
Hepatitis B Virus X Protein Stimulates Proliferation, Wound Closure and Inhibits Apoptosis of HuH-7 Cells via CDC42

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
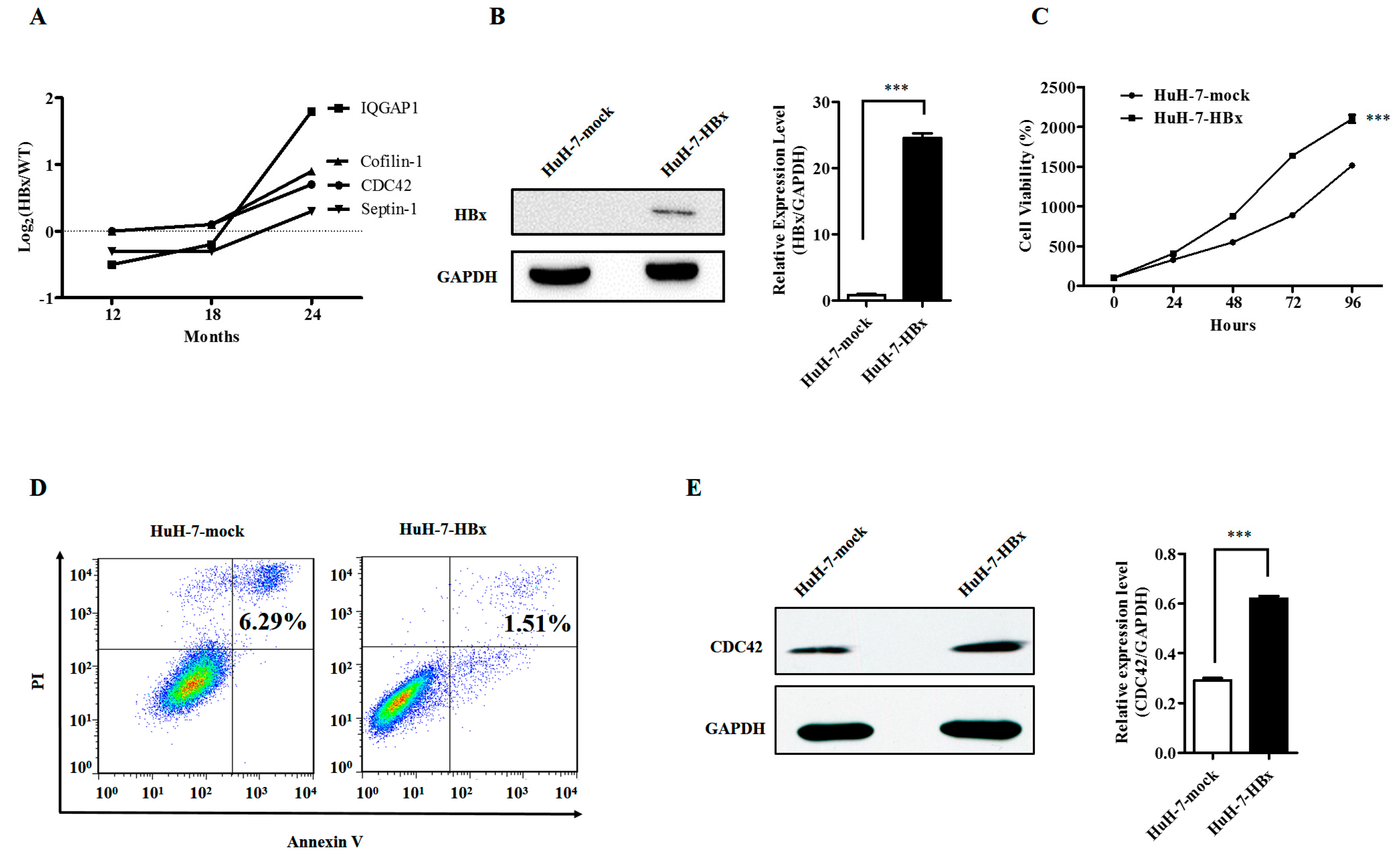
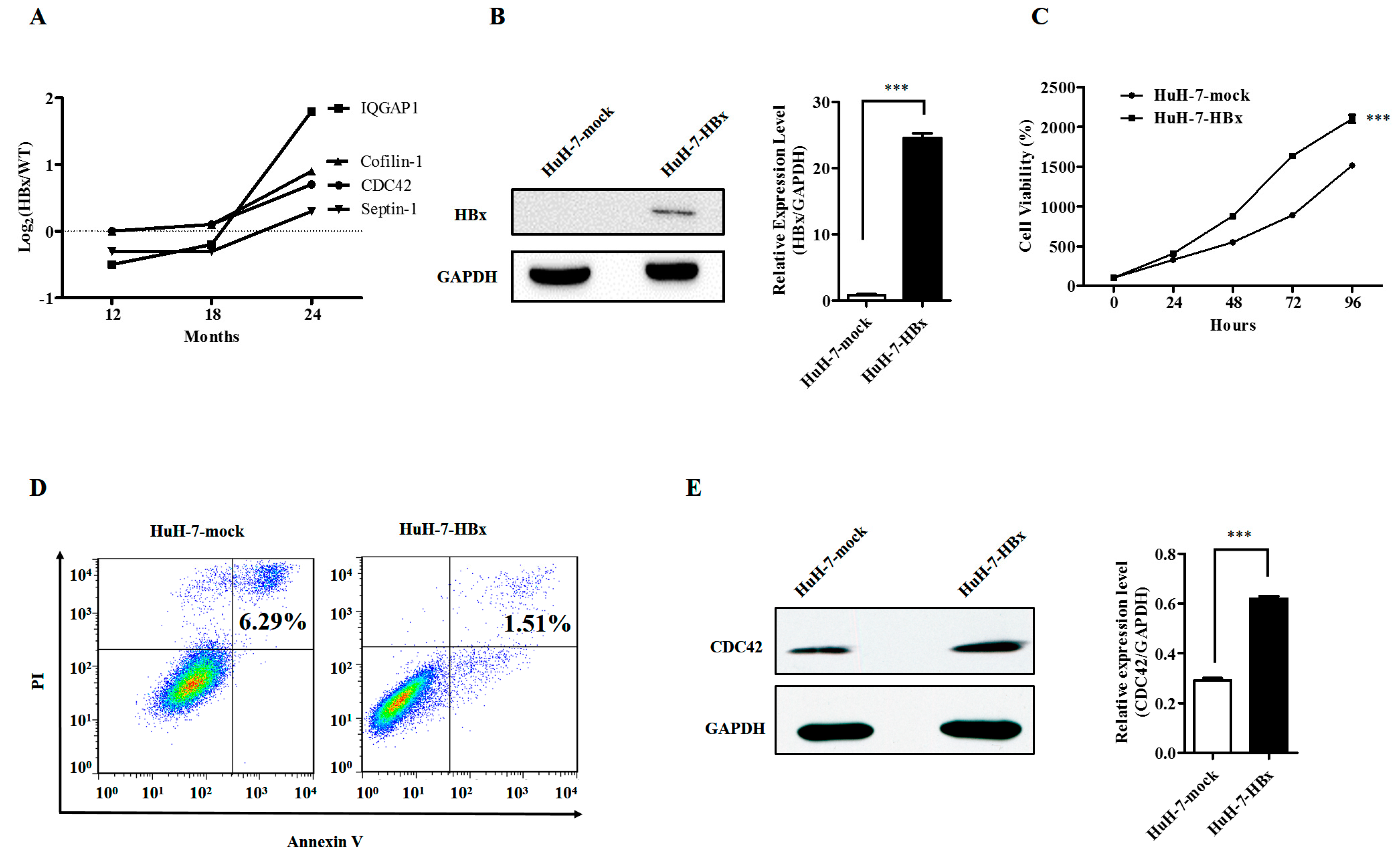
2.1. Hepatitis B Virus X Protein Promoted Proliferation and Inhibited Apoptosis of HuH-7 Cells with Up-Regulated Expression of CDC42
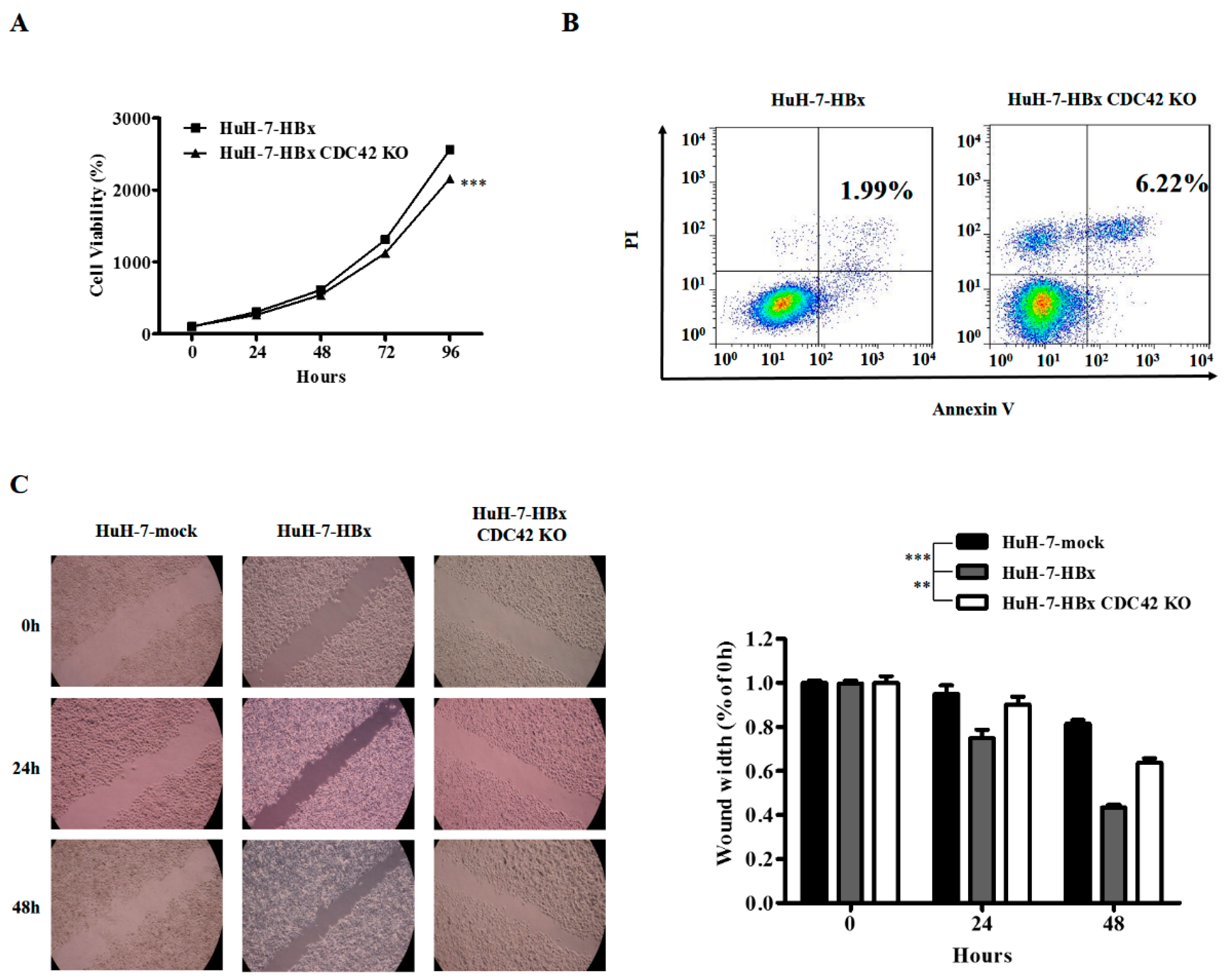
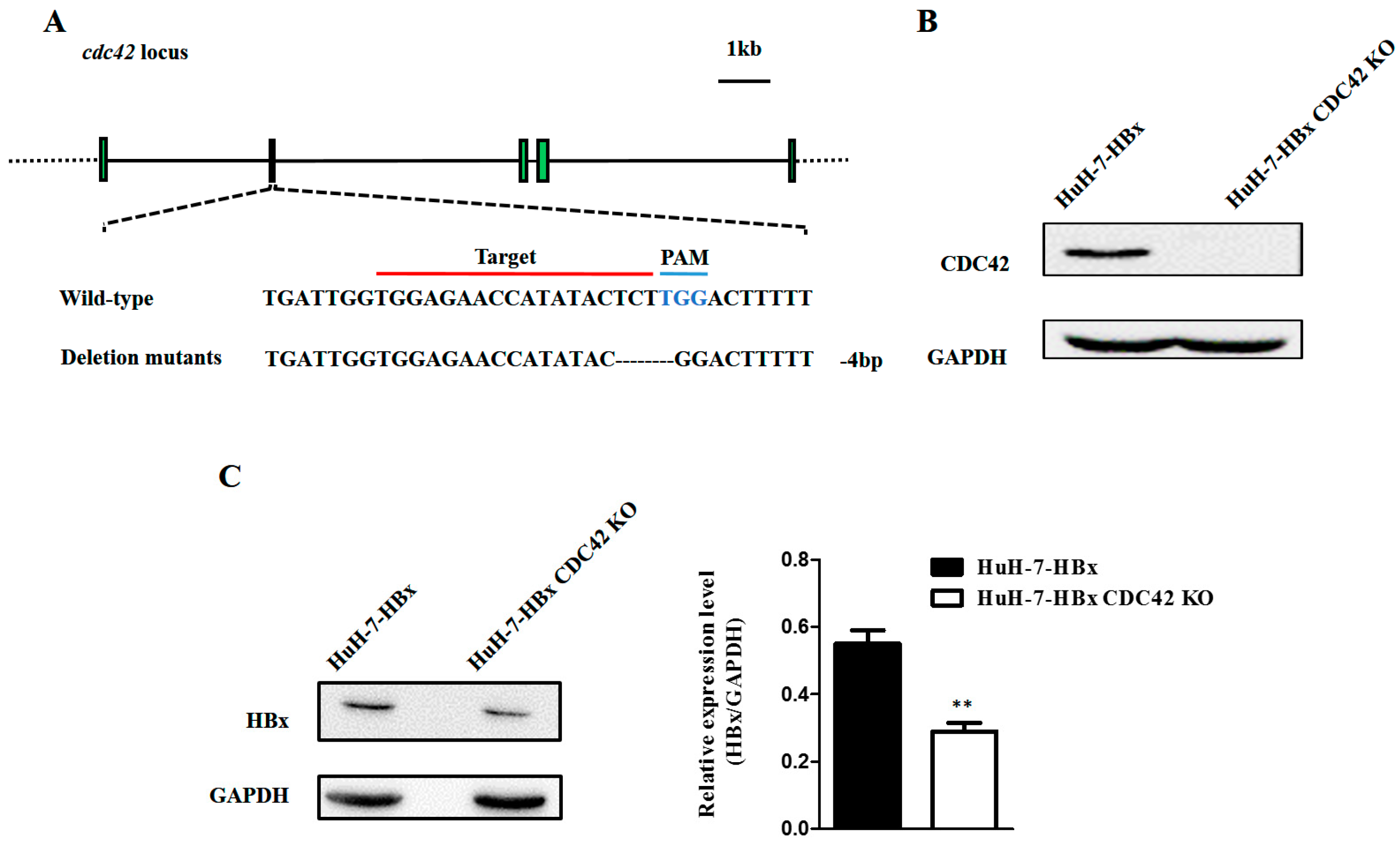
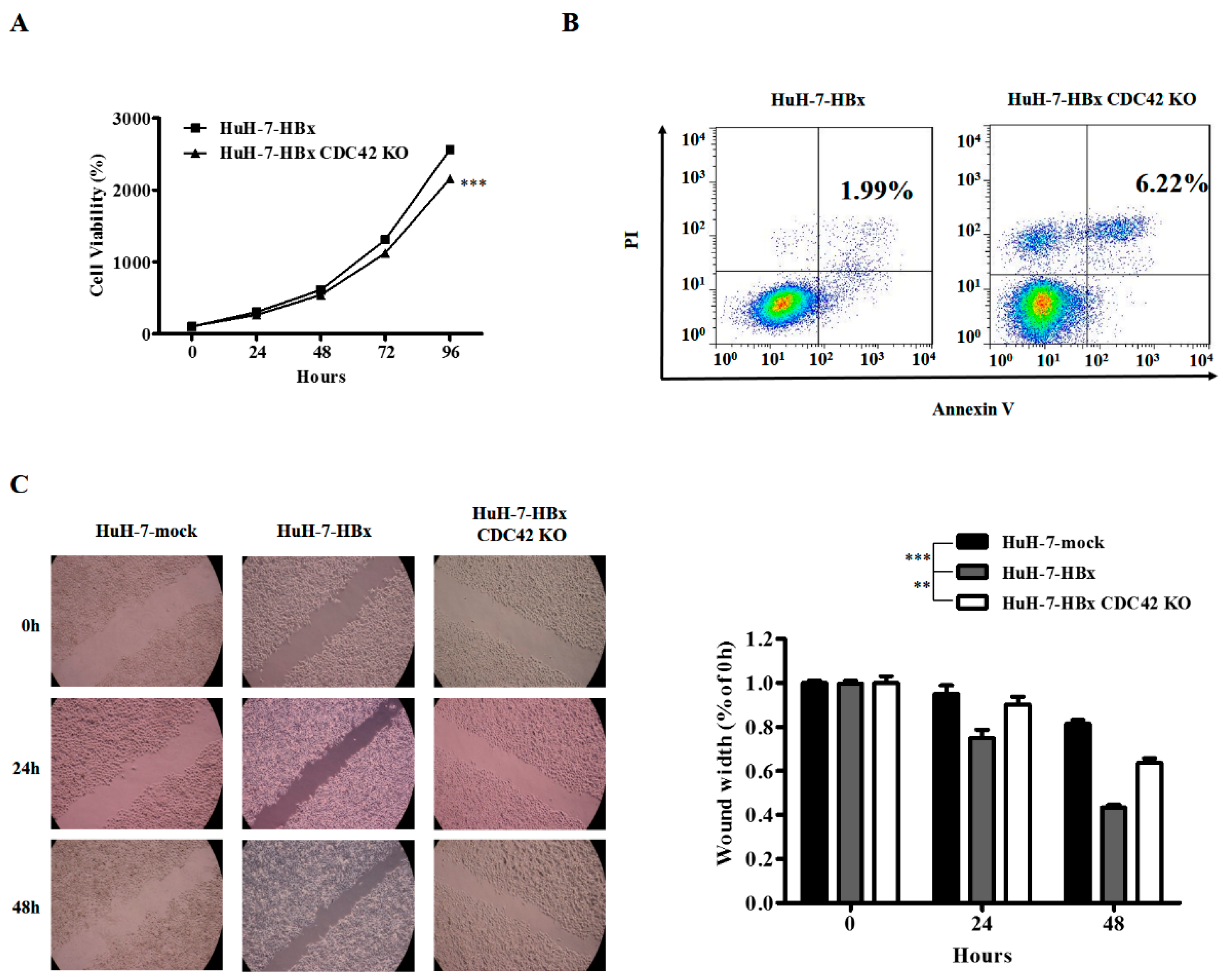
2.2. CDC42 Was Required by HBx to Stimulate Proliferation, Wound Closure, and Inhibit Apoptosis of HuH-7 Cells
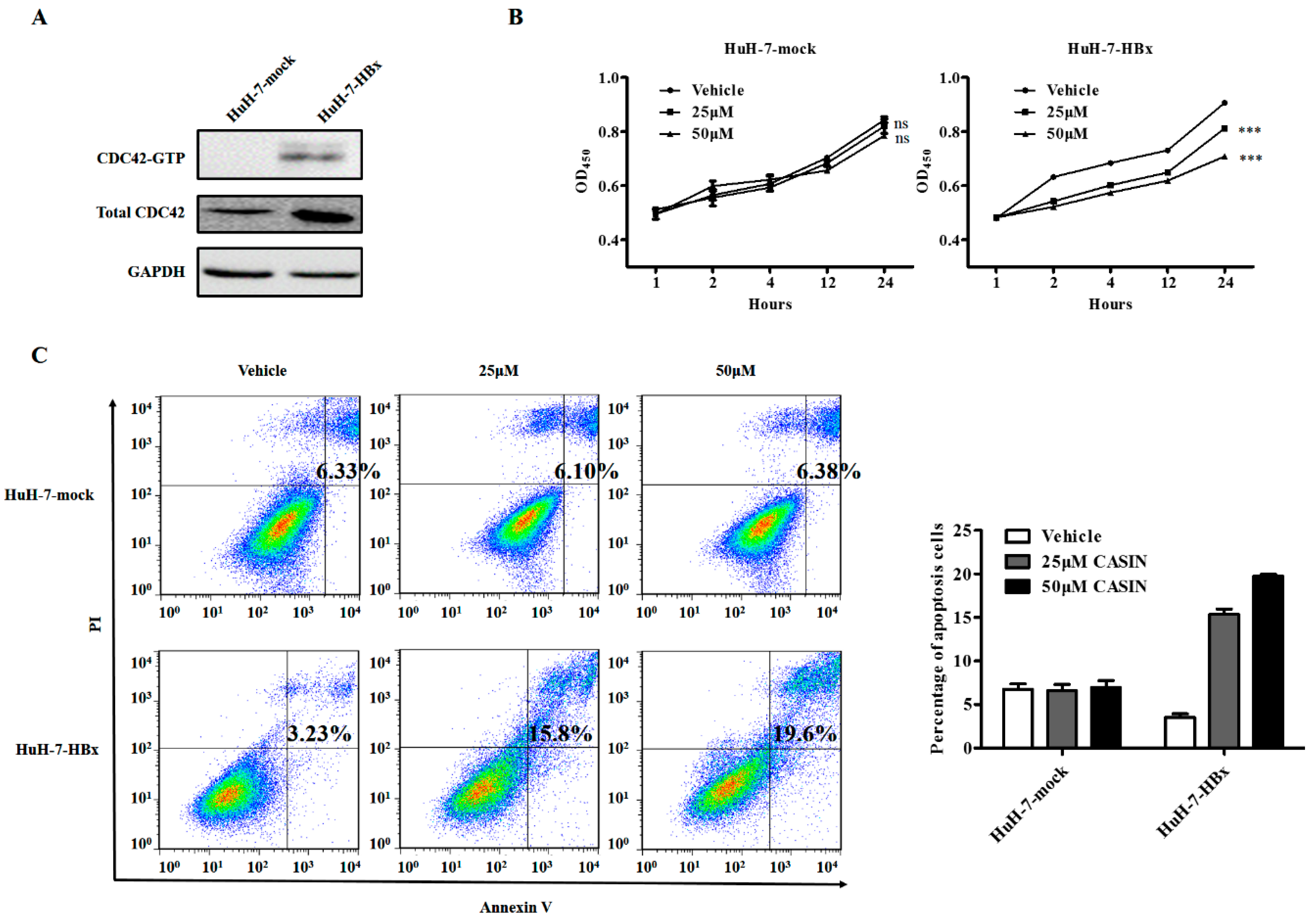
2.3. CDC42 Inhibitor Reduced Proliferation and Promoted Apoptosis Only in HBx-Expressing HuH-7 Cells
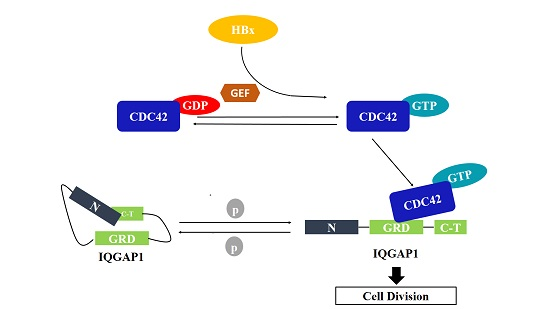
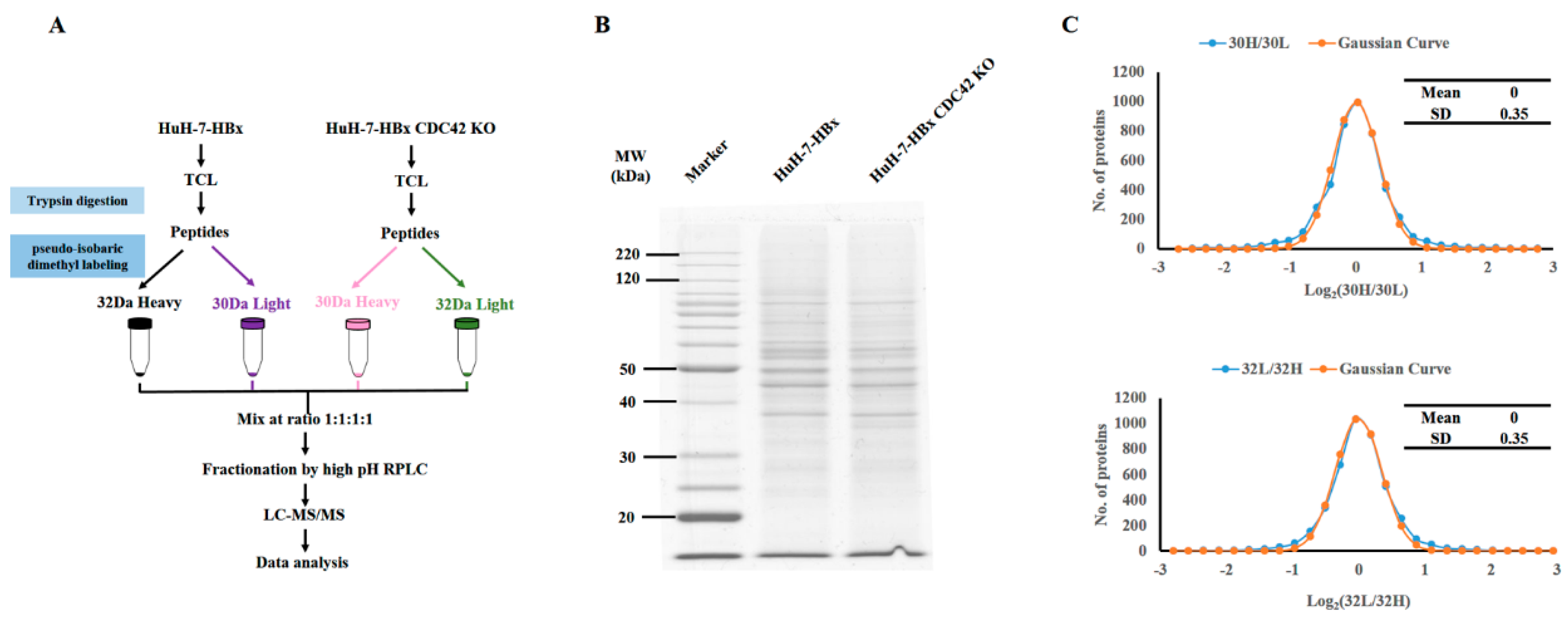
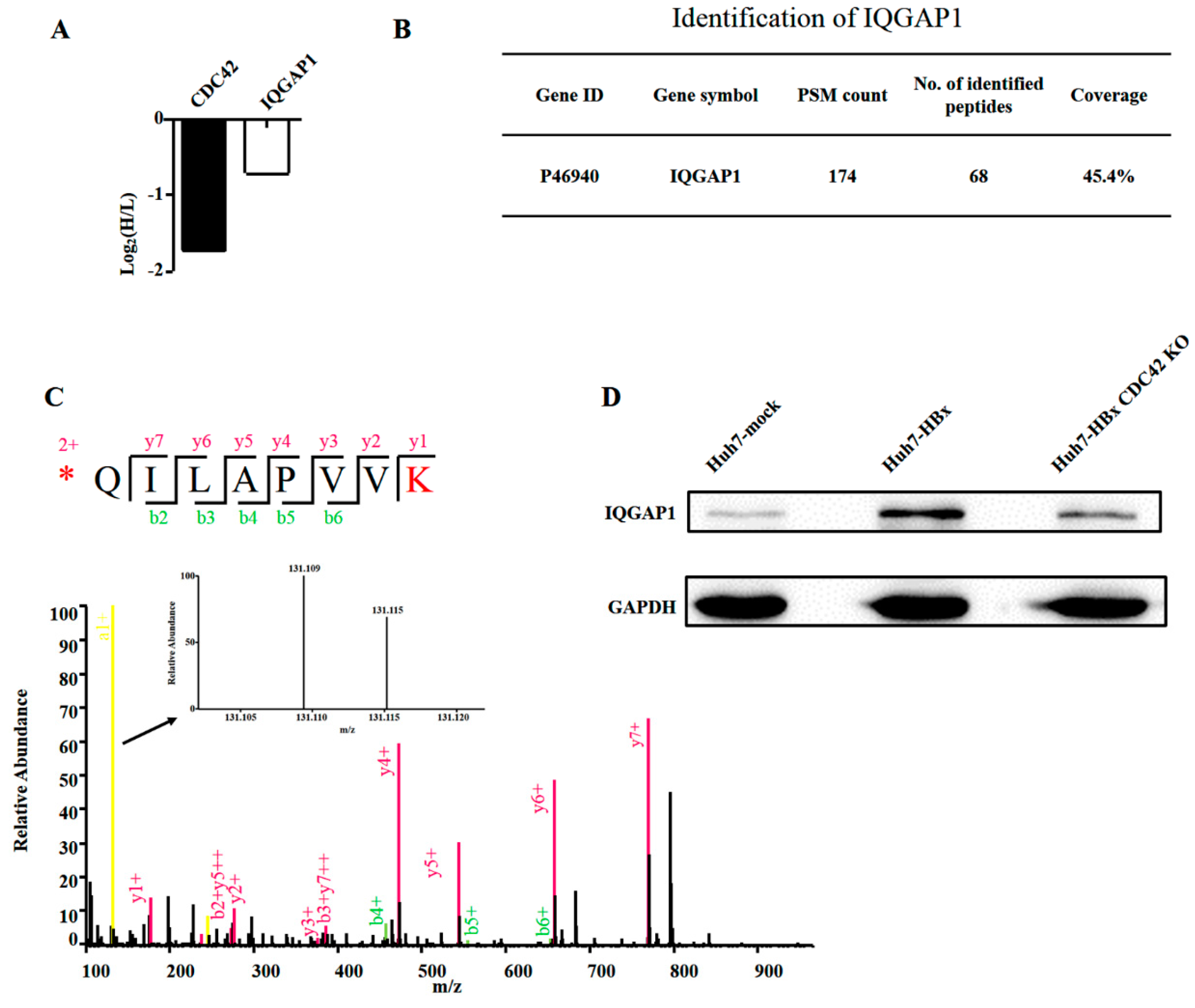
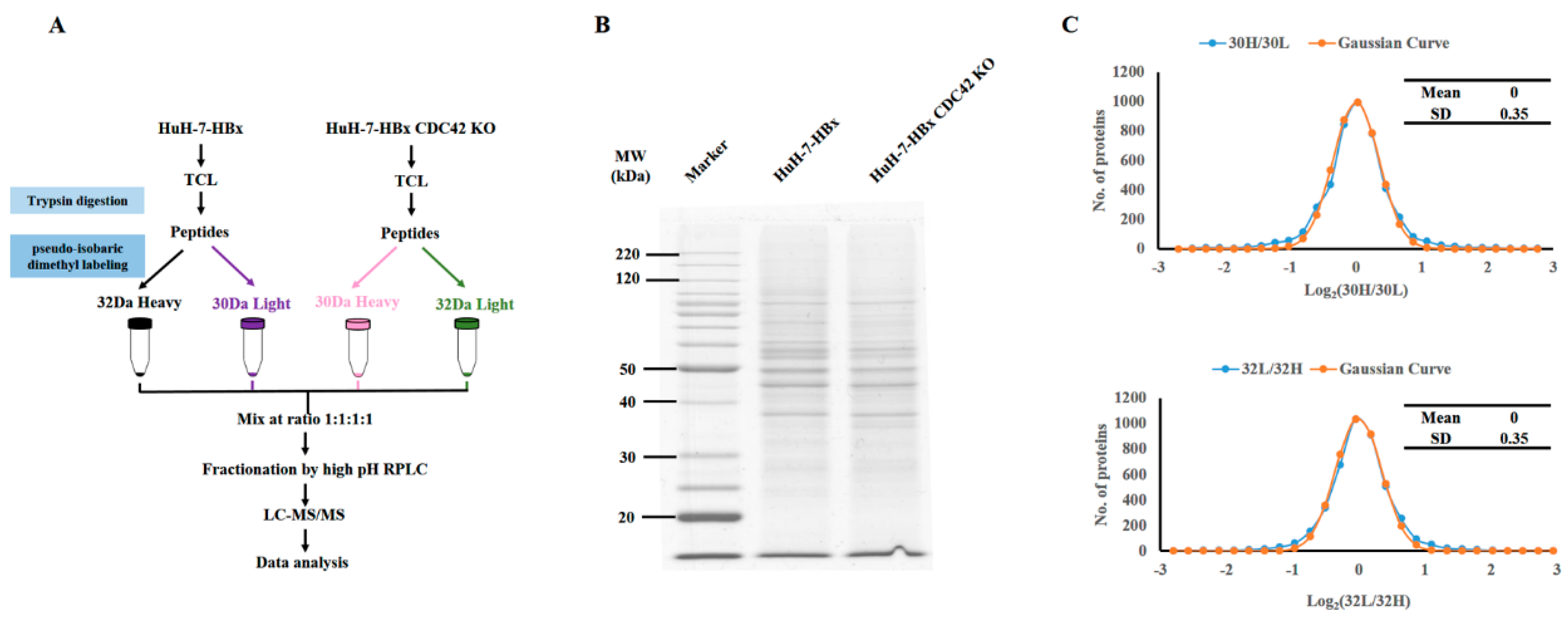
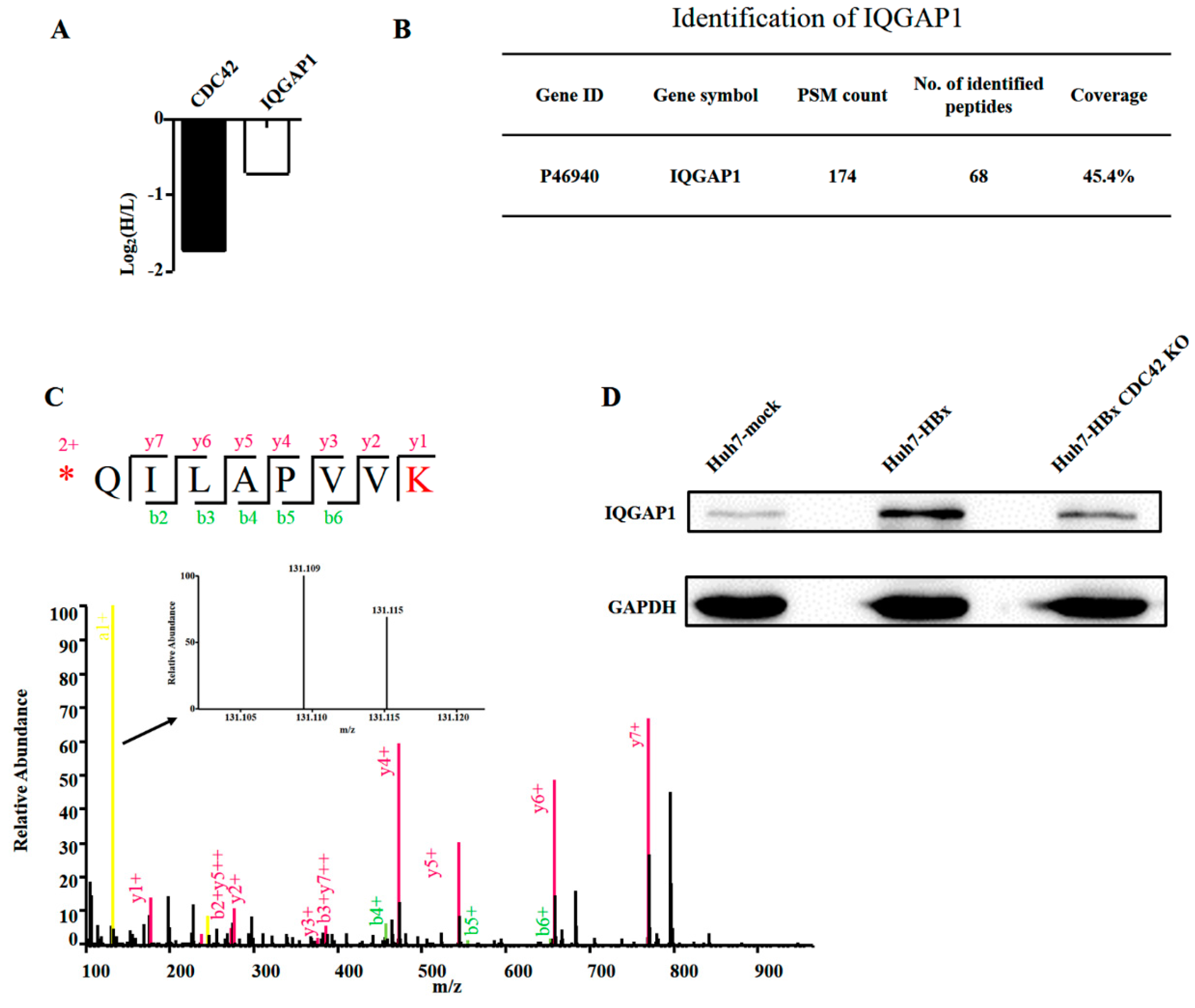
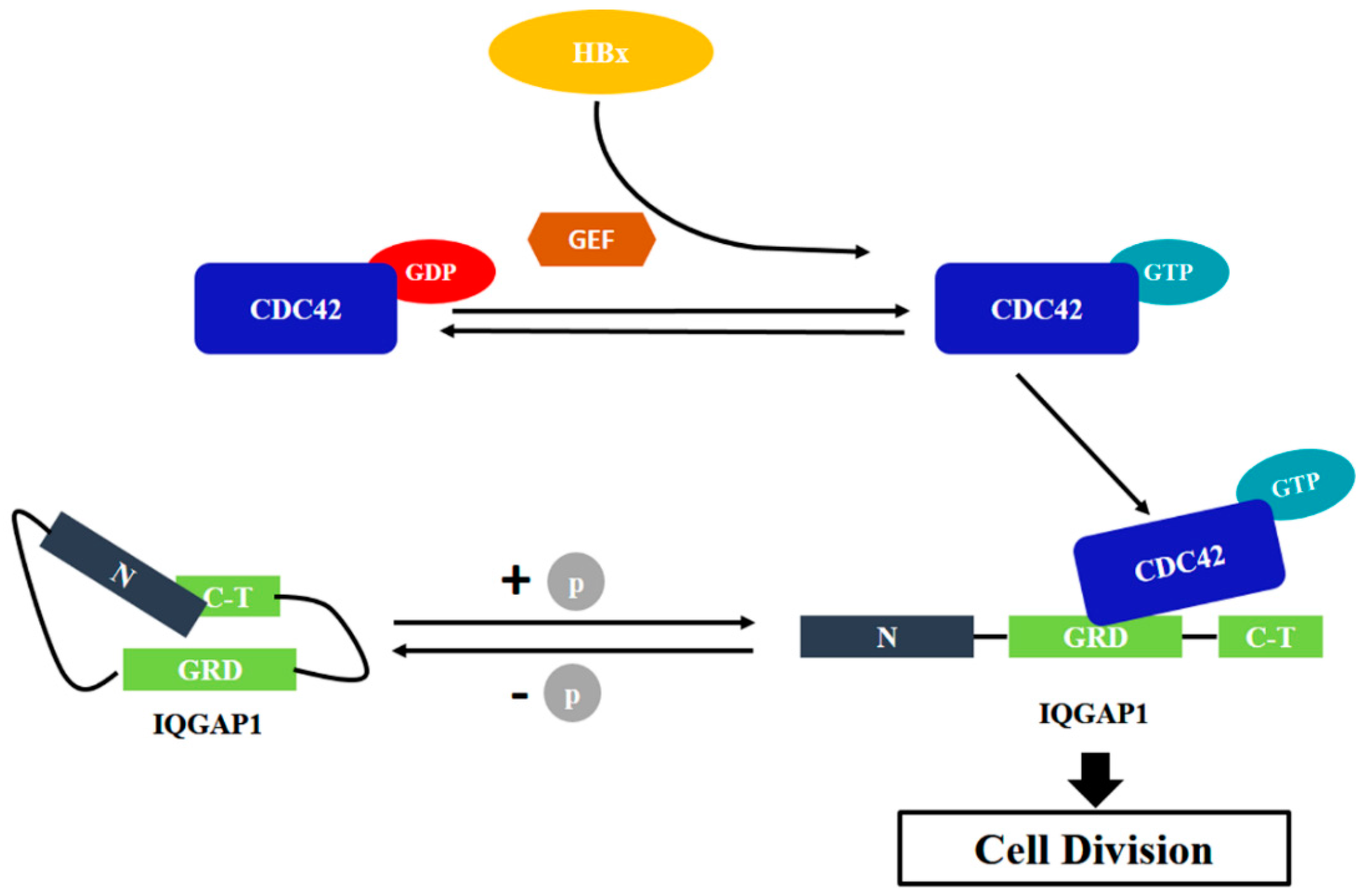
2.4. Quantitative Proteomics Implied That IQGAP1 Participated in the HBx-Mediated Proliferation of HuH-7 Cells
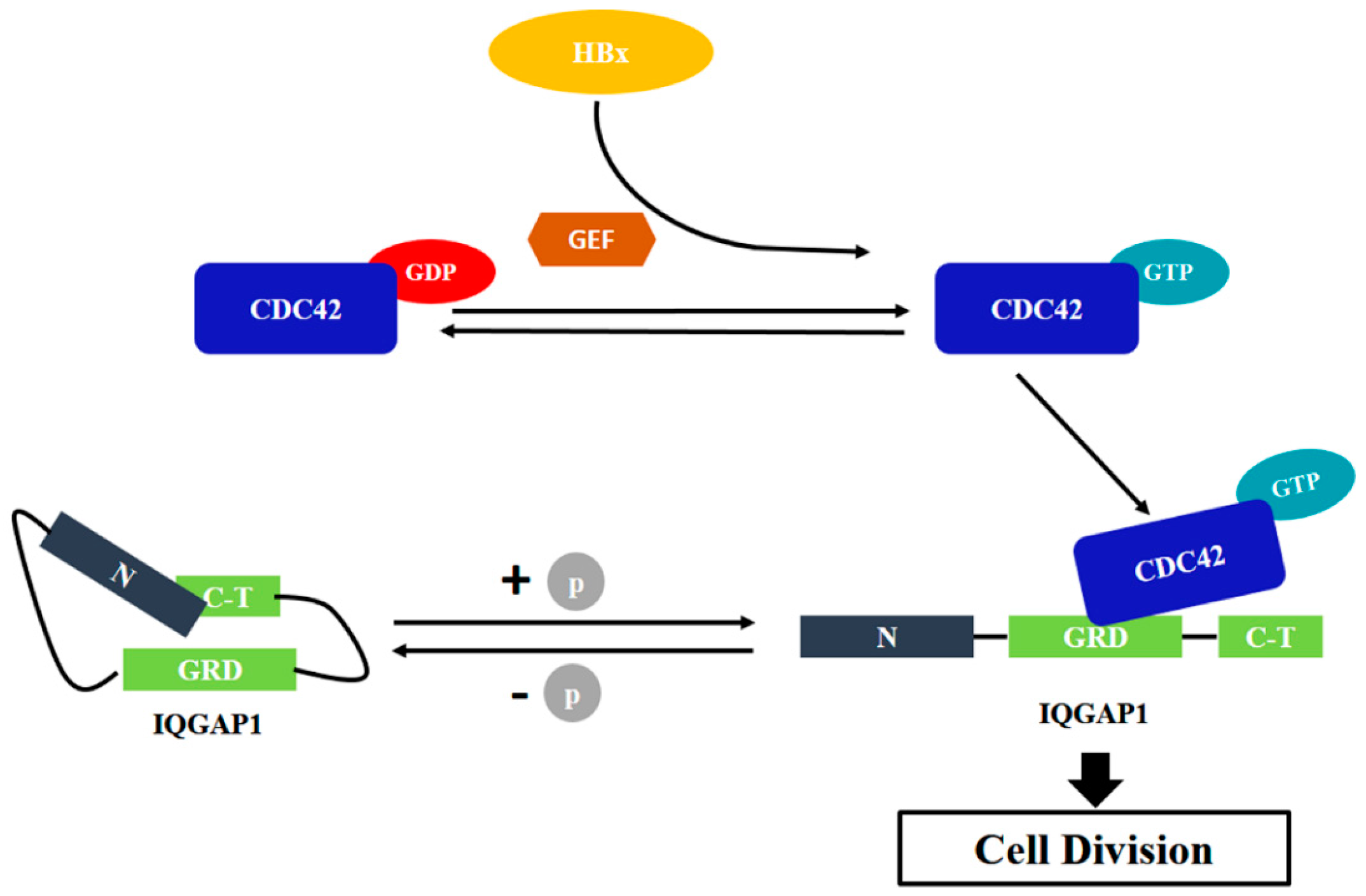
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Cell Lines, and Reagents
4.2. Cell Viability Assay
4.3. Cell Apoptosis Assay
4.4. Wound Healing Assay
4.5. Quantitative Real-Time RT-PCR
