
Synthesis and Biological Evaluation of 7-Deoxy-Epothilone Analogues

 ,
,
Abstract
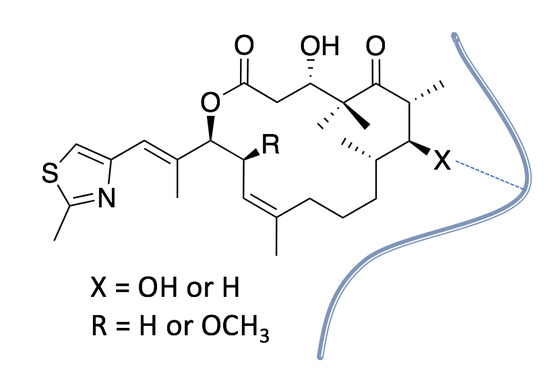
:
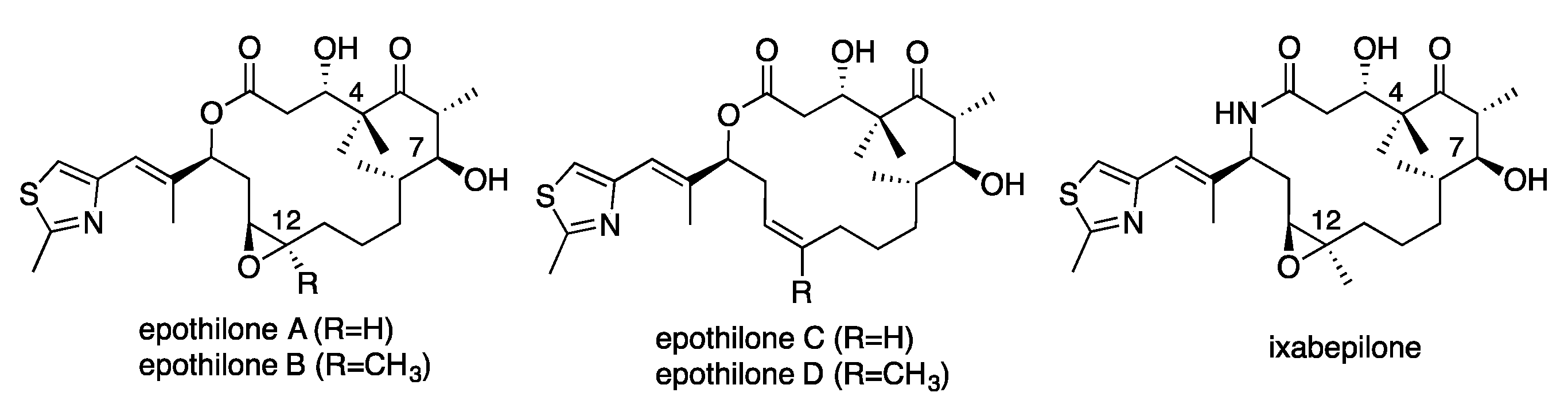
