
Zinc Prevents the Development of Diabetic Cardiomyopathy in db/db Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
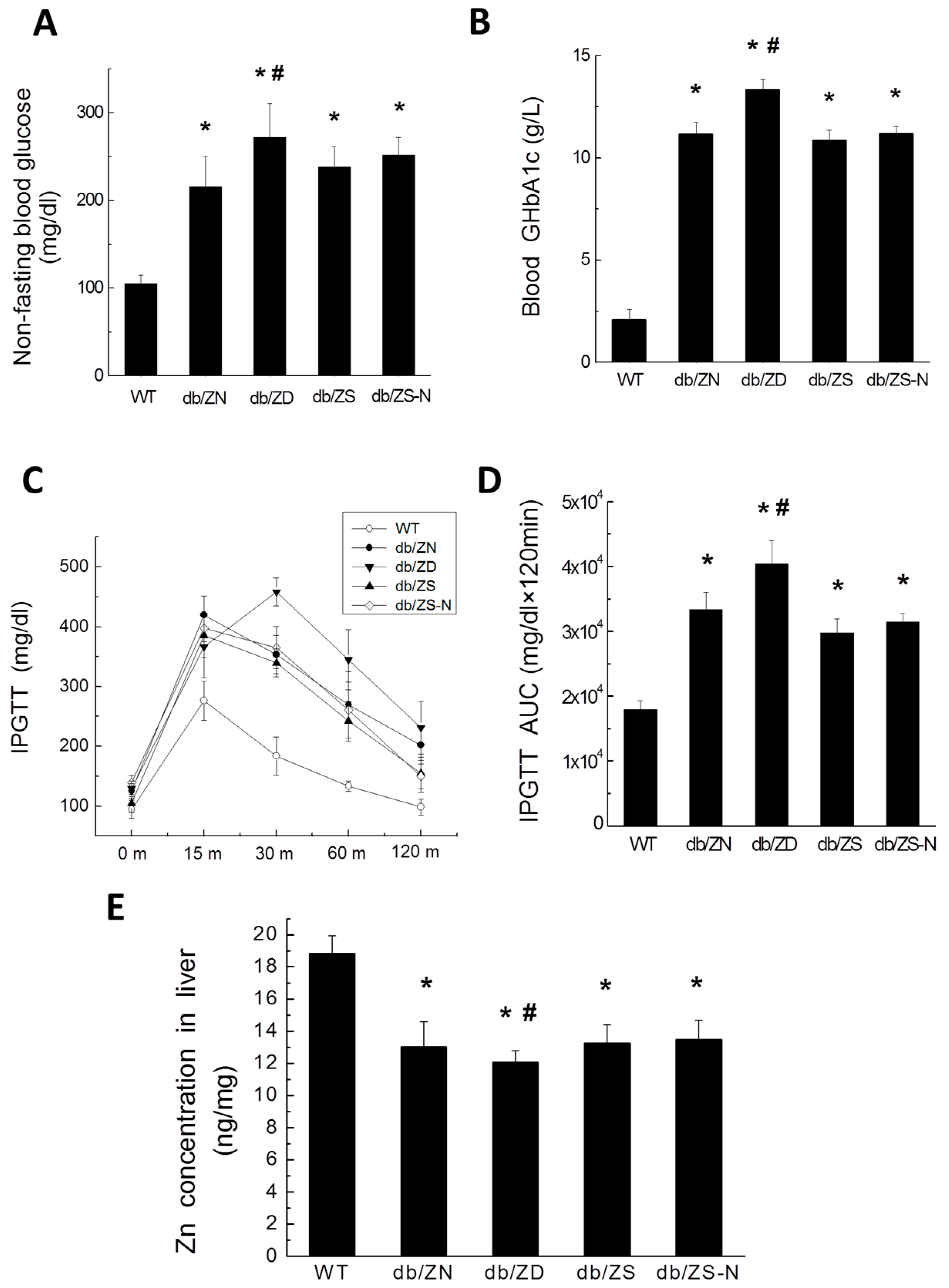
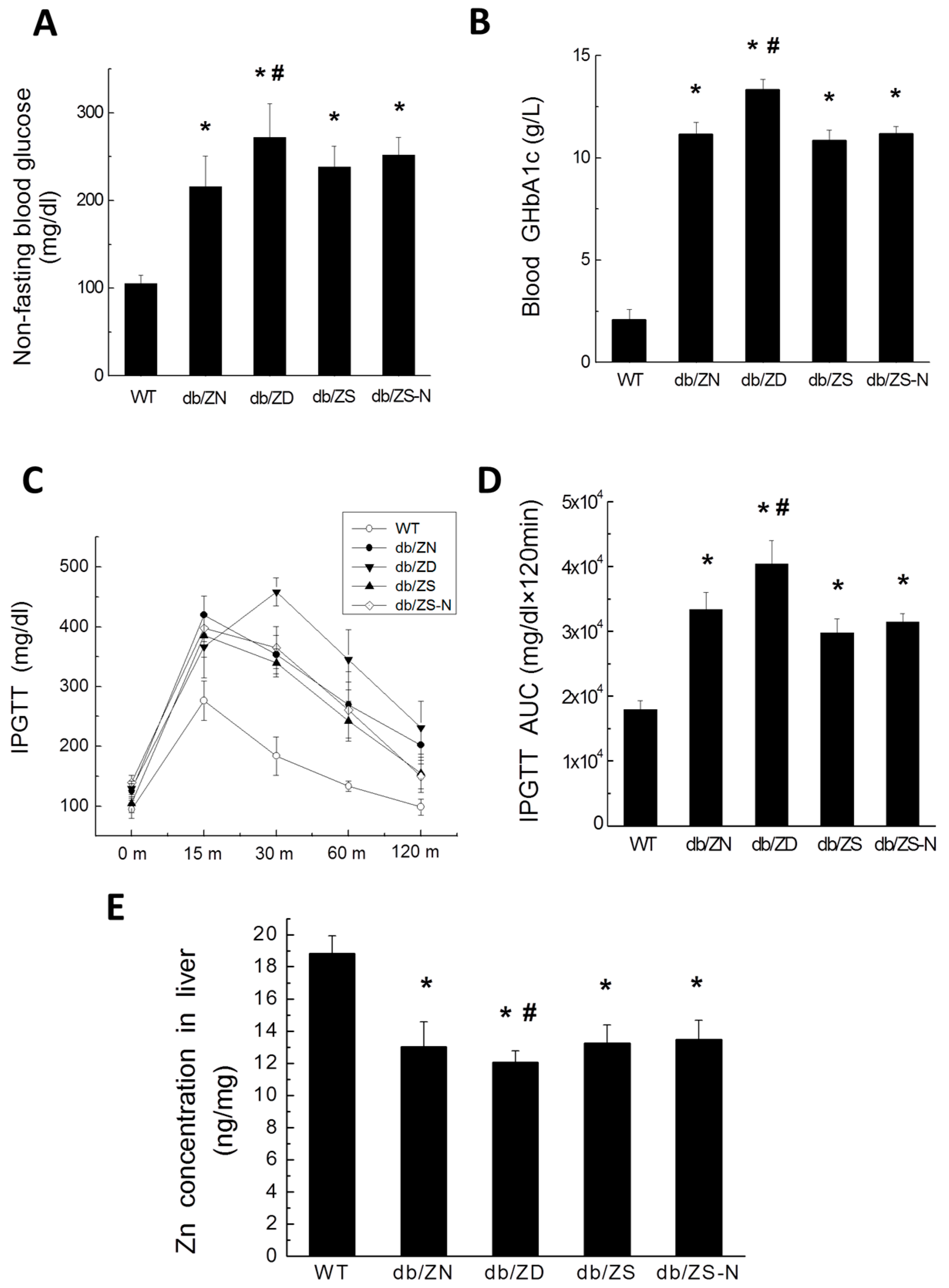
2.1. General Features of db/db Mice after Treatment with Different Zn Amounts
2.2. Zn Levels in Cardiac and Liver Tissues
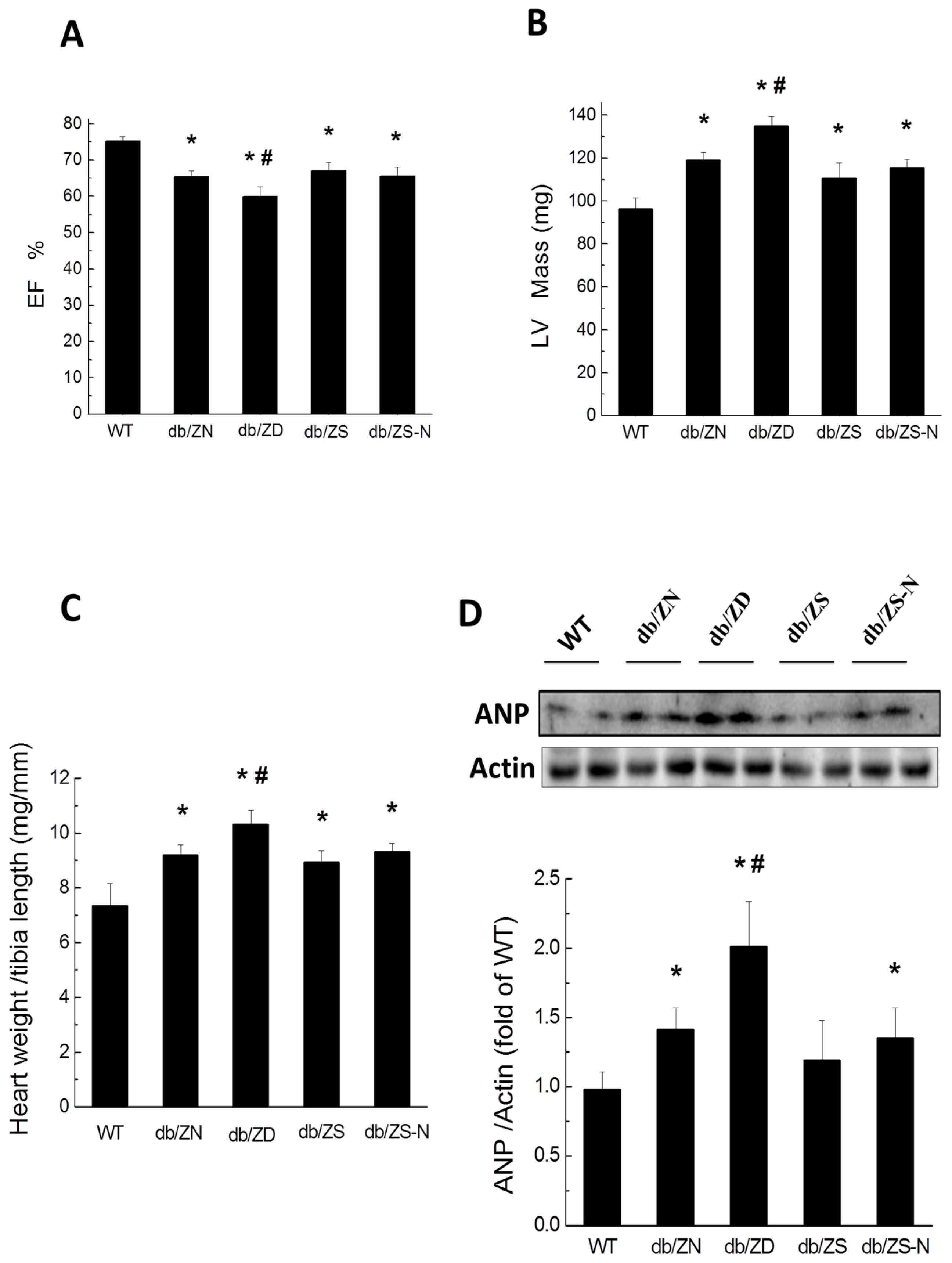
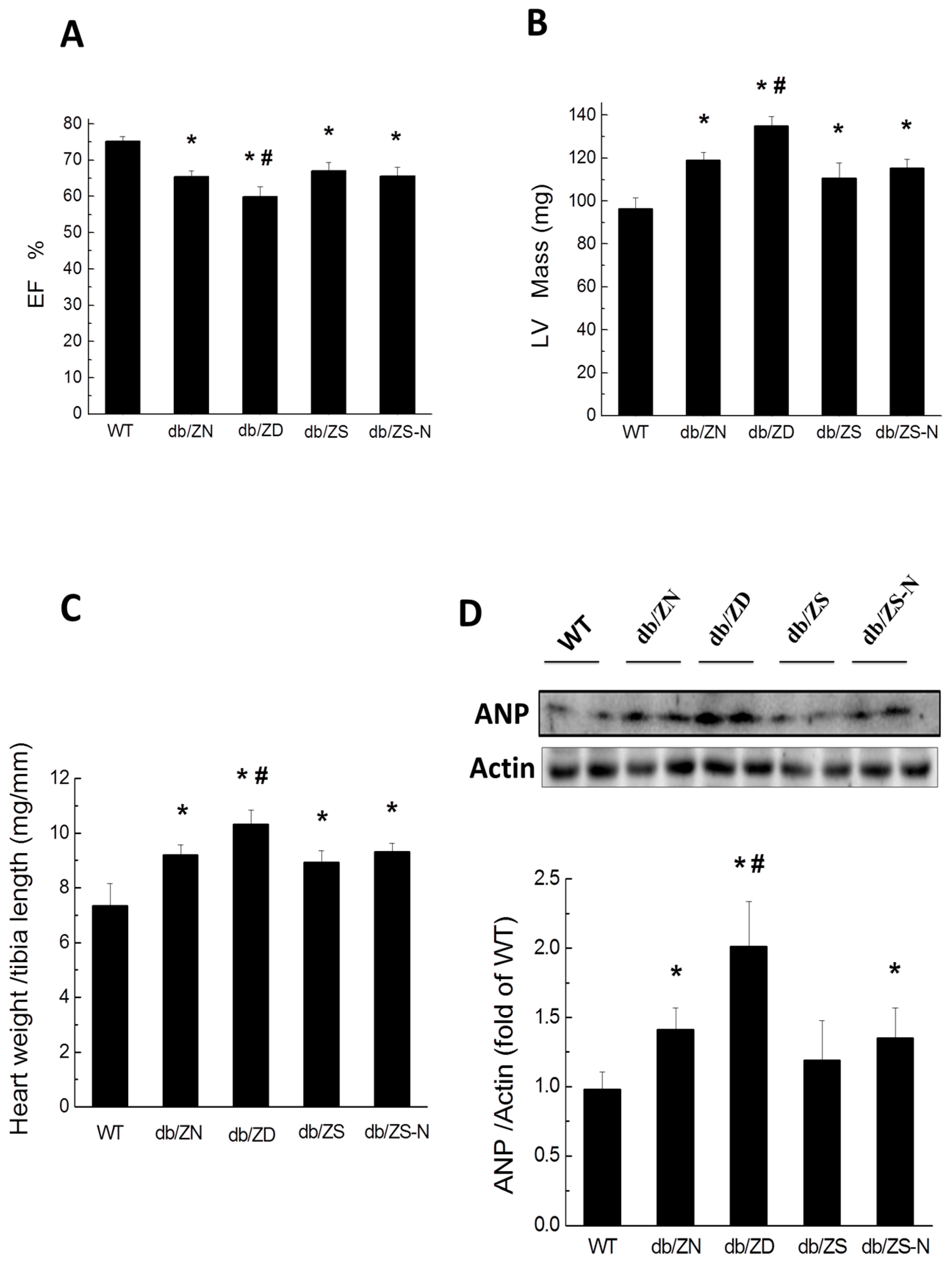
2.3. Cardiac Hypertrophy and Function
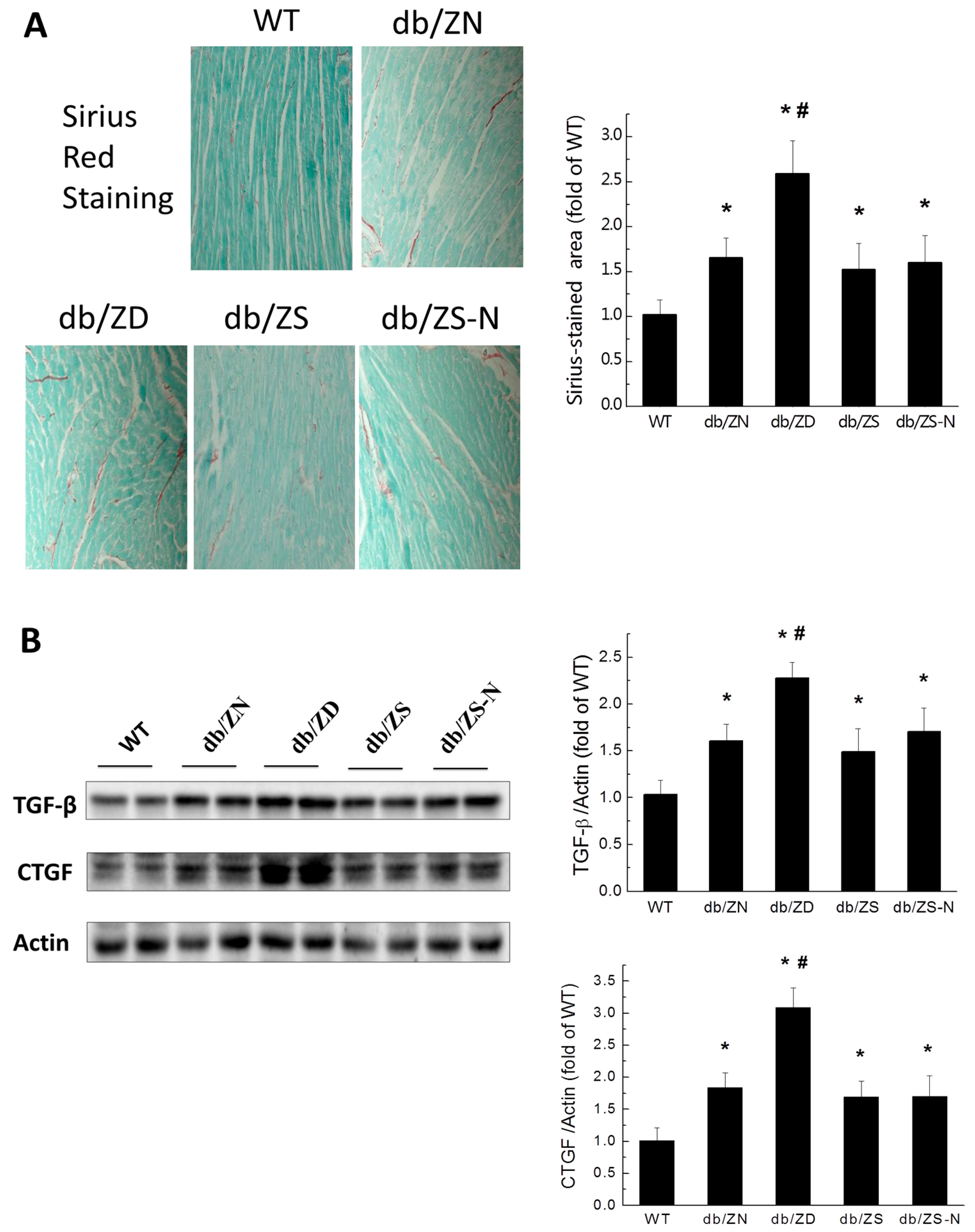
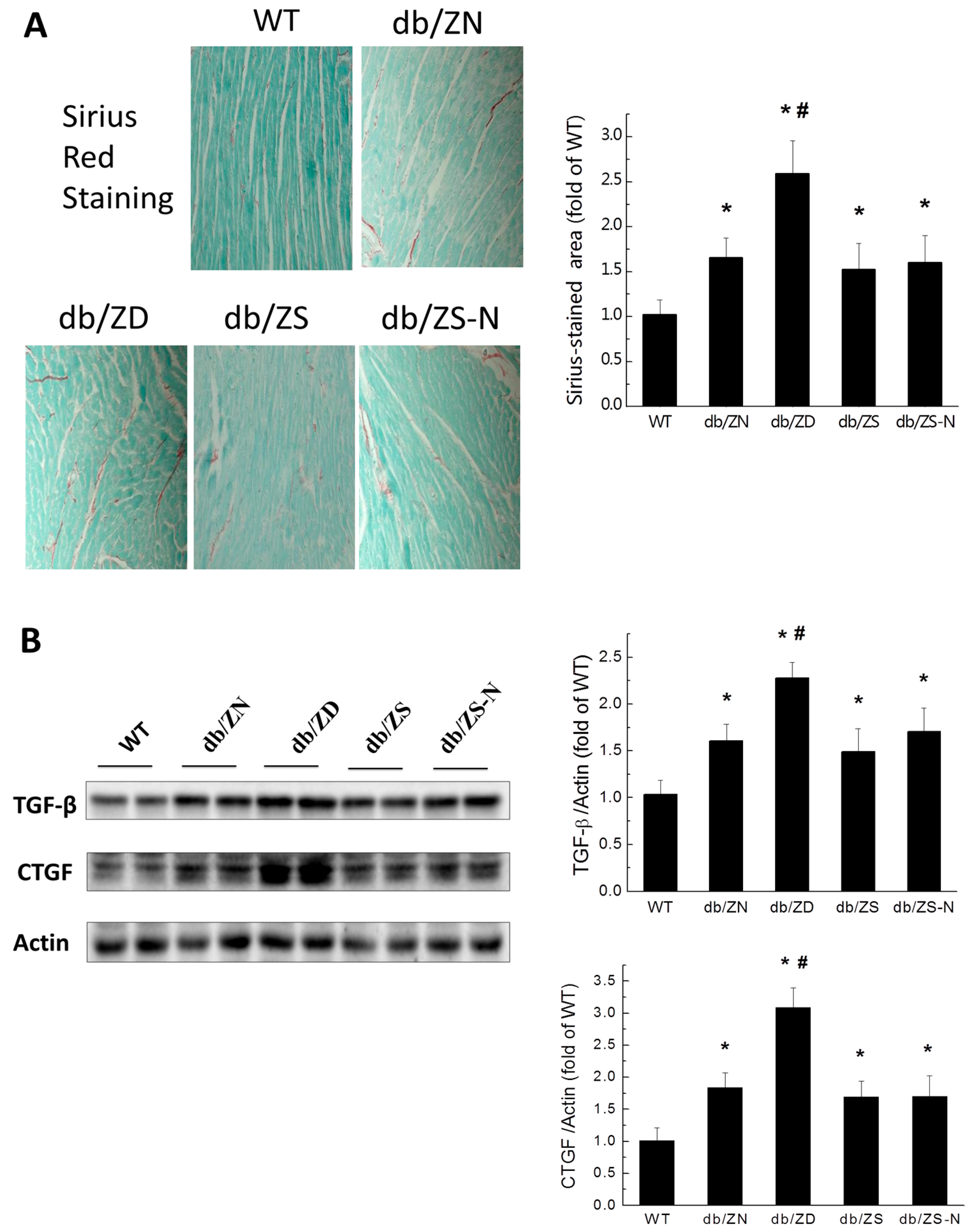
2.4. Zn Prevents Cardiac Fibrosis in db/db Mice
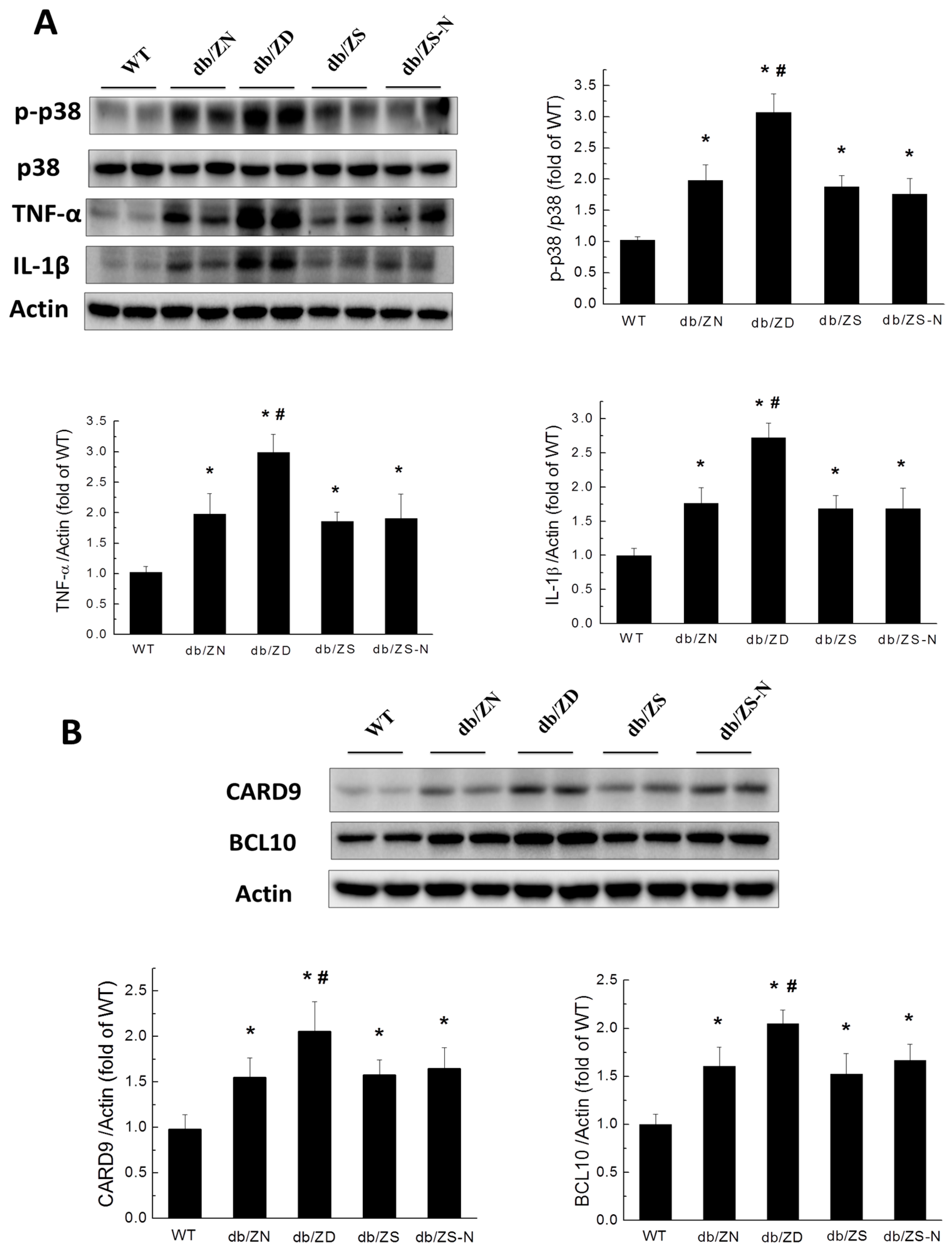
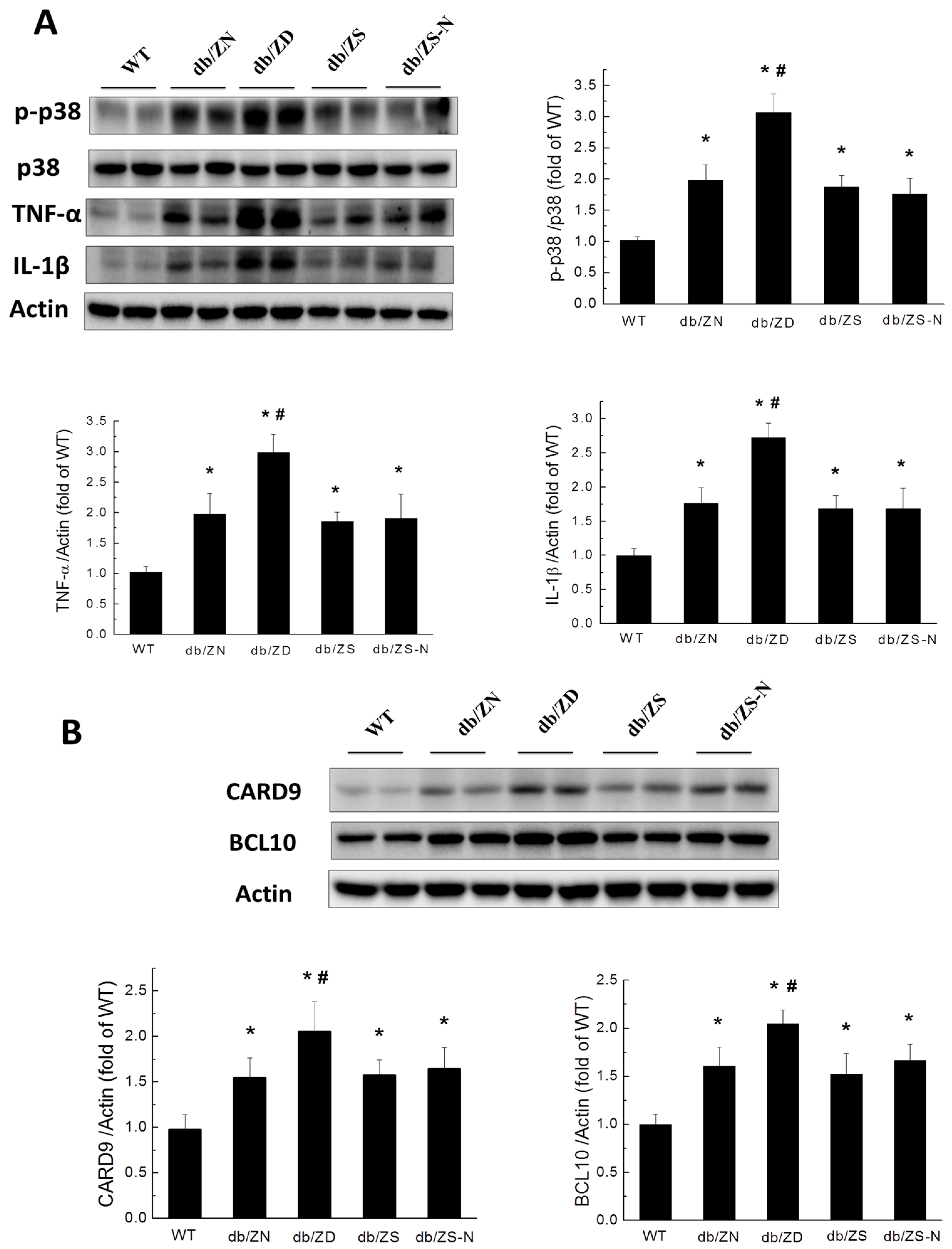
2.5. Zn Prevents Cardiac Inflammation in db/db Mice
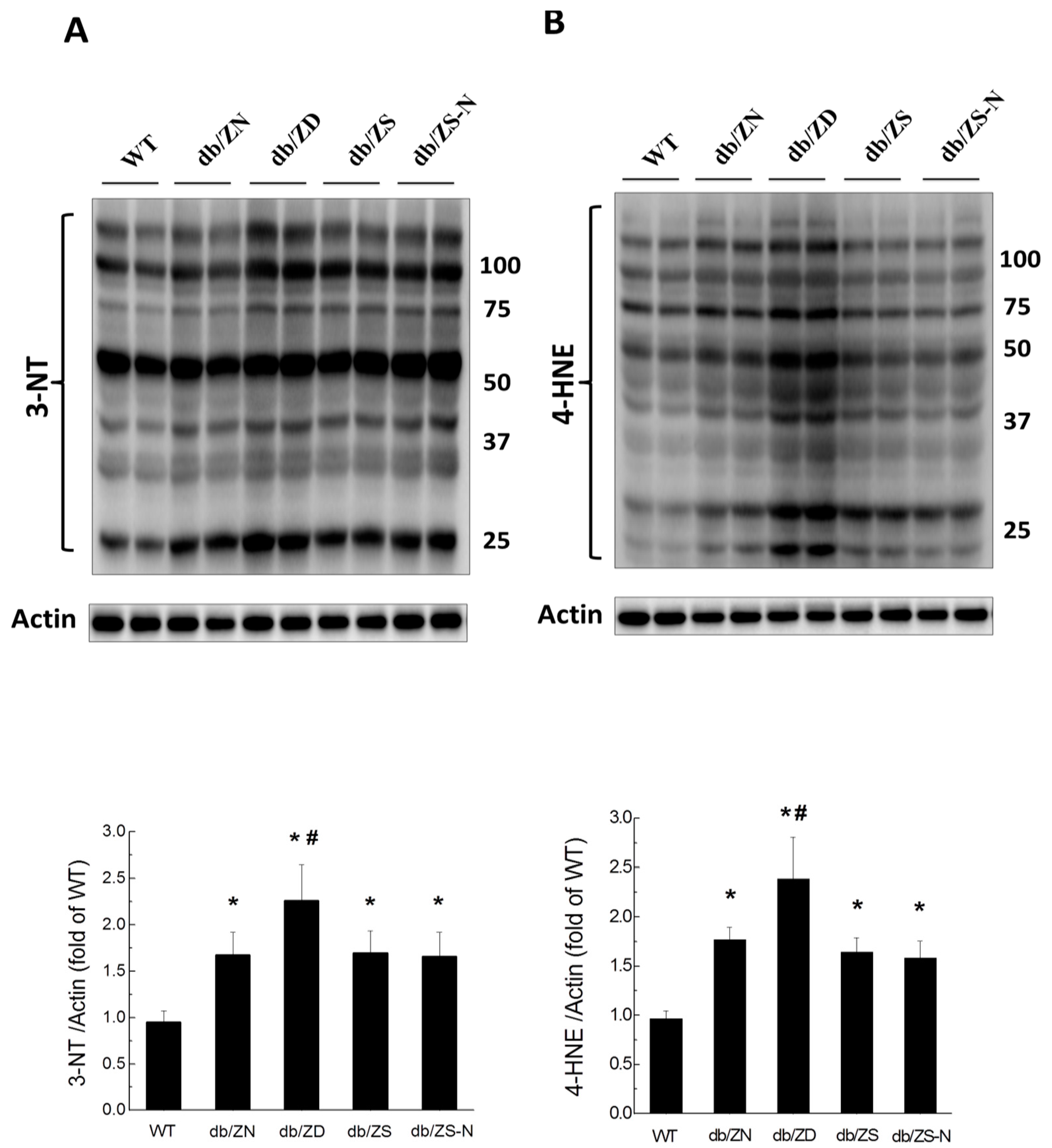
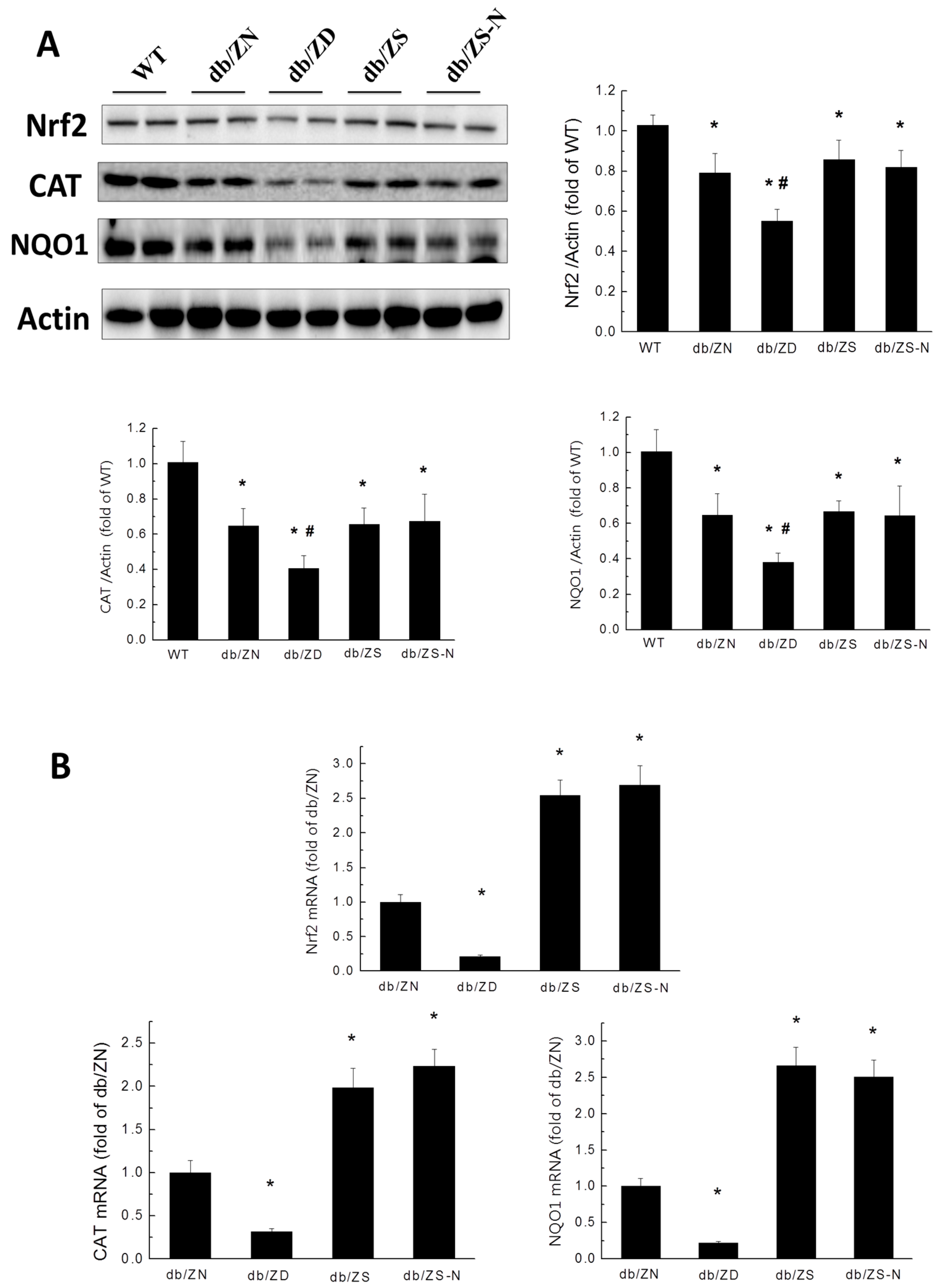
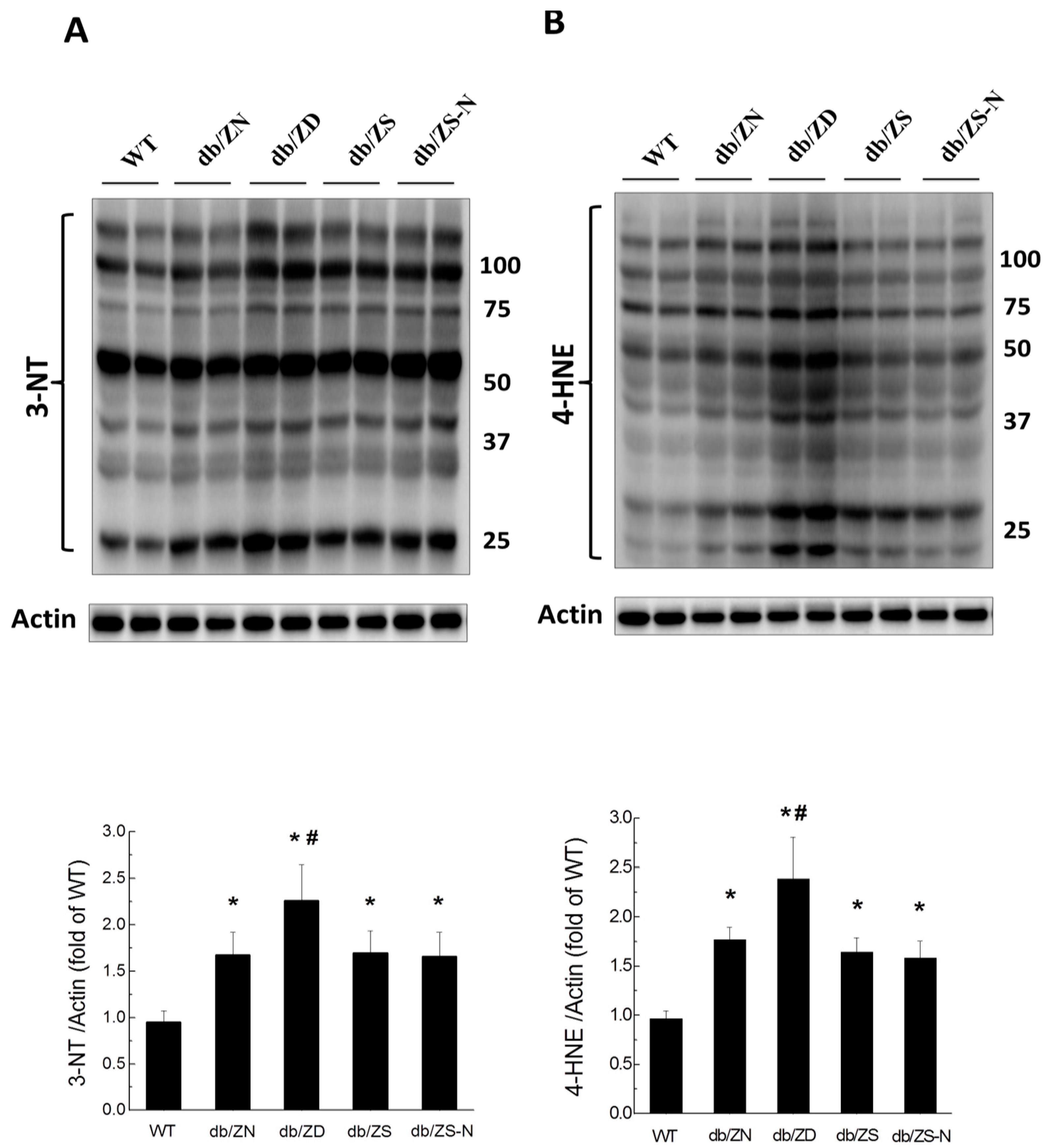
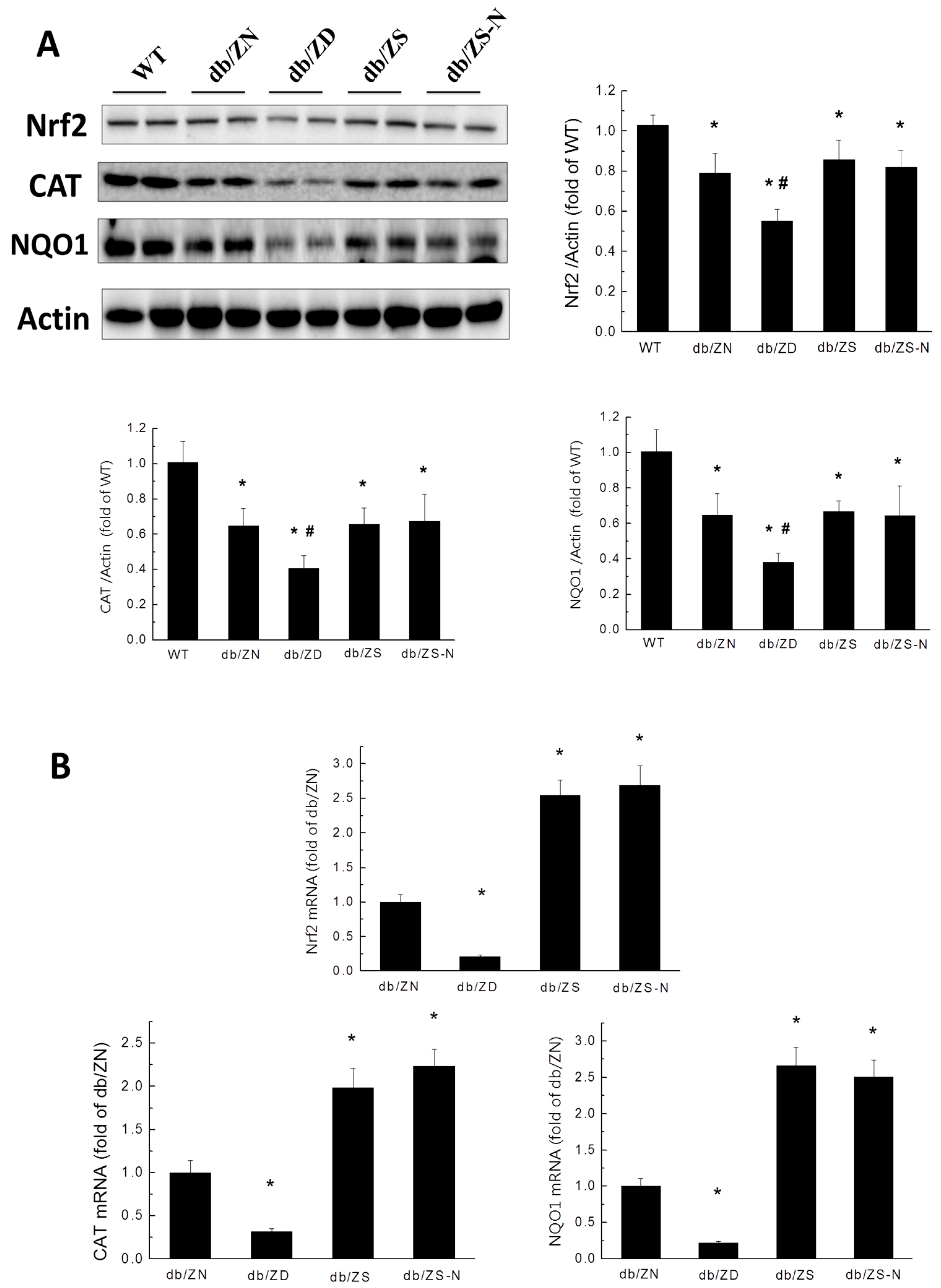
2.6. Zn Attenuation of Cardiac Oxidative Stress Is Probably Associated with Nrf2 Activation to Upregulate Downstream Antioxidants
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Intraperitoneal Glucose Tolerance Test (IPGTT)
4.3. Echocardiography for EF% and LV Mass Assessment
4.4. Zn Concentration Assessment in the Liver Tissue
4.5. Sirius Red Staining
4.6. Quantitative Real-Time PCR
4.7. Western Blot
