
The Essential Role of Pin1 via NF-κB Signaling in Vascular Inflammation and Atherosclerosis in ApoE−/− Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
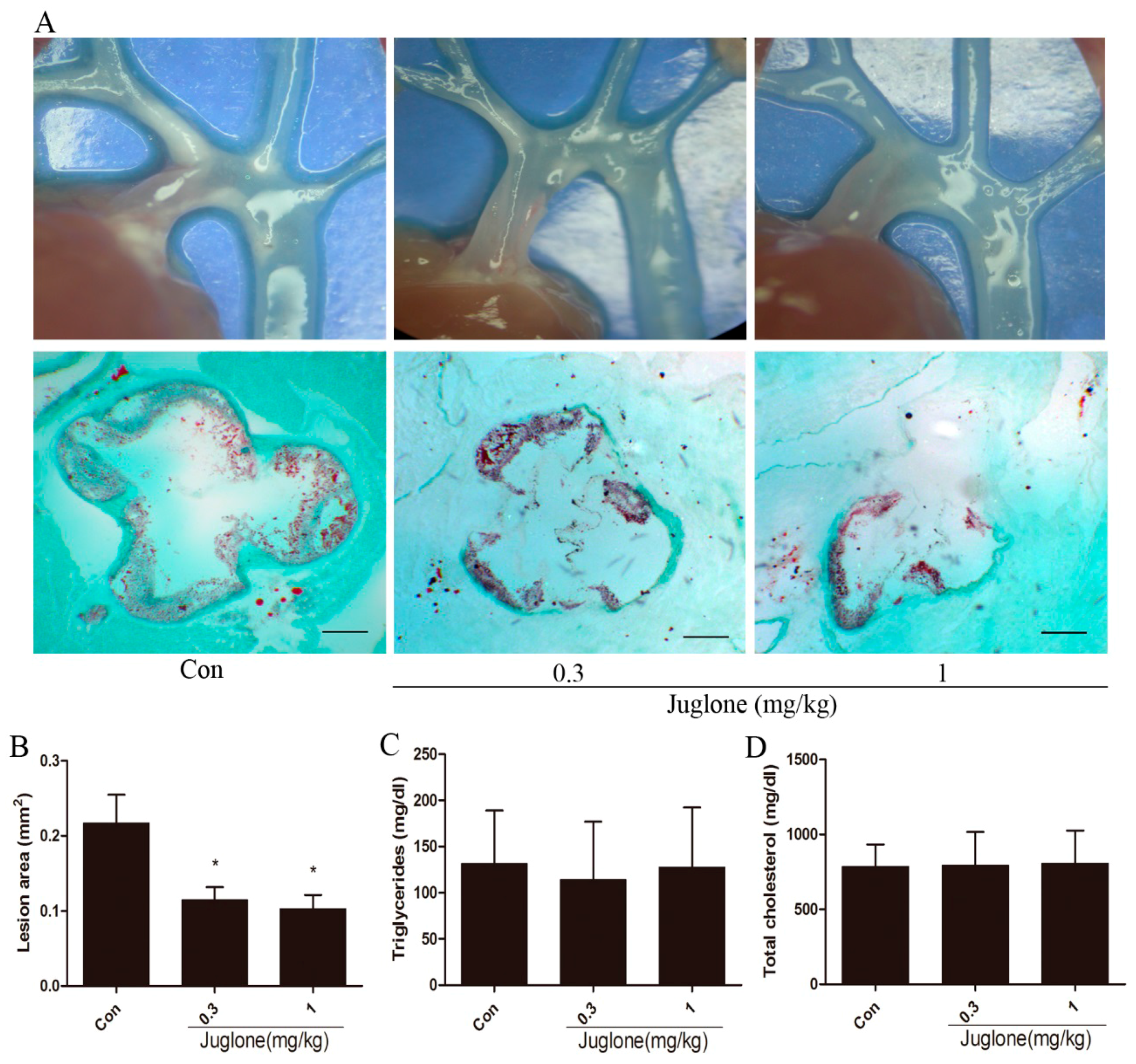
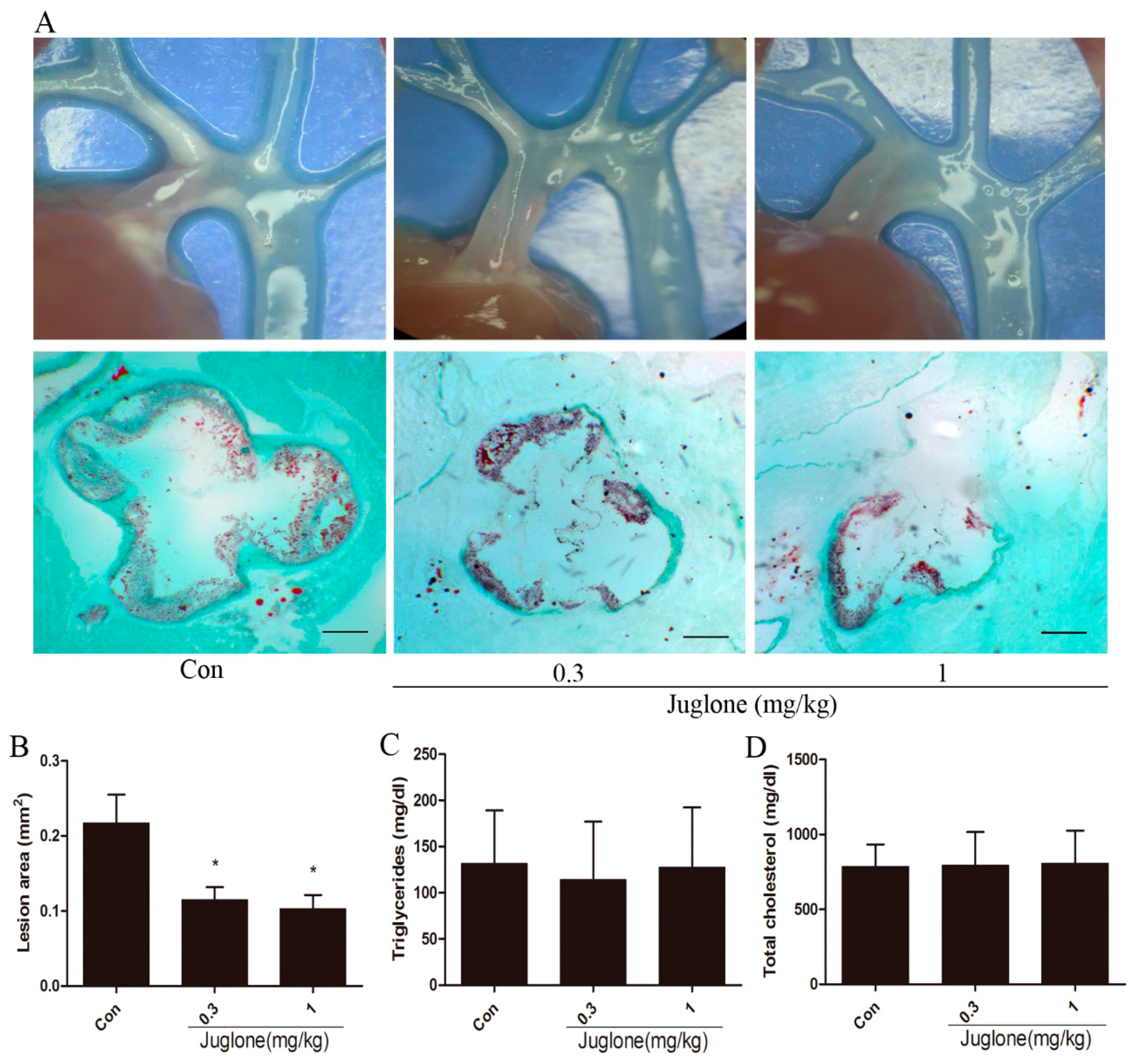
2.1. Inhibition of Pin1 Attenuates Atherosclerosis in ApoE−/− Mice
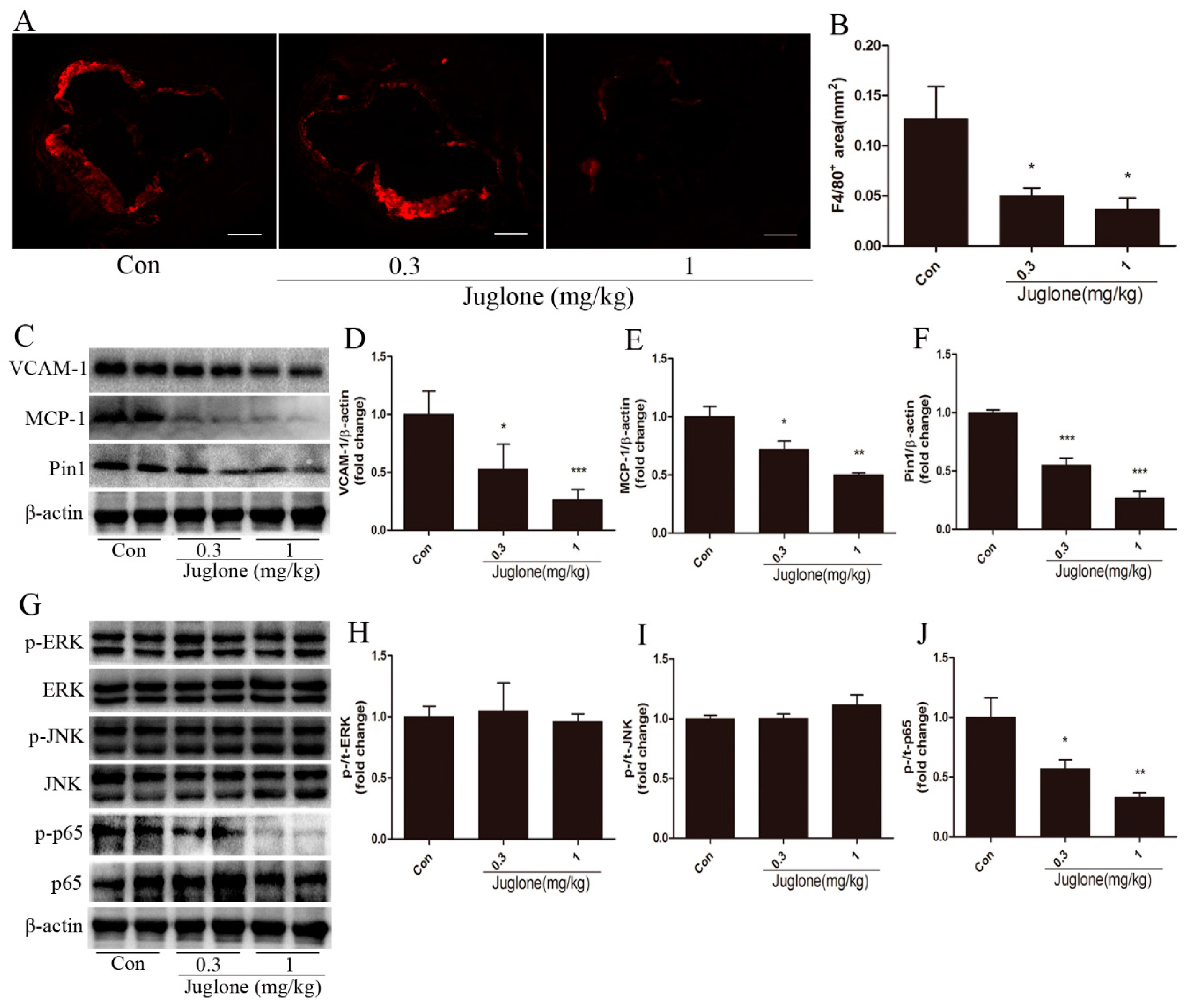
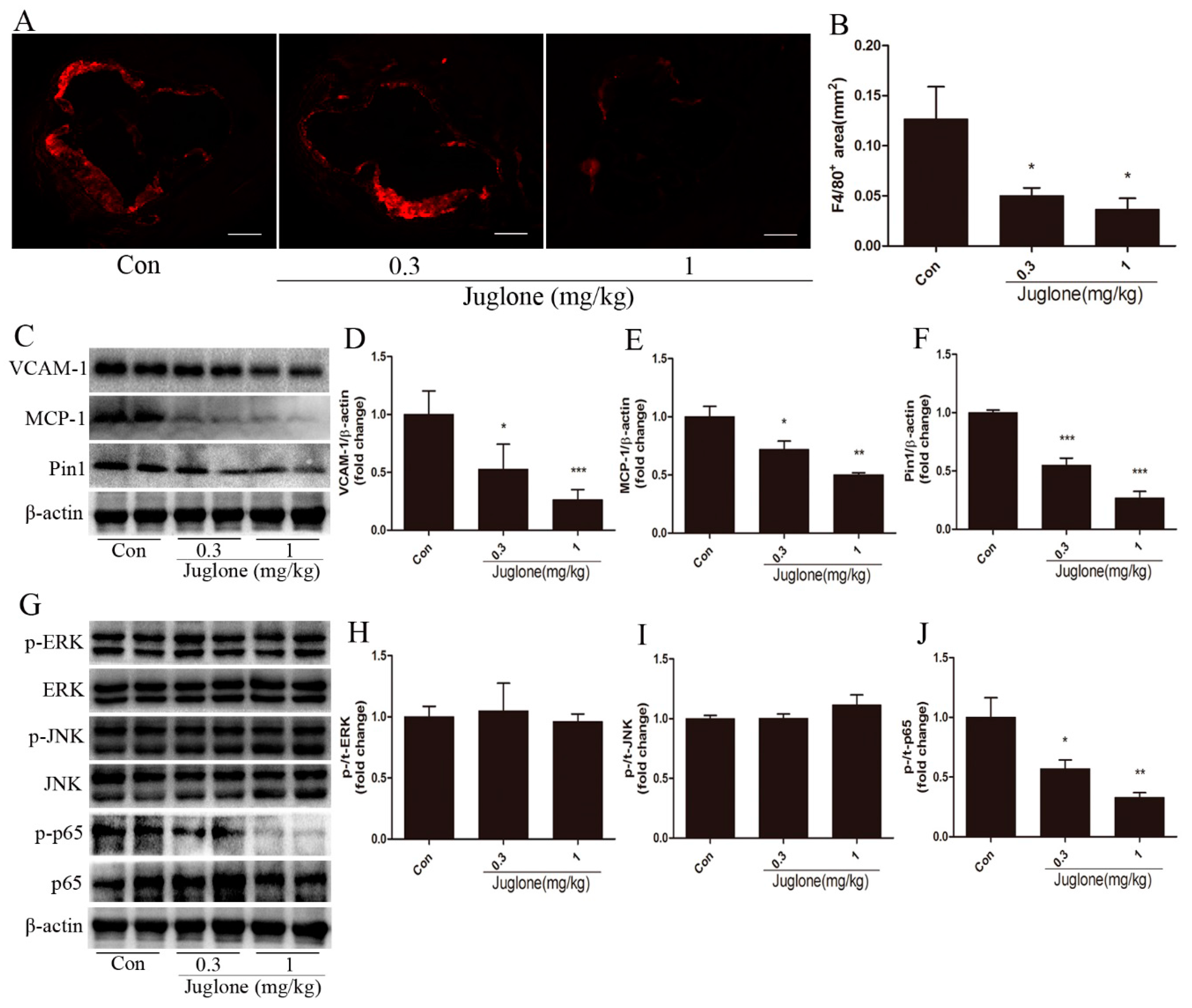
2.2. Inhibition of Pin1 Suppresses Vascular Inflammation in ApoE−/− Mice
2.3. NF-κB Activation is Regulated by Pin1 in ApoE−/− Mice
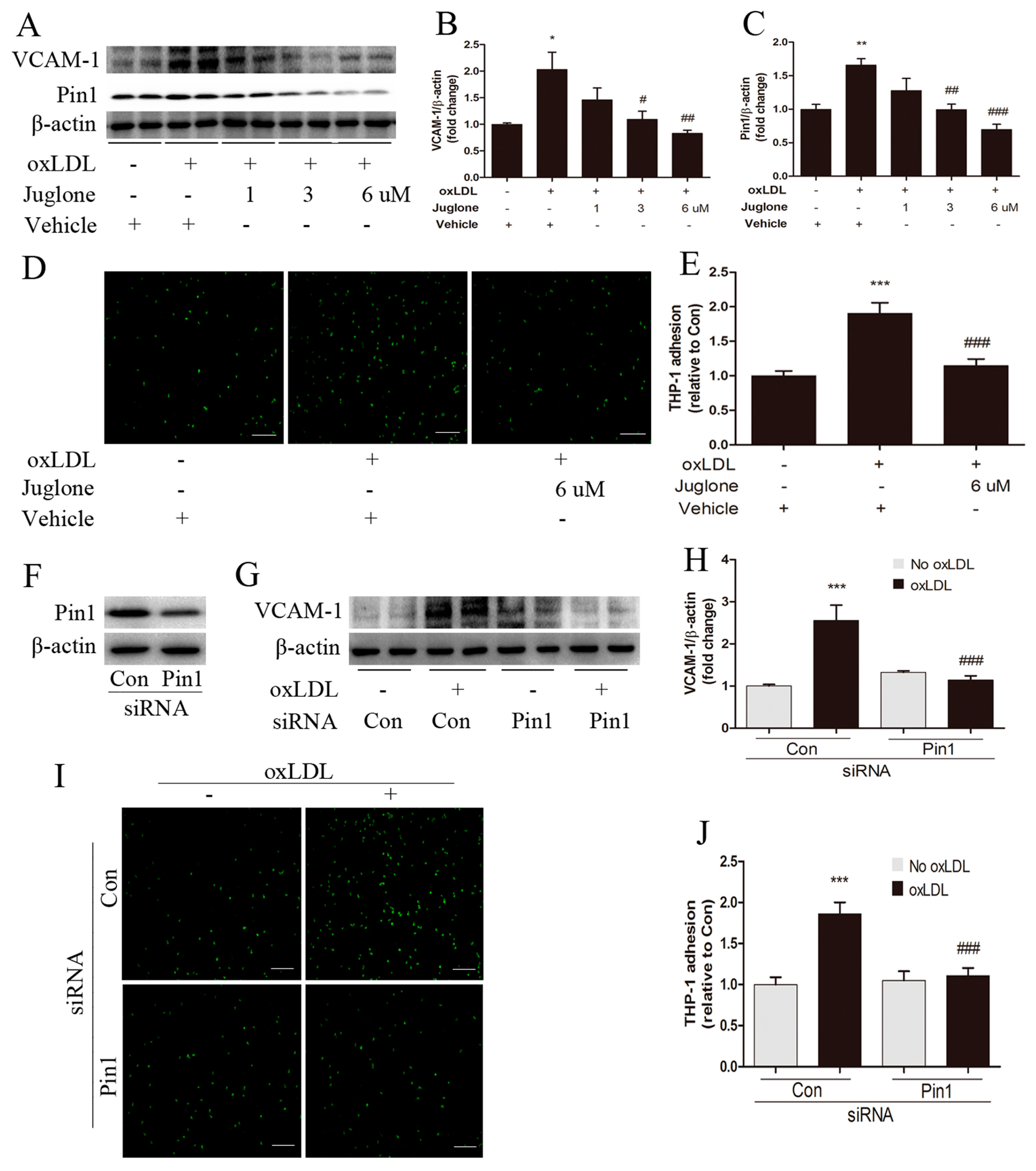
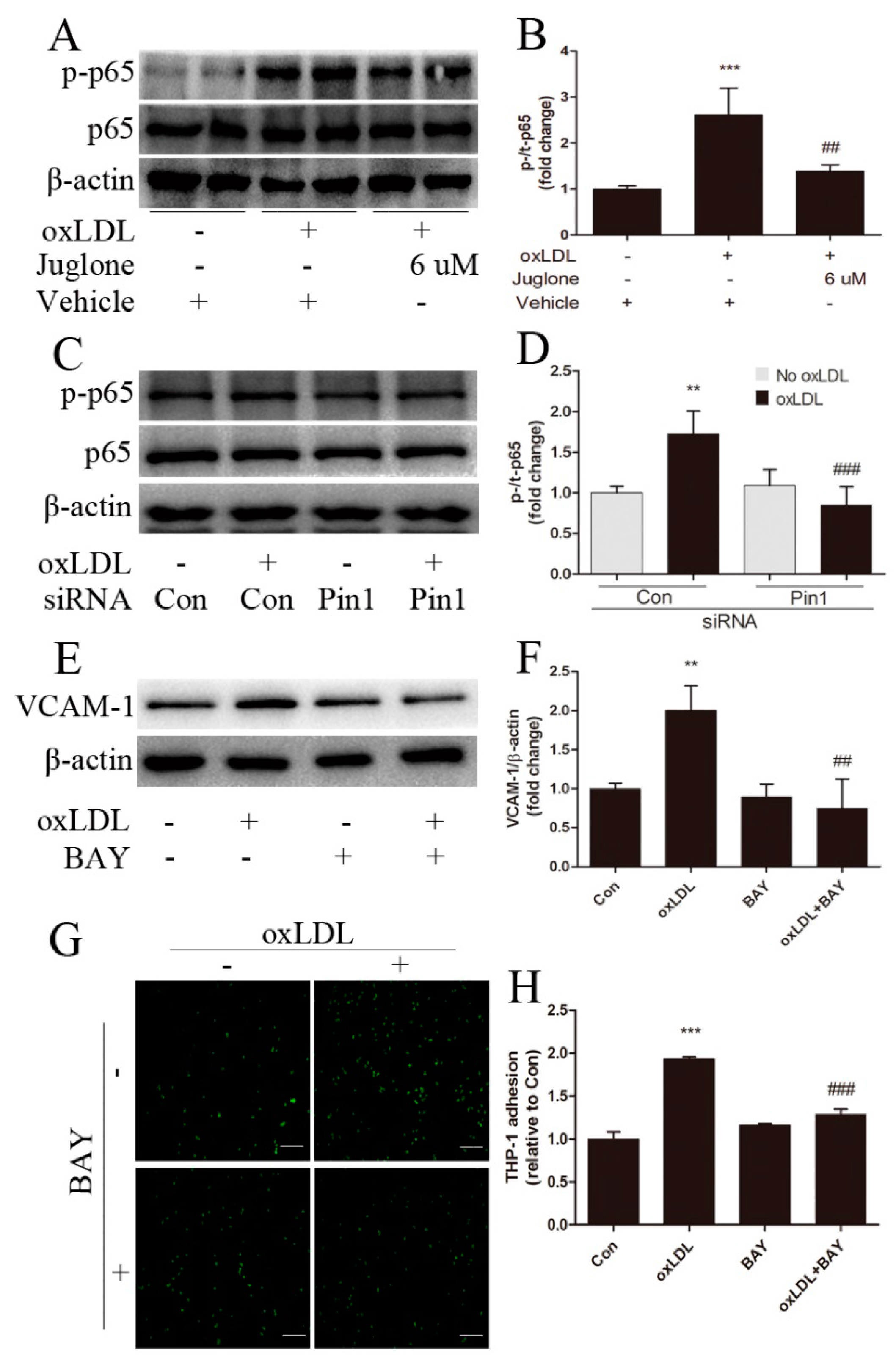
2.4. Pin1 Regulates oxLDL-Induced Inflammatory Response in EA.hy926 Cells
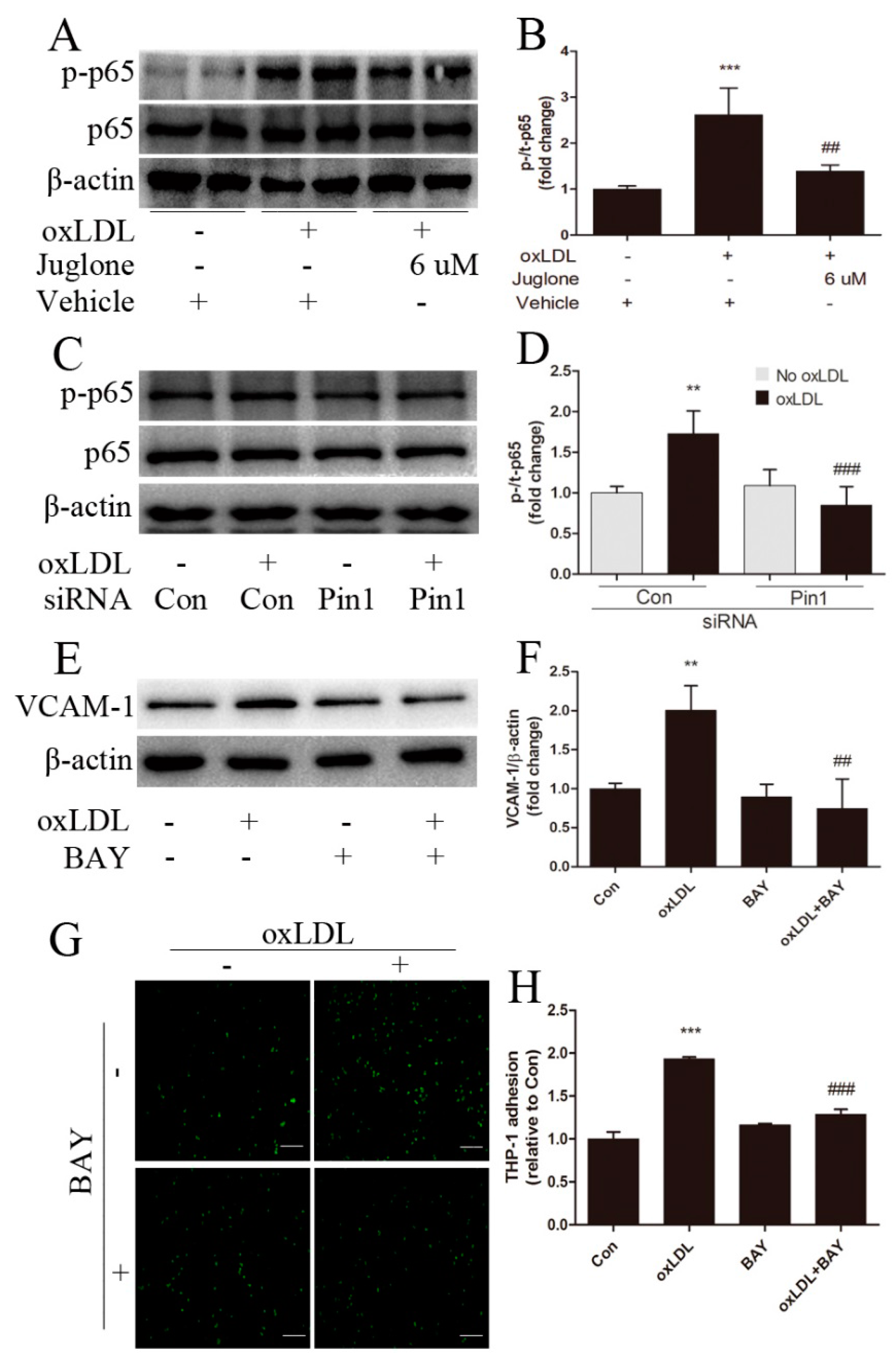
