
Systematic Design of Trypsin Cleavage Site Mutated Exendin4-Cysteine 1, an Orally Bioavailable Glucagon-Like Peptide-1 Receptor Agonist

Abstract
:1. Introduction
2. Results
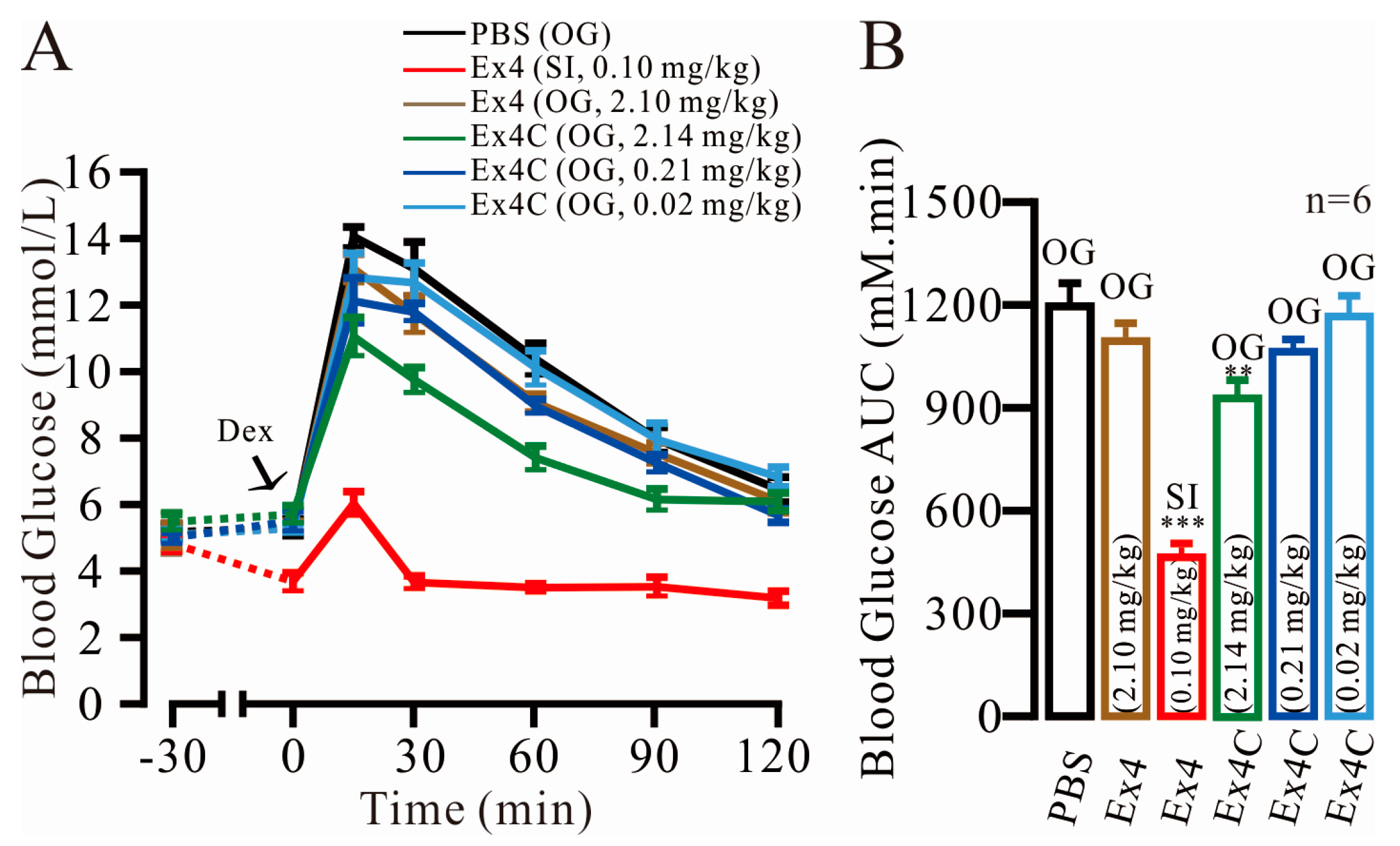
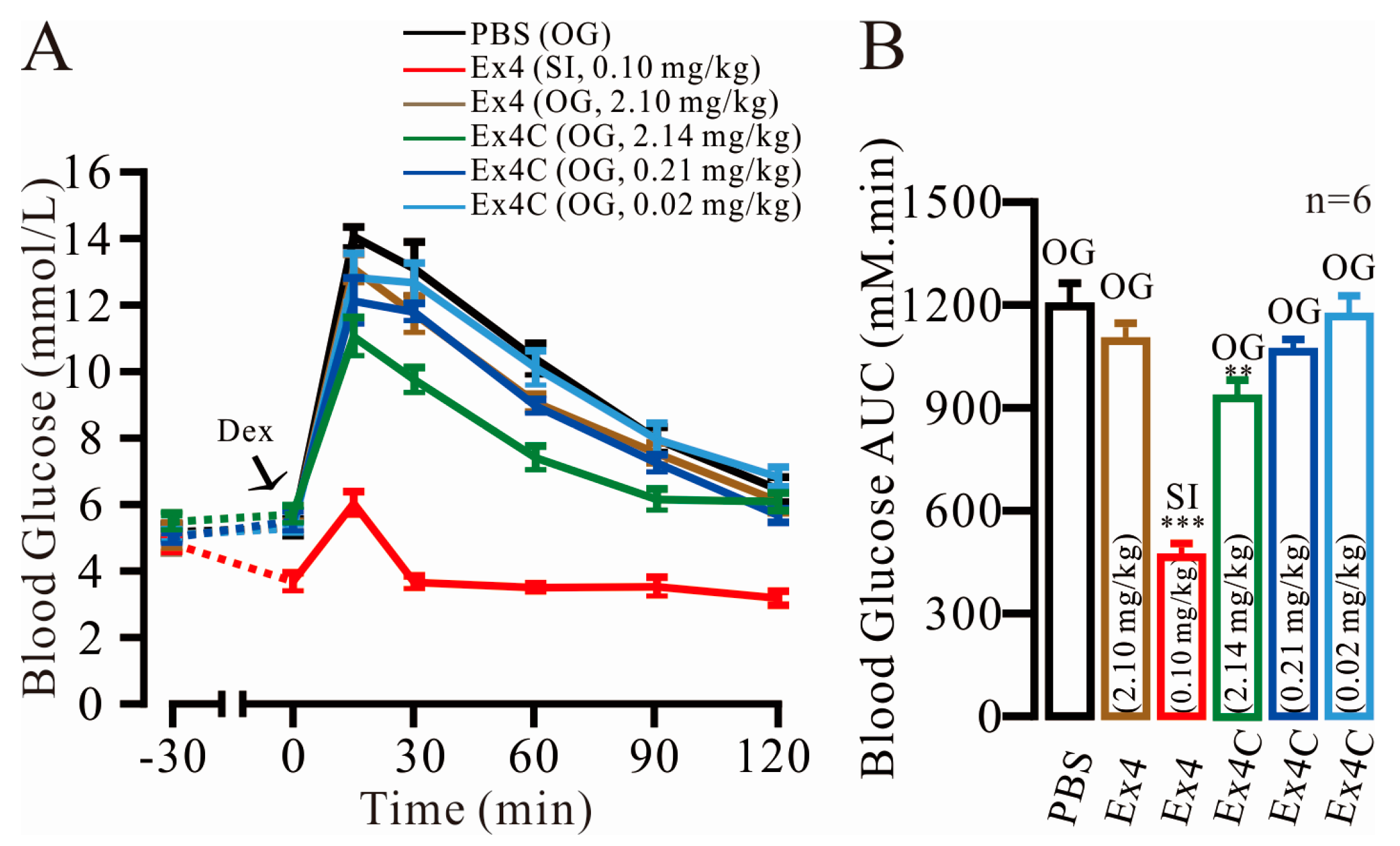
2.1. Modest Normalization of Blood Glucose Levels by Exendin4-Cysteine (Ex4C) in Intraperitoneal Glucose Tolerance Tests (IPGTTs)
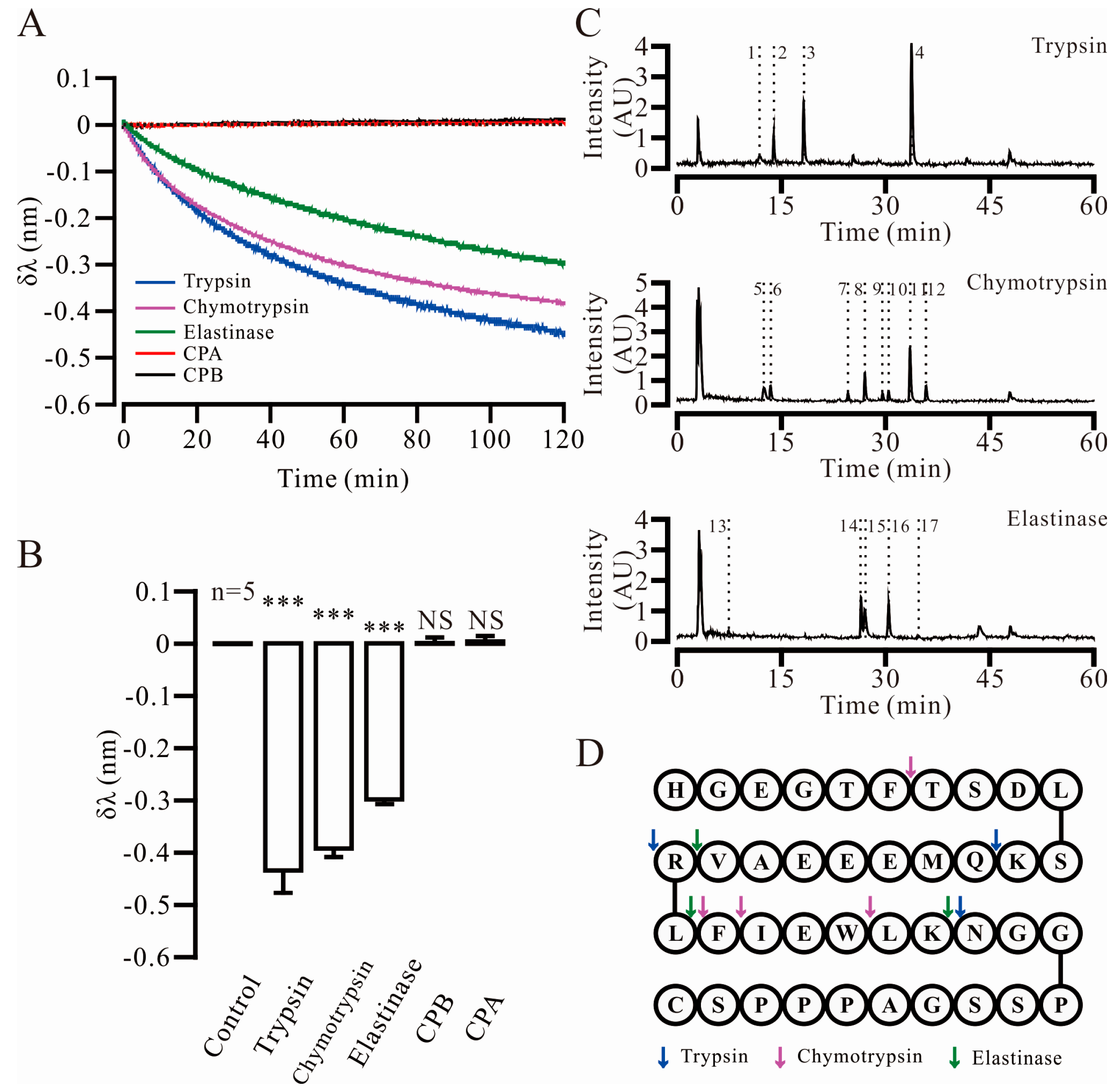
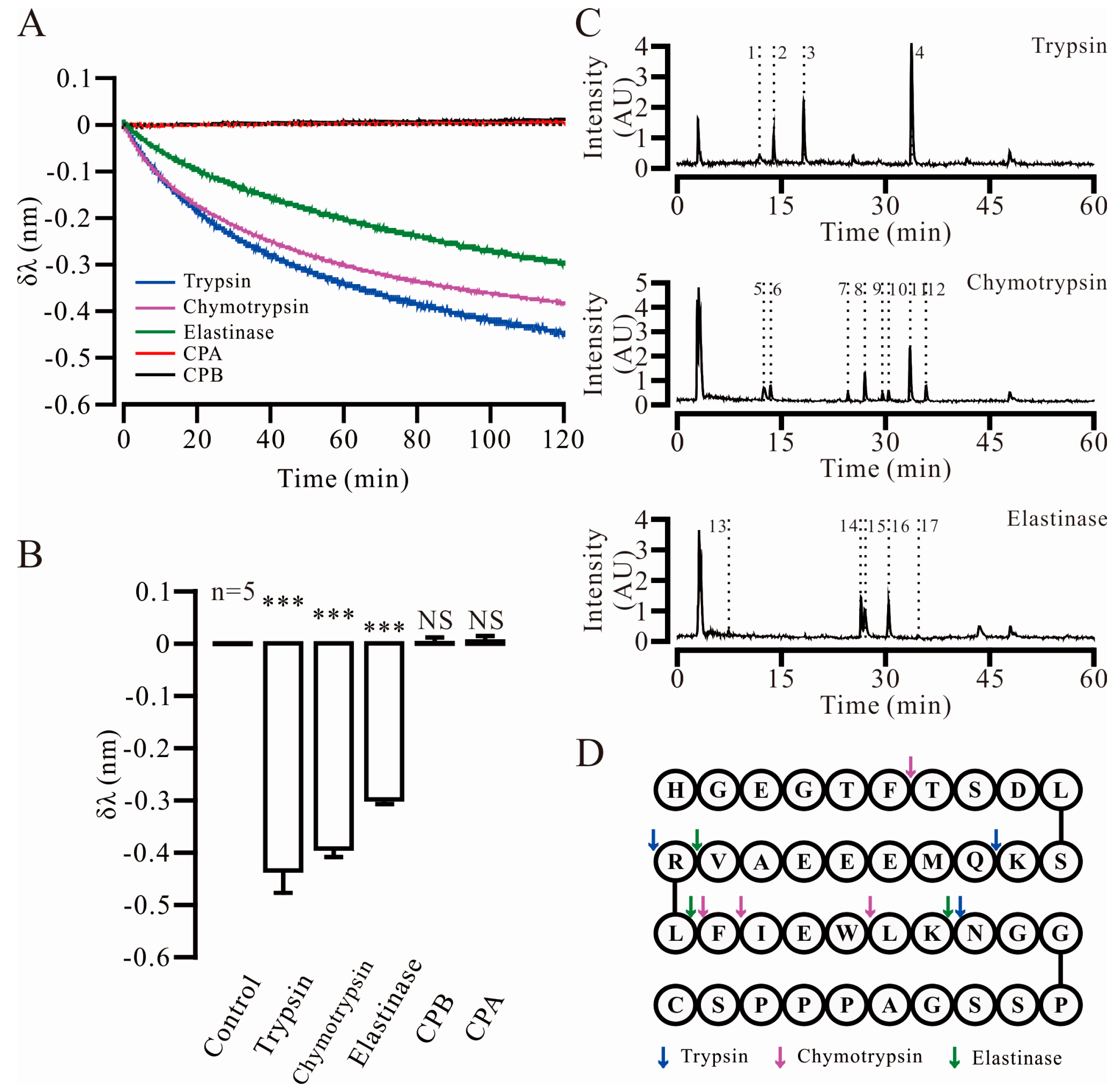
2.2. Three Key Proteases and Seven Specific Cleavage Sites in Ex4C Were Confirmed
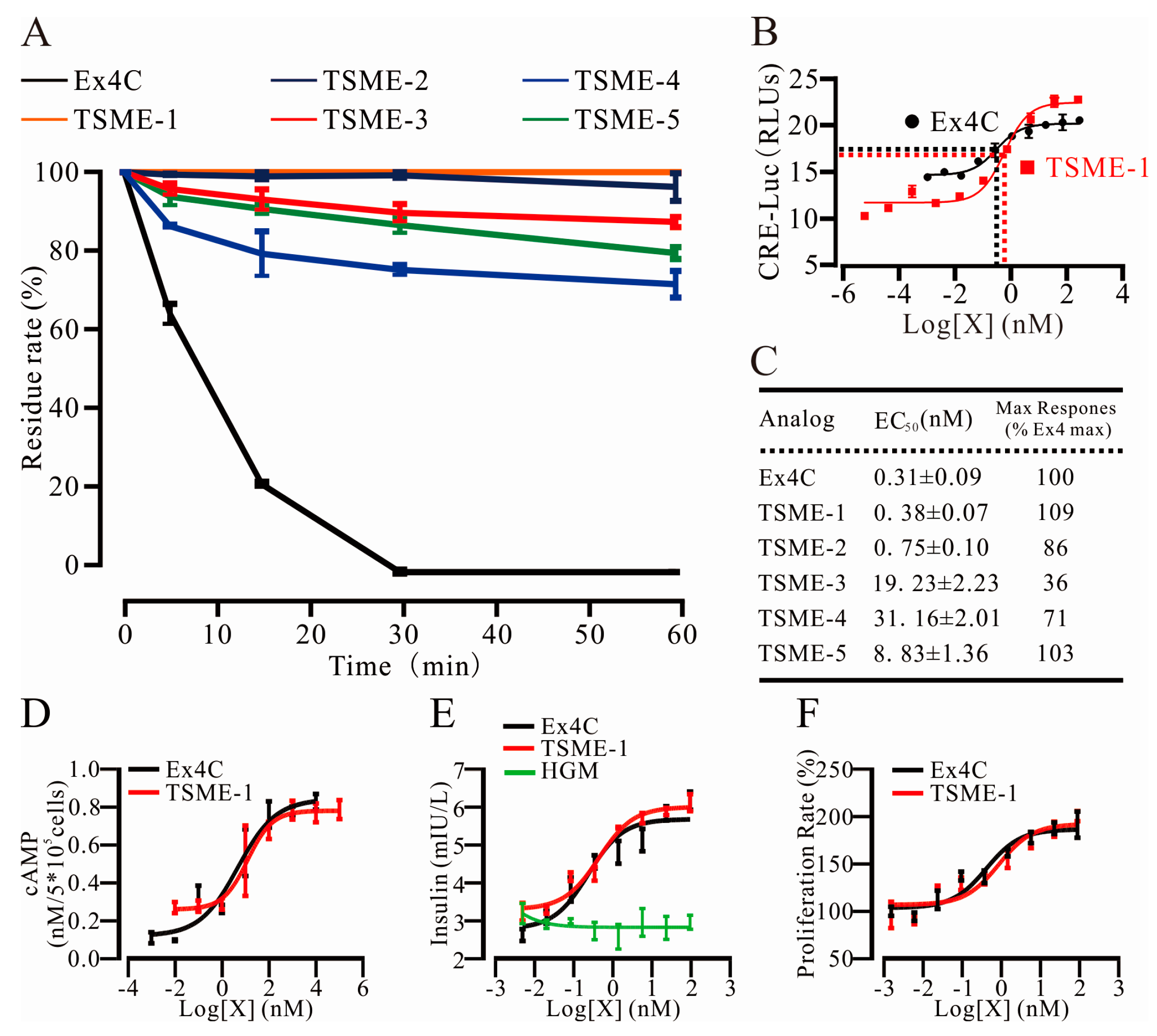
2.3. Trypsin Cleavage Site Mutated Exendin4-Cysteine 1 (TSME-1) Was Designed and Screened under the Assistance of Computer
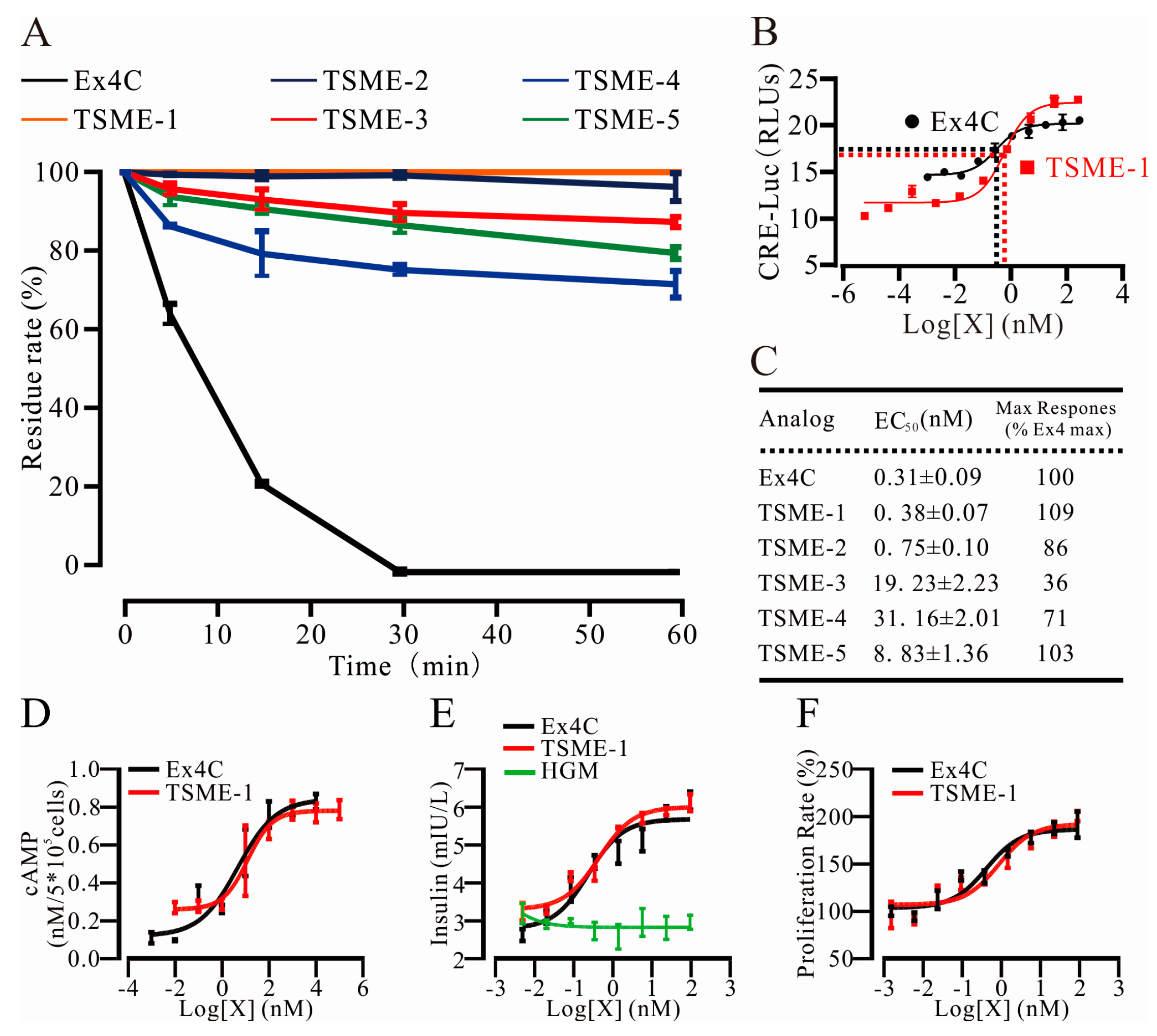