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bertoni, A.G.; Tsai, A.; Kasper, E.K.; Brancati, F.L. Diabetes and idiopathic cardiomyopathy: A nationwide case-control study. Diabetes Care 2003, 26, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Romero, F.; Rodriguez-Moran, M. Hypomagnesemia, oxidative stress, inflammation, and metabolic syndrome. Diabetes Metab. Res. Rev. 2006, 22, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Shannahan, J.H.; Schladweiler, M.C.; Richards, J.H.; Ledbetter, A.D.; Ghio, A.J.; Kodavanti, U.P. Pulmonary oxidative stress, inflammation, and dysregulated iron homeostasis in rat models of cardiovascular disease. J. Toxicol. Environ. Health A 2010, 73, 641–656. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 2008, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.F.; Qi, W.; Feng, B.; Mu, J.; Zeng, W.; Guo, Y.H.; Pang, Q.; Ye, Z.L.; Liu, L.; Yuan, F.H. Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sci. 2011, 88, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Jurowski, K.; Szewczyk, B.; Nowak, G.; Piekoszewski, W. Biological consequences of zinc deficiency in the pathomechanisms of selected diseases. J. Biol. Inorg. Chem. 2014, 19, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Oesterling, E.; Stromberg, A.; Toborek, M.; MacDonald, R.; Hennig, B. Zinc deficiency induces