4.6. CRISPR/Cas9 System
4.7. DNA Extraction
4.8. Western Blot Analysis
4.9. Quantitative Proteomics Analysis
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nguyen, V.T.; Law, M.G.; Dore, G.J. Hepatitis B-related hepatocellular carcinoma: Epidemiological characteristics and disease burden. J. Viral Hepat. 2009, 16, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Katayama, F.; Kato, H.; Tanaka, H.; Wang, J.; Qiao, Y.L.; Inoue, M. Hepatitis B and C virus infection and hepatocellular carcinoma in China: A review of epidemiology and control measures. J. Epidemiol. Jpn. Epidemiol. Assoc. 2011, 21, 401–416. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus X protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vilchez, S.; Lara-Pezzi, E.; Trapero-Marugan, M.; Moreno-Otero, R.; Sanz-Cameno, P. The molecular and pathophysiological implications of hepatitis B X antigen in chronic hepatitis B virus infection. Rev. Med. Virol. 2011, 21, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.; Zheng, Y. CDC42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.G.; Lucarini, G.; Brancorsini, D.; Zizzi, A.; Pugnaloni, A.; Giacchetti, A.; Ricotti, G.; Biagini, G. Involvement of E-cadherin, β-catenin, CDC42 and CXCR4 in the progression and prognosis of cutaneous melanoma. Br. J. Dermatol. 