2.5. NF-κB Signaling Involves in Pin1-Dependent Regulation of Inflammatory Response in EA.hy926 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Mice
4.3. Quantification of Atherosclerotic Lesions
4.4. Cell Culture and Treatment
4.5. Monocyte-Endothelial Cell Adhesion Assay
4.6. Western Blot Analysis
4.7. Immunohistochemistry
4.8. Serum Lipid Analysis
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contribution
Conflicts of Interest
Abbreviations
| Pin1 | prolyl-isomerase-1 |
| ApoE−/− | apolipoprotein E deficient |
| VCAM-1 | vascular cell adhesion molecule-1 |
| MCP-1 | monocyte chemoattractant protein-1 |
| siRNA | small interfering RNA |
| OxLDL | oxidative low density lipoproteins |
| NF-κB | nuclear factor kappa-B |
| ERK | extracellular regulated protein kinases |
| JNK | c-Jun N-terminal kinase |
| THP1 | human acute monocytic leukemia cell line |
References
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.P.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in ApoE−/− mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Rostam, M.A.; Piva, T.J.; Rezaei, H.B.; Kamato, D.; Little, P.J.; Zheng, W.; Osman, N. Peptidyl-prolyl isomerases: Functionality and potential therapeutic targets in cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2015, 42, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.G.; Pokharel, Y.R.; Lim, S.C.; Hwang, Y.P.; Han, E.H.; Yoon, J.H.; Ahn, S.G.; Lee, K.Y.; Kang, K.W. Novel role of Pin1 induction in type II collagen-mediated rheumatoid arthritis. J. Immunol. 