vascular pro-inflammatory parameters associated with NF-κB and PPAR signaling. J. Am. Coll. Nutr. 2008, 27, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.M.; Suschek, C.V.; Wetzel, W.; Kroncke, K.D.; Kolb-Bachofen, V. Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radic. Biol. Med. 2008, 44, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.J.; Joshi, P.C.; Fan, X.; Brown, L.A.; Ritzenthaler, J.D.; Roman, J.; Guidot, D.M. Zinc supplementation restores PU.1 and Nrf2 nuclear binding in alveolar macrophages and improves redox balance and bacterial clearance in the lungs of alcohol-fed rats. Alcohol. Clin. Exp. Res. 2011, 35, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, M.; Zhang, Z.; Gu, J.; Chen, J.; Payne, K.M.; Tan, Y.; Wang, Y.; Yin, X.; Zhang, X.; et al. Zinc deficiency exacerbates while zinc supplement attenuates cardiac hypertrophy in high-fat diet-induced obese mice through modulating p38 MAPK-dependent signaling. Toxicol. Lett. 2016, 258, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Q.; Sun, W.; Xin, Y.; Zhang, Z.; Tan, Y.; Zhou, S.; Zhang, C.; Cai, L.; Lu, X.; et al. Zinc treatment prevents type 1 diabetes-induced hepatic oxidative damage, endoplasmic reticulum stress, and cell death, and even prevents possible steatohepatitis in the OVE26 mouse model: Important role