2007, 157, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 2002, 87, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Gomez Del Pulgar, T.; Valdes-Mora, F.; Bandres, E.; Perez-Palacios, R.; Espina, C.; Cejas, P.; Garcia-Cabezas, M.A.; Nistal, M.; Casado, E.; Gonzalez-Baron, M.; et al. CDC42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int. J. Oncol. 2008, 33, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.S.; Huang, S.M.; Lin, H.H.; Wu, C.C.; Wang, C.J. Different expression of apoptotic proteins between HBV-infected and non-HBV-infected hepatocellular carcinoma. Hepato Gastroenterol. 2007, 54, 2061–2068. [Google Scholar]
- Cooper, A.B.; Wu, J.; Lu, D.; Maluccio, M.A. Is autotaxin (ENPP2) the link between hepatitis C and hepatocellular cancer? J. Gastrointest. Surg. 2007, 11, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhai, L.; Yi, T.; Gao, H.; Fan, F.; Li, Y.; Wang, Y.; Li, N.; Xing, X.; Su, N.; et al. Hepatitis B virus x induces inflammation and cancer in mice liver through dysregulation of cytoskeletal remodeling and lipid metabolism. Oncotarget 2016, 7, 70559–70574. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Dorr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. CDC42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Lebensohn, A.M.; Pelish, H.E.; Kirschner, M.W. Biochemical suppression