2009, 183, 6689–6697. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.J.; Esnault, S.; Rosenthal, L.A.; Szakaly, R.J.; Sorkness, R.L.; Westmark, P.R.; Sandor, M.; Malter, J.S. Pin1 regulates TGF-β1 production by activated human and murine eosinophils and contributes to allergic lung fibrosis. J. Clin. Investig. 2008, 118, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Hien, T.T.; Lim, S.C.; Jun, D.W.; Choi, H.S.; Yoon, J.H.; Cho, I.J.; Kang, K.W. Pin1 induction in the fibrotic liver and its roles in TGF-β1 expression and Smad2/3 phosphorylation. J. Hepatol. 2014, 60, 1235–1241. [Google Scholar] [CrossRef]
- Ruan, L.; Torres, C.M.; Qian, J.; Chen, F.; Mintz, J.D.; Stepp, D.W.; Fulton, D.; Venema, R.C. Pin1 prolyl isomerase regulates endothelial nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Luscher, T.F.; Cosentino, F. Pin1 inhibitor Juglone prevents diabetic vascular dysfunction. Int. J. Cardiol. 2016, 203, 702–707. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yu, Y.; Jiang, H.; Zhang, L.; Zhang, P.P.; Yu, P.; Jia, J.G.; Chen, R.Z.; Zou, Y.Z.; Ge, J.B. Simvastatin suppresses vascular inflammation and atherosclerosis in ApoE−/− mice by downregulating the HMGB1-RAGE axis. Acta Pharmacol. Sin. 2013, 34, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Toko, H.; Konstandin, M.H.; Doroudgar, S.; Ormachea, L.; Joyo, E.; Joyo, A.Y.; Din, S.; Gude, N.A.; Collins, B.; Volkers, M.; et