of metallothionein. Toxicol. Lett. 2015, 233, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cui, W.; Tan, Y.; Luo, P.; Chen, Q.; Zhang, C.; Qu, W.; Miao, L.; Cai, L. Zinc is essential for the transcription function of Nrf2 in human renal tubule cells in vitro and mouse kidney in vivo under the diabetic condition. J. Cell. Mol. Med. 2014, 18, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhao, Y.; Sun, X.; Song, Z.; McClain, C.J.; Zhou, Z. Dietary zinc deficiency exaggerates ethanol-induced liver injury in mice: Involvement of intrahepatic and extrahepatic factors. PLoS ONE 2013, 8, e76522. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.D.; Lin, P.Y. Zinc-induced hyperleptinemia relates to the amelioration of sucrose-induced obesity with zinc repletion. Obes. Res. 2000, 8, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Marreiro, D.N.; Fisberg, M.; Cozzolino, S.M. Zinc nutritional status and its relationships with hyperinsulinemia in obese children and adolescents. Biol. Trace Elem. Res. 2004, 100, 137–149. [Google Scholar] [CrossRef]
- Basaki, M.; Saeb, M.; Nazifi, S.; Shamsaei, H.A. Zinc, copper, iron, and chromium concentrations in young patients with type 2 diabetes mellitus. Biol. Trace Elem. Res. 2012, 148, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Rosenkranz, E.; Overbeck, S.; Warmuth, S.; Mocchegiani, E.; Giacconi, R.; Weiskirchen, R.; Karges, W.; Rink, L. Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J. Nutr. Biochem. 2012, 23, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Kazi, T.G.; Afridi, H.I.; Kazi, N.; Jamali, M.K.; Arain, M.B.; Jalbani, N.; Kandhro, G.A. Copper, chromium, manganese, iron, nickel, and zinc levels in biological samples of diabetes mellitus patients. Biol. Trace Elem. Res. 2008, 122, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.M.; Yoon, J.S. Glycemic control of type 2 diabetic patients after short-term zinc supplementation. Nutr. Res. Pract. 2008, 2, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, R.; Ranasinghe, P.; Galappatthy, P.; Malkanthi, R.; Constantine, G.; Katulanda, P. Effects of zinc supplementation on diabetes mellitus: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2012, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Renal Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Bai, Y.; Miao, X.; Luo, P.; Chen, Q.; Tan, Y.; Rane, M.J.; Miao, L.; Cai, L. Prevention of diabetic nephropathy by sulforaphane: Possible role of nrf2 upregulation and activation. Oxid. Med. Cell Longev. 2012, 2012, 821936. [Google Scholar] [CrossRef] [PubMed]
- Khullar, M.; Al-Shudiefat, A.A.; Ludke, A.; Binepal, G.; Singal, P.K. Oxidative stress: A key contributor to diabetic cardiomyopathy. Can. J. Physiol. Pharmacol. 2010, 88, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Bansode, R.R.; Bal, N.C.; Mehta, M.; Mehta, K.D. Protein kinase CBETA deficiency attenuates obesity syndrome of ob/ob mice by promoting white adipose tissue remodeling. J. Lipid Res. 2012, 53, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Qin, X.; Peterson, M.R.; Haller, S.E.; Wilson, K.A.; Hu, N.; Lin, X.; Nair, S.; Ren, J.; He, G. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J. Mol. Cell. Cardiol. 2016, 92, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Bertin, J.; Guo, Y.; Wang, L.; Srinivasula, S.M.; Jacobson, M.D.; Poyet, J.L.; Merriam, S.; Du, M.Q.; Dyer, M.J.; Robison, K.E.; et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-κB. J. Biol. Chem. 2000, 275, 41082–41086. [Google Scholar] [CrossRef] [PubMed]
- Marko, L.; Henke, N.; Park, J.K.; Spallek, B.; Qadri, F.; Balogh, A.; Apel, I.J.; Oravecz-Wilson, K.I.; Choi, M.; Przybyl, L.; et al. Bcl10 mediates angiotensin II-induced cardiac damage and electrical remodeling. Hypertension 2014, 64, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.M.; Zhang, Y.; You, Y.; Wang, D.; Li, H.; Duramad, O.; Qin, X.F.; Dong, C.; Lin, X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 2007, 8, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Ulevitch, R.J. Limiting inflammatory responses during activation of innate immunity. Nat. Immunol. 