of small-molecule inhibitors: A strategy to identify inhibitor targets and signaling pathway components. Chem. Biol. 2006, 13, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, F.; Lv, Y.; Li, C.; Xu, X.; Deng, C.; Wang, D.; Sun, Y.; Hu, G.; Lang, Z.; et al. HBsAg and HBx knocked into the p21 locus causes hepatocellular carcinoma in mice. Hepatology 2004, 39, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Hepatitis B virus x protein: A multifunctional viral regulator. J. Gastroenterol. 2001, 36, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Clippinger, A.J.; Bouchard, M.J. Modulation of apoptotic signaling by the hepatitis B virus x protein. Viruses 2012, 4, 2945–2972. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Liu, Y.; Zhang, Q.; Gao, L.; Han, L.; Ma, C.; Zhang, L.; Chen, Y.H.; Sun, W. Hepatitis B virus sensitizes hepatocytes to TRAIL-induced apoptosis through bax. J. Immunol. 2007, 178, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, X.; Hu, D.; Feng, T.; Li, H.; Lu, Y.; Huang, J. Hepatitis B virus X (HBx) play an anti-apoptosis role in hepatic progenitor cells by activating Wnt/β-catenin pathway. Mol. Cell. Biochem. 2013, 383, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Expression of hepatitis B virus x protein does not alter the accumulation of spontaneous mutations in transgenic mice. J. Virol. 