al. Regulation of cardiac hypertrophic signaling by prolyl isomerase Pin1. Circ. Res. 2013, 112, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.; Kao, D.; Blom, H.J.; Tavares, D.A.I.; Castro, R.; Loscalzo, J.; Handy, D.E. S-adenosylhomocysteine induces inflammation through NF-κB: A possible role for EZH2 in endothelial cell activation. Biochim. Biophys. Acta 2016, 1862, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Indra, M.R.; Karyono, S.; Ratnawati, R.; Malik, S.G. Quercetin suppresses inflammation by reducing ERK1/2 phosphorylation and NF-κB activation in leptin-induced human umbilical vein endothelial cells (HUVECs). BMC Res. Notes 2013, 6, 275. [Google Scholar] [CrossRef] [PubMed]
- Liu, M. Pin1 inhibition by either inhibitor treatment or gene knockdown did not change the activation of ERK or JNK signal pathways induced by oxLDL in EA.hy926 cells. Fudan University: Shanghai, China, 2017. [Google Scholar]
- Vandersmissen, I.; Craps, S.; Depypere, M.; Coppiello, G.; van Gastel, N.; Maes, F.; Carmeliet, G.; Schrooten, J.; Jones, E.A.; Umans, L.; et al. Endothelial Msx1 transduces hemodynamic changes into an arteriogenic remodeling response. J. Cell Biol. 2015, 210, 1239–1256. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Yu, P.; Jiang, H.; Yang, X.; Zhao, J.; Zou, Y.; Ge, J. The Essential Role of Pin1 via NF-κB Signaling in Vascular Inflammation and Atherosclerosis in ApoE−/− Mice. Int. J. Mol. Sci. 2017, 18, 644. https://doi.org/10.3390/ijms18030644
Liu M, Yu P, Jiang H, Yang X, Zhao J, Zou Y, Ge J. The Essential Role of Pin1 via NF-κB Signaling in Vascular Inflammation and Atherosclerosis in ApoE−/− Mice. International Journal of Molecular Sciences. 2017; 18(3):644. https://doi.org/10.3390/ijms18030644
Chicago/Turabian StyleLiu, Ming, Peng Yu, Hong Jiang, Xue Yang, Ji Zhao, Yunzeng Zou, and Junbo Ge. 2017. "The Essential Role of Pin1 via NF-κB Signaling in Vascular Inflammation and Atherosclerosis in ApoE−/− Mice" International Journal of Molecular Sciences 18, no. 3: 644. https://doi.org/10.3390/ijms18030644