2.4. All Analogs Exhibited Significantly Increased Trypsin Resistance but Retained Their Bioactivities to Varying Degrees
2.4.1. Trypsin Resistance Was Measured by HPLC
2.4.2. The Analogs Activated GPL-1R to Varying Degrees
2.4.3. cAMP Measurements Were Used to Determine the Ability of TSME-1 to Activate Proximal GLP-1R Signaling
2.5. TSME-1 Significantly Enhanced Insulin Release and Promoted RINm5f Cell Proliferation
2.5.1. TSME-1 Enhanced Insulin Release by RINm5f Cells
2.5.2. TSME-1 Promoted RINm5f Cell Proliferation
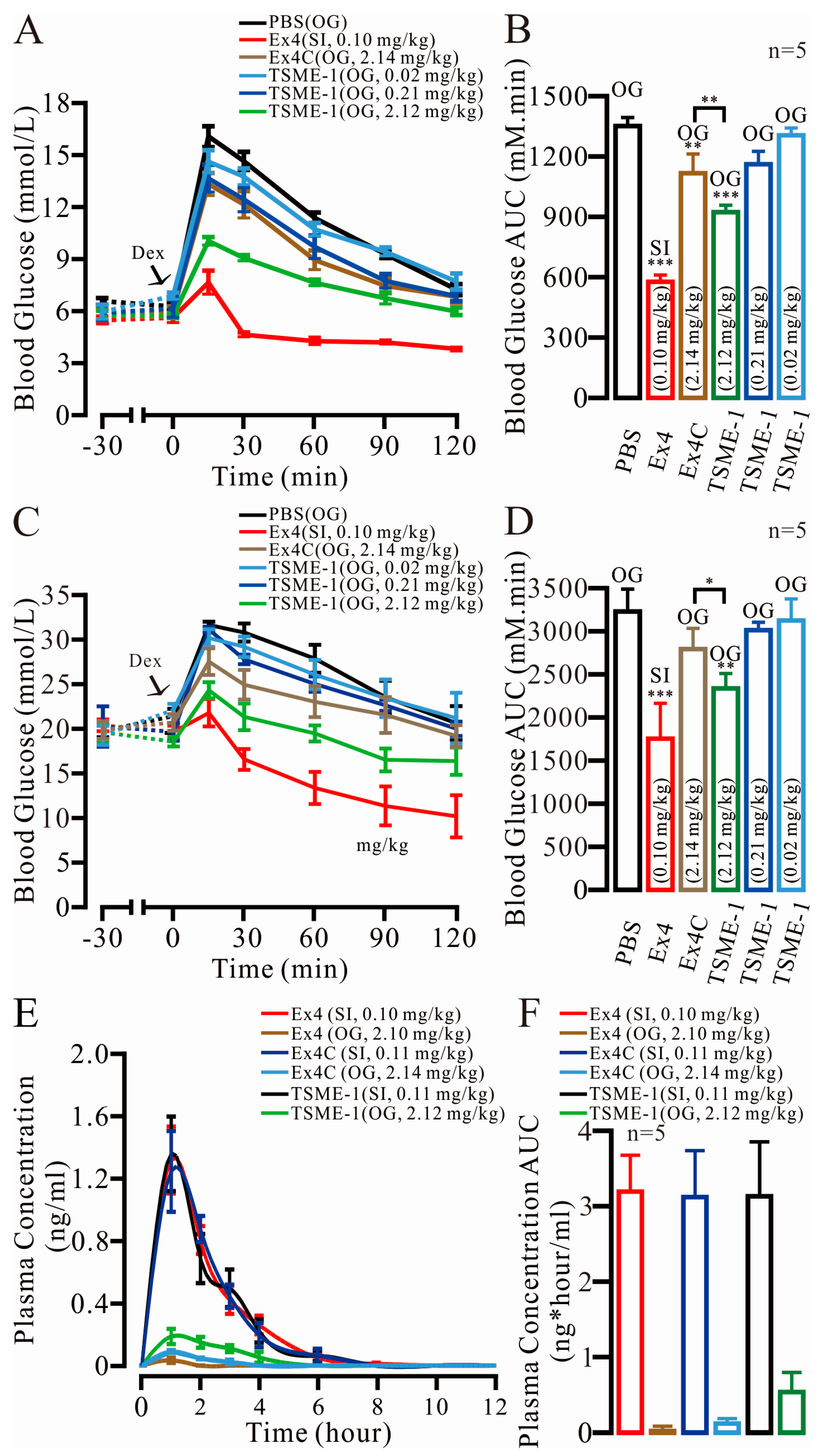
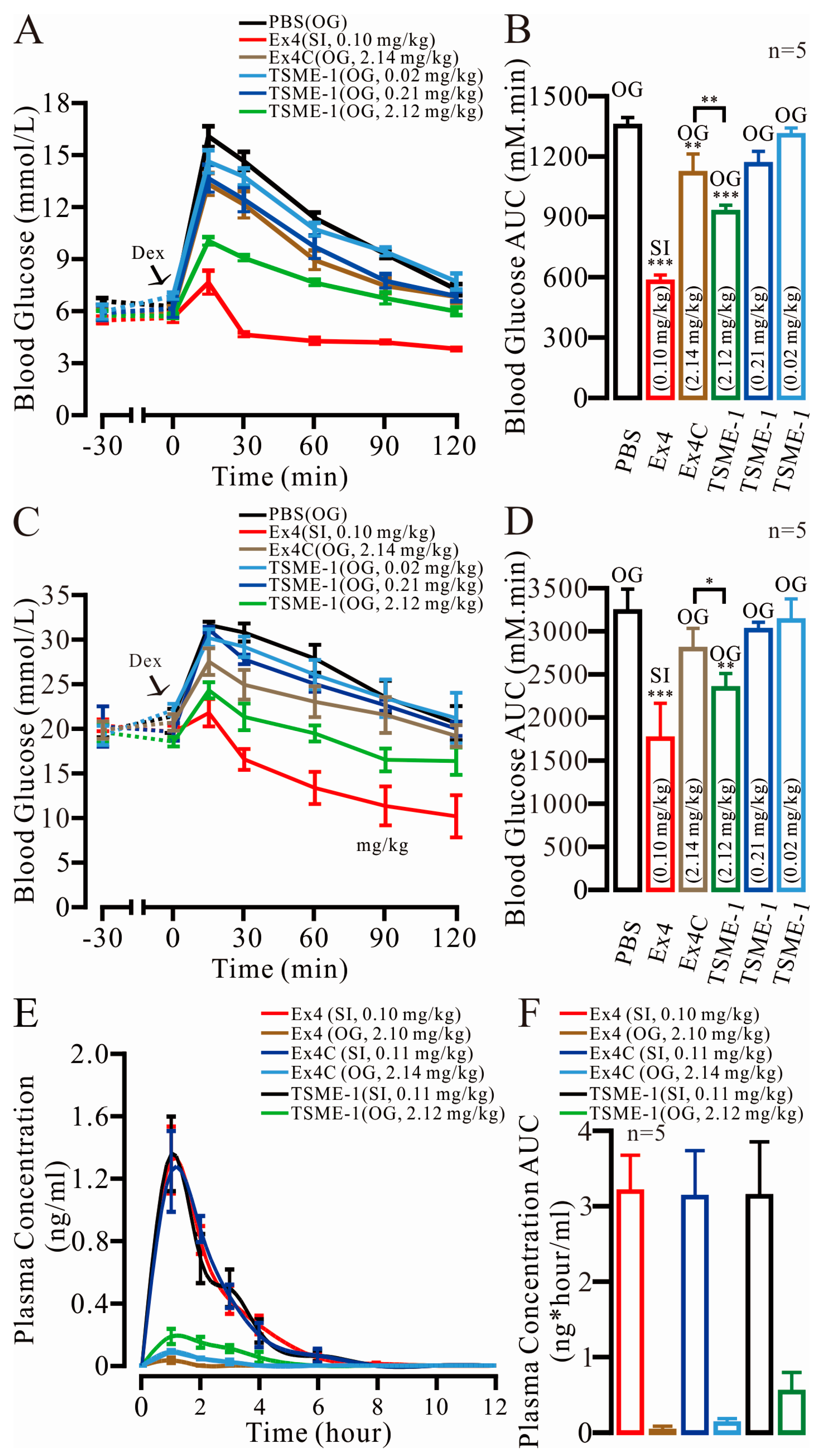
2.6. TSME-1 Significantly Normalized the BGLs of the Normal and Streptozotocin (STZ)/High Fat Diet (HFD)-Induced T2D Mice When Administered by Oral Gavage
2.7. The Relative Bioavailability of TSME-1 Was Higher Than That of Ex4 and Ex4C