2000, 74, 5266–5272. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Hall, A. Rho, Rac and CDC42 gtpases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997, 9, 86–92. [Google Scholar] [CrossRef]
- Ma, L.; Rohatgi, R.; Kirschner, M.W. The Arp2/3 complex mediates actin polymerization induced by the small GTP-binding protein CDC42. Proc. Natl. Acad. Sci. USA 1998, 95, 15362–15367. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Ma, L.; Miki, H.; Lopez, M.; Kirchhausen, T.; Takenawa, T.; Kirschner, M.W. The interaction between N-WASP and the Arp2/3 complex links CDC42-dependent signals to actin assembly. Cell 1999, 97, 221–231. [Google Scholar] [CrossRef]
- Zugasti, O.; Rul, W.; Roux, P.; Peyssonnaux, C.; Eychene, A.; Franke, T.F.; Fort, P.; Hibner, U. Raf-MEK-Erk cascade in anoikis is controlled by Rac1 and CDC42 via Akt. Mol. Cell. Biol. 2001, 21, 6706–6717. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.P.; Hawley, S.P.; Ruston, J.; Du, J.; Brakebusch, C.; Jones, N.; Pawson, T. Podocyte-specific loss of CDC42 leads to congenital nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the Rho GTPases Rac1 and CDC42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Yang, W. Cellular signaling for activation of Rho GTPase CDC42. Cell. Signal. 