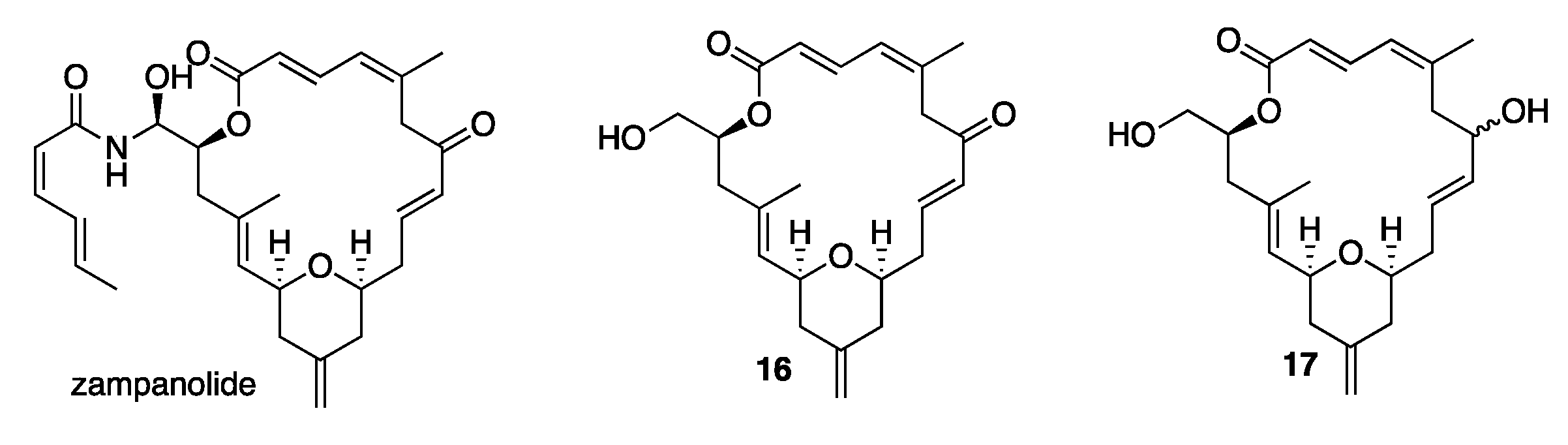
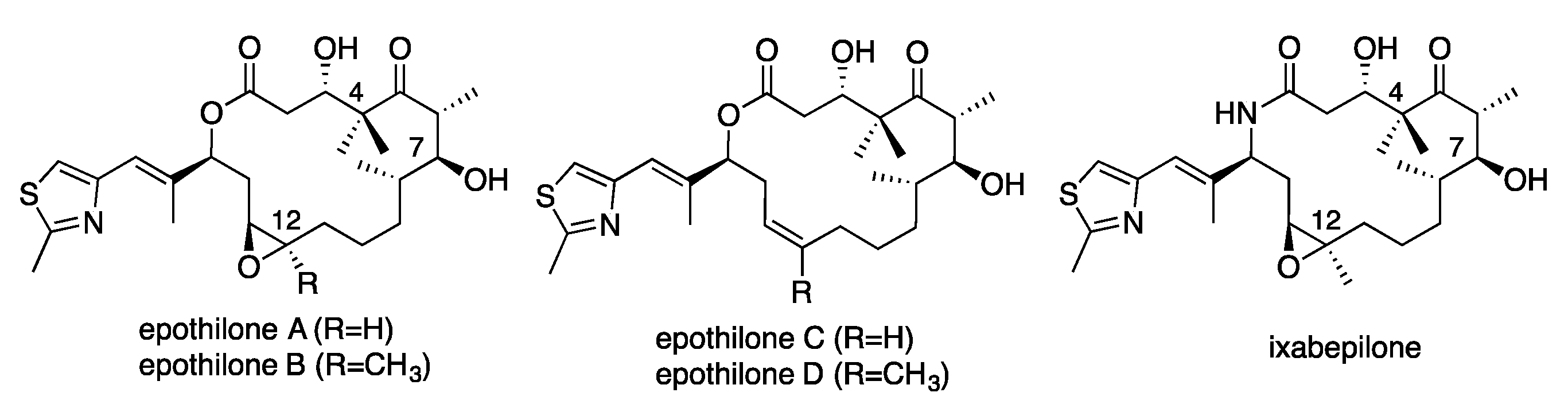
1. Introduction
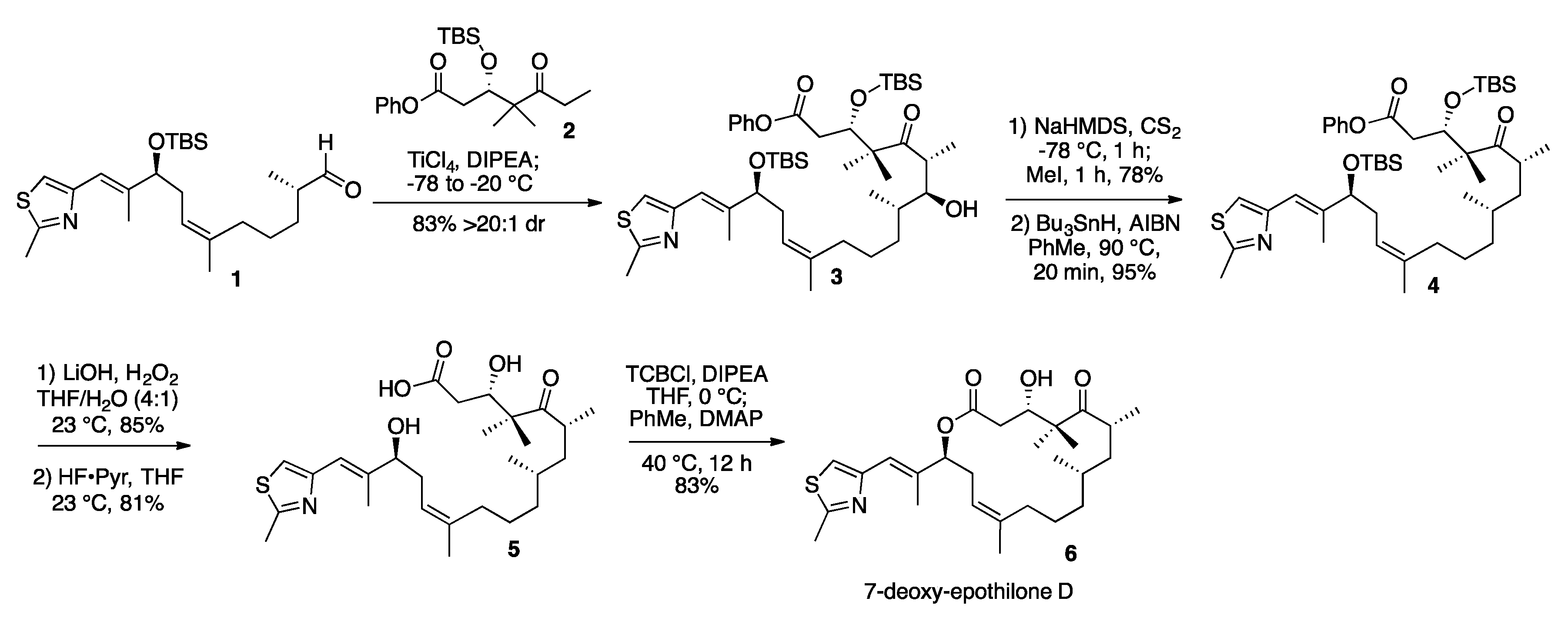