2005, 6, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J. CARD9 signaling in the innate immune response. Ann. N. Y. Acad. Sci. 2008, 1143, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Yang, M.; Qi, G.; Zheng, J.; Jia, L.; Cheng, J.; Tian, C.; Li, H.; Lin, X.; Du, J. Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by Angiotensin II infusion. Am. J. Hypertens. 2011, 24, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wang, Y.; Zhou, G.; Chen, T.; Song, Y.; Li, X.; Kang, Y.J. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Carlson, E.C.; Donthi, R.V.; Kralik, P.M.; Shen, X.; Epstein, P.N. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes 2002, 51, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kinugawa, S.; Ide, T.; Matsusaka, H.; Inoue, N.; Ohta, Y.; Yokota, T.; Sunagawa, K.; Tsutsui, H. Overexpression of glutathione peroxidase attenuates myocardial remodeling and preserves diastolic function in diabetic heart. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zheng, S.; Metreveli, N.S.; Epstein, P.N. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006, 55, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Guo, W.; Sun, W.; Miao, X.; Wu, H.; Cong, X.; Wintergerst, K.A.; Kong, X.; Cai, L. Sulforaphane reduction of testicular apoptotic cell death in diabetic mice is associated with the upregulation of Nrf2 expression and function. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E14–E23. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, S.; Zhou, S.; Yan, X.; Wang, Y.; Chen, J.; Mellen, N.; Kong, M.; Gu, J.; Tan, Y.; et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell. Cardiol. 2014, 77, 42–52. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Wang, B.; Wang, Y.; Tong, Q.; Liu, Q.; Sun, J.; Zheng, Y.; Cai, L. Zinc Prevents the Development of Diabetic Cardiomyopathy in db/db Mice. Int. J. Mol. Sci. 2017, 18, 580. https://doi.org/10.3390/ijms18030580
Wang S, Wang B, Wang Y, Tong Q, Liu Q, Sun J, Zheng Y, Cai L. Zinc Prevents the Development of Diabetic Cardiomyopathy in db/db Mice. International Journal of Molecular Sciences. 2017; 18(3):580. https://doi.org/10.3390/ijms18030580
Chicago/Turabian StyleWang, Shudong, Bowei Wang, Yuehui Wang, Qian Tong, Quan Liu, Jian Sun, Yang Zheng, and Lu Cai. 2017. "Zinc Prevents the Development of Diabetic Cardiomyopathy in db/db Mice" International Journal of Molecular Sciences 18, no. 3: 580. https://doi.org/10.3390/ijms18030580