2008, 20, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, H.; Wu, X.; Zhang, Z.; Du, D.; Zhou, W.; Zhou, S.; Brakebusch, C.; Chen, Z. Hepatocyte-specific deletion of CDC42 results in delayed liver regeneration after partial hepatectomy in mice. Hepatology 2009, 49, 240–249. [Google Scholar] [CrossRef] [PubMed]
- van Hengel, J.; D'Hooge, P.; Hooghe, B.; Wu, X.; Libbrecht, L.; de Vos, R.; Quondamatteo, F.; Klempt, M.; Brakebusch, C.; van Roy, F. Continuous cell injury promotes hepatic tumorigenesis in CDC42-deficient mouse liver. Gastroenterology 2008, 134, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, Y.; Zhang, Y.; Liu, F.; Cheng, X.; Hou, N.; Zhao, X.; Yang, X. Expression profiling reveals dysregulation of cellular cytoskeletal genes in HBx-induced hepatocarcinogenesis. Cancer Biol. Ther. 2007, 6, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wu, X.; Plemenitas, A.; Yu, H.; Sawai, E.T.; Abo, A.; Peterlin, B.M. CDC42 and Rac1 are implicated in the activation of the NEF-associated kinase and replication of HIV-1. Curr. Biol. 1996, 6, 1677–1684. [Google Scholar] [CrossRef]
- Weissbach, L.; Settleman, J.; Kalady, M.F.; Snijders, A.J.; Murthy, A.E.; Yan, Y.X.; Bernards, A. Identification of a human RasGAP-related protein containing calmodulin-binding motifs. J. Biol. Chem. 1994, 269, 20517–20521. [Google Scholar] [PubMed]