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Peptide Synthesis
4.4. Intraperitoneal Glucose Tolerance Test (IPGTT)
4.5. Key Proteases Screen Assay
4.6. Specific Cleavage Sites Screen Assay
4.7. Molecular Dynamics Simulation
4.8. GLP-1 Receptor Gene Activation Assays
4.9. CHO Cell cAMP Accumulation Assay
4.10. Cellular Activity of TSME-1 on Rat Insulinoma β-Cells (RINm5f)
4.11. Relative Bioavailability in C57BL/6J Mice
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of ly2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed]
- McSharry, J.; McGowan, L.; Farmer, A.J.; French, D.P. Perceptions and experiences of taking oral medications for the treatment of type 2 diabetes mellitus: A systematic review and meta-synthesis of qualitative studies. Diabet. Med. 2016, 33, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Homberger, E.; Siegel, E.G.; Allen, R.C.; Eaton, R.P.; Ebert, R.; Creutzfeldt, W. Incretin effects of increasing glucose loads in man calculated from venous insulin and c-peptide responses. J. Clin. Endocrinol. Metab. 1986, 63, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Kolterman, O.G.; Kim, D.D.; Shen, L.; Ruggles, J.A.; Nielsen, L.L.; Fineman, M.S.; Baron, A.D. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. AJHP 2005, 62, 173–181. [Google Scholar] [PubMed]
- John, B.; Buse, E.A. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: A 26-week randomised, parallel-group, multinational, open-label trial (lead-6). Lancet 2009, 374, 39–47. [Google Scholar]
