org/10.3390/ijms18030534