- Noritake, J.; Watanabe, T.; Sato, K.; Wang, S.; Kaibuchi, K. IQGAP1: A key regulator of adhesion and migration. J. Cell Sci. 2005, 118, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Jadeski, L.; Mataraza, J.M.; Jeong, H.W.; Li, Z.; Sacks, D.B. IQGAP1 stimulates proliferation and enhances tumorigenesis of human breast epithelial cells. J. Biol. Chem. 2008, 283, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.L.; O'Sullivan, M.G.; Parkinson, J.F.; Shaw, J.M.; Payne, C.A.; Brewer, J.M.; Young, L.; Reader, D.J.; Wheeler, H.T.; Cook, R.J.; et al. IQGAP1 and IGFBP2: Valuable biomarkers for determining prognosis in glioma patients. J. Neuropathol. Exp. Neurol. 2007, 66, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Balenci, L.; Clarke, I.D.; Dirks, P.B.; Assard, N.; Ducray, F.; Jouvet, A.; Belin, M.F.; Honnorat, J.; Baudier, J. IQGAP1 protein specifies amplifying cancer cells in glioblastoma multiforme. Cancer Res. 2006, 66, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Swart-Mataraza, J.M.; Li, Z.; Sacks, D.B. IQGAP1 is a component of CDC42 signaling to the cytoskeleton. J. Biol. Chem. 2002, 277, 24753–24763. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Sonn, R.; Tekletsadik, Y.K.; Samorodnitsky, D.; Osman, M.A. IQGAP1 regulates cell proliferation through a novel CDC42-mtor pathway. J. Cell Sci. 2009, 122, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Valletta, S.; Dolatshad, H.; Bartenstein, M.; Yip, B.H.; Bello, E.; Gordon, S.; Yu, Y.; Shaw, J.; Roy, S.; Scifo, L.; et al. Asxl1 mutation correction by CRISPR/Cas9 restores gene function in leukemia cells and increases survival in mouse xenografts. Oncotarget 2015, 6, 44061–44071. [Google Scholar] [PubMed]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Qi, Y.; Luo, J.; Yang, J.; Xie, Q.; Deng, C.; Su, N.; Wei, W.; Shi, D.; Xu, F.; et al. Hepatitis B Virus X Protein Stimulates Proliferation, Wound Closure and Inhibits Apoptosis of HuH-7 Cells via CDC42. Int. J. Mol. Sci. 2017, 18, 586. https://doi.org/10.3390/ijms18030586
Xu Y, Qi Y, Luo J, Yang J, Xie Q, Deng C, Su N, Wei W, Shi D, Xu F, et al. Hepatitis B Virus X Protein Stimulates Proliferation, Wound Closure and Inhibits Apoptosis of HuH-7 Cells via CDC42. International Journal of Molecular Sciences. 2017; 18(3):586. https://doi.org/10.3390/ijms18030586
Chicago/Turabian StyleXu, Yongru, Yingzi Qi, Jing Luo, Jing Yang, Qi Xie, Chen Deng, Na Su, Wei Wei, Deshun Shi, Feng Xu, and et al. 2017. "Hepatitis B Virus X Protein Stimulates Proliferation, Wound Closure and Inhibits Apoptosis of HuH-7 Cells via CDC42" International Journal of Molecular Sciences 18, no. 3: 586. https://doi.org/10.3390/ijms18030586