- Quoyer, J.; Longuet, C.; Broca, C.; Linck, N.; Costes, S.; Varin, E.; Bockaert, J.; Bertrand, G.; Dalle, S. GLP-1 mediates antiapoptotic effect by phosphorylating bad through a β-arrestin 1-mediated ERK1/2 activation in pancreatic β-cells. J. Biol. Chem. 2010, 285, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liang, W.; Guan, H.; Liu, J.; Liu, L.; Li, H.; He, X.; Zheng, J.; Chen, J.; Cao, X.; et al. Exendin-4 promotes pancreatic β-cell proliferation via inhibiting the expression of Wnt5a. Endocrine 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Roth, J.A.; Nguyen, H.; Felber, E.; Furnback, W.; Garrison, L.P. The short-term cost-effectiveness of once-daily liraglutide versus once-weekly exenatide for the treatment of type 2 diabetes mellitus in the united states. PLoS ONE 2015, 10, e0121915. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.; Holscher, C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.N.; Ndefo, U.A.; Oliver, A.; Payton, E. Albiglutide: A once-weekly glucagon-like peptide-1 receptor agonist for type 2 diabetes mellitus. Am. J. Health Syst. Pharm. 2015, 72, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Xie, J.; Fernandez Lando, L.; Kabul, S.; Swindle, R.W. Liraglutide versus exenatide once weekly: Persistence, adherence, and early discontinuation. Clin. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rulis, M. Novo Nordisk Announces Positive Results for Phase 2 Trial with Oral Semaglutide in People with Type 2 Diabetes. Available online: http://www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=16A23FB6–7B4C-47E7–90C4-A0790A1B8732 (accessed on February 20 2015).
- Leone-Bay, A.; Sato, M.; Paton, D.; Hunt, A.H.; Sarubbi, D.; Carozza, M.; Chou, J.; McDonough, J.; Baughman, R.A. Oral delivery of biologically active parathyroid hormone. Pharm. Res. 2001, 18, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Mahato, R.I.; Narang, A.S.; Thoma, L.; Miller, D.D. Emerging trends in oral delivery of peptide and protein drugs. Crit. Rev. Ther. Drug Carrier Syst. 2003, 20, 153–214. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.B.; Braun, G.B.; She, Z.G.; Kotamraju, V.R.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. A free cysteine prolongs the half-life of a homing peptide and improves its tumor-penetrating activity. J. Control. Release 2014, 175, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnurch, A. The use of inhibitory agents to overcome the enzymatic barrier to perorally administered therapeutic peptides and proteins. J. Control. Release 1998, 52, 1–16. [Google Scholar] [CrossRef]
- Rinderknecht, H.; Renner, I.G.; Douglas, A.P.; Adham, N.F. Profiles of pure pancreatic secretions obtained by direct pancreatic duct cannulation in normal healthy human subjects. Gastroenterology 1978, 75, 1083–1089. [Google Scholar] [PubMed]
- Karsdal, M.A.; Riis, B.J.; Mehta, N.; Stern, W.; Arbit, E.; Christiansen, C.; Henriksen, K. Lessons learned from the clinical development of oral peptides. Br. J. Clin. Pharmacol. 2015, 79, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Gunnar Skoglund, M.A.H.; Holz, G.G. Glucagon-like peptide 1 stimulates insulin gene promoter activity by protein kinase A–independent activation of the rat insulin I gene camp response element. Diabetes 2000, 49, 1156–1164. [Google Scholar] [CrossRef]
- Chepurny, O.G.; Holz, G.G. Over-expression of the glucagon-like peptide-1 receptor on INS-1 cells confers autocrine stimulation of insulin gene promoter activity: A strategy for production of pancreatic β-cell lines for use in transplantation. Cell Tissue Res. 2002, 307, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Alama, T.; Kusamori, K.; Katsumi, H.; Sakane, T.; Yamamoto, A. Absorption-enhancing effects of gemini surfactant on the intestinal absorption of poorly absorbed hydrophilic drugs including peptide and protein drugs in rats. Int. J. Pharm. 2016, 499, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, P.; Artursson, P. Oral absorption of peptides and nanoparticles across the human intestine: Opportunities, limitations and studies in human tissues. Adv. Drug Deliv. Rev. 2016, 106, 256–276. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C.; Lowe, M.E. Human pancreatic digestive enzymes. Digest. Dis. Sci. 2007, 52, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Braunwald, E.; Raz, I.; Cavender, M.A.; Morrow, D.A.; Jarolim, P.; Udell, J.A.; Mosenzon, O.; Im, K.; Umez-Eronini, A.A.; et al. Heart failure, saxagliptin, and diabetes mellitus: Observations from the savor-timi 53 randomized trial. Circulation 2014, 130, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef] [PubMed]
- Irie, S.; Matsumura, Y.; Zdravkovic, M.; Jacobsen, L.V.; Kageyama, S. Tolerability, pharmacokinetics and pharmacodynamics of the once-daily human GLP-1 analog liraglutide in japanese healthy subjects: A randomized, double-blind, placebo-controlled dose-escalation study. Int. J. Clin. Pharmacol. Ther. 2008, 46, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.M.; Nuffer, W. Albiglutide: A new GLP-1 receptor agonist for the treatment of type 2 diabetes. Ann. Pharmacother. 2014, 48, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, R.; Tan, Z.; Huang, B.; Zhang, S. Computational polypharmacology: A new paradigm for drug discovery. Expert Opin. Drug Discov. 2017, 12, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Schroeder, C.I.; Mitchell, J.M.; Fairlie, D.P.; Edmonds, D.J.; Griffith, D.A.; Ruggeri, R.B.; Derksen, D.R.; Loria, P.M.; Price, D.A.; et al. Truncated glucagon-like peptide-1 and exendin-4 α-conotoxin PL14A peptide chimeras maintain potency and α-helicity and reveal interactions vital for camp signaling in vitro. J. Biol. Chem. 2016, 291, 15778–15787. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Parthier, C.; Reedtz-Runge, S.; Rudolph, R.; Stubbs, M.T. Passing the baton in class B GPCRs: Peptide hormone activation via helix induction? Trends Biochem. Sci. 2009, 34, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Gromada, J.; Brock, B.; Schmitz, O.; Rorsman, P. Glucagon-like peptide-1: Regulation of insulin secretion and therapeutic potential. Basic Clin. Pharmacol. Toxicol. 2004, 95, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.E.; Egan, J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Therap. 2007, 113, 546–593. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Modi, H.; Kawamori, D.; Kulkarni, R.N.; Joffraud, M.; Thorens, B. Glucagon-like peptide-1 increases β-cell glucose competence and proliferation by translational induction of insulin-like growth factor-1 receptor expression. J. Biol. Chem. 2010, 285, 10538–10545. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease | No. | Retention Time | Apparent Molecular Weight | Theoretical Molecular Weight | Fragment |
|---|---|---|---|---|---|
| Trypsin | 1 | 11.910 | 1024.4663 | 1024.4694 | 28–39 |
| 2 | 13.936 | 991.4494 | 991.4513 | 13–20 | |
| 3 | 18.260 | 1278.5927 | 1278.5960 | 1–12 | |
| 4 | 33.747 | 948.5579 | 948.5553 | 21–27 | |
| Chymotrypsin | 5 | 12.570 | 1265.6440 | 1265.6484 | 26–39 |
| 6 | 13.420 | 647.2809 | 647.2784 | 1–6 | |
| 7 | 24.620 | 447.2233 | 447.2238 | 23–25 | |
| 8 | 27.026 | 1735.8964 | 1735.8531 | 7–21 | |
| 9 | 29.595 | 594.2947 | 594.2922 | 22–25 | |
| 10 | 30.445 | 2364.1284 | 2364.1137 | 1–21 | |
| 11 | 33.593 | 1882.9354 | 1882.9216 | 7–22 | |
| 12 | 35.891 | 2511.1896 | 2511.1822 | 1–22 | |
| Elastase | 13 | 7.424 | 288.2029 | 288.2029 | 20–21 |
| 14 | 26.439 | 2094.9582 | 2094.9286 | 1–19 | |
| 15 | 26.982 | 1841.0082 | 1840.9228 | 22–39 | |
| 16 | 30.438 | 2364.1284 | 2364.1137 | 1–21 | |
| 17 | 34.816 | 707.3790 | 707.3763 | 22–26 |
| Site | Amino Acid | Mutation | Score |
|---|---|---|---|
| Wild Type | –21.4 | ||
| 12 | Lysine | Methionine | –22.5 |
| Valine | –22.3 | ||
| Isoleucine | –22.3 | ||
| 20 | Arginine | Phenylalanine | –20.2 |
| Leucine | –20.1 | ||
| Tyrosine | –20.1 | ||
| 27 | Lysine | Methionine | –20.2 |
| Isoleucine | –19.2 | ||
| Valine | –18.7 |
| Analogue | Sepuence | Binding Energy |
|---|---|---|
| Exendin4-C | HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPSC | −50.60 |
| TSME-1 | HGEGTFTSDLSMQMEEEAVLLFIEWLMNGGPSSGAPPPSC | −45.88 |
| TSME-2 | HGEGTFTSDLSMQMEEEAVFLFIEWLMNGGPSSGAPPPSC | −44.88 |
| TSME-3 | HGEGTFTSDLSVQMEEEAVV LFIEWLVNGG PSSGAPPPSC | −43.81 |
| TSME-4 | HGEGTFTSDLSMQMEEEAVYLFIEWLMNGGPSSGAPPPSC | −42.93 |
| TSME-5 | HGEGTFTSDLSIQMEEEAVY LFIEWLINGGPSSGAPPPSC | −44.33 |
| TSME-6 | HGEGTFTSDLSIQMEEEAVV LFIEWLFNGGPSSGAPPPSC | −41.92 |
| TSME-7 | HGEGTFTSDLS(D–I)QMEEEAV(D-Y)LFIEWL(D-V)NGGPSSGAPPPSC | −40.27 |
| TSME-8 | HGEGTFTSDLS(D–V)QMEEEAV(D-L)LFIEWL(D-V)NGGPSSGAPPPSC | −40.91 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sai, W.; Tian, H.; Yang, K.; Tang, D.; Bao, J.; Ge, Y.; Song, X.; Zhang, Y.; Luo, C.; Gao, X.; et al. Systematic Design of Trypsin Cleavage Site Mutated Exendin4-Cysteine 1, an Orally Bioavailable Glucagon-Like Peptide-1 Receptor Agonist. Int. J. Mol. Sci. 2017, 18, 578. https://doi.org/10.3390/ijms18030578
Sai W, Tian H, Yang K, Tang D, Bao J, Ge Y, Song X, Zhang Y, Luo C, Gao X, et al. Systematic Design of Trypsin Cleavage Site Mutated Exendin4-Cysteine 1, an Orally Bioavailable Glucagon-Like Peptide-1 Receptor Agonist. International Journal of Molecular Sciences. 2017; 18(3):578. https://doi.org/10.3390/ijms18030578
Chicago/Turabian StyleSai, Wenbo, Hong Tian, Kangmin Yang, Daoqi Tang, Jinxiao Bao, Yang Ge, Xiaoda Song, Yu Zhang, Cheng Luo, Xiangdong Gao, and et al. 2017. "Systematic Design of Trypsin Cleavage Site Mutated Exendin4-Cysteine 1, an Orally Bioavailable Glucagon-Like Peptide-1 Receptor Agonist" International Journal of Molecular Sciences 18, no. 3: 578. https://doi.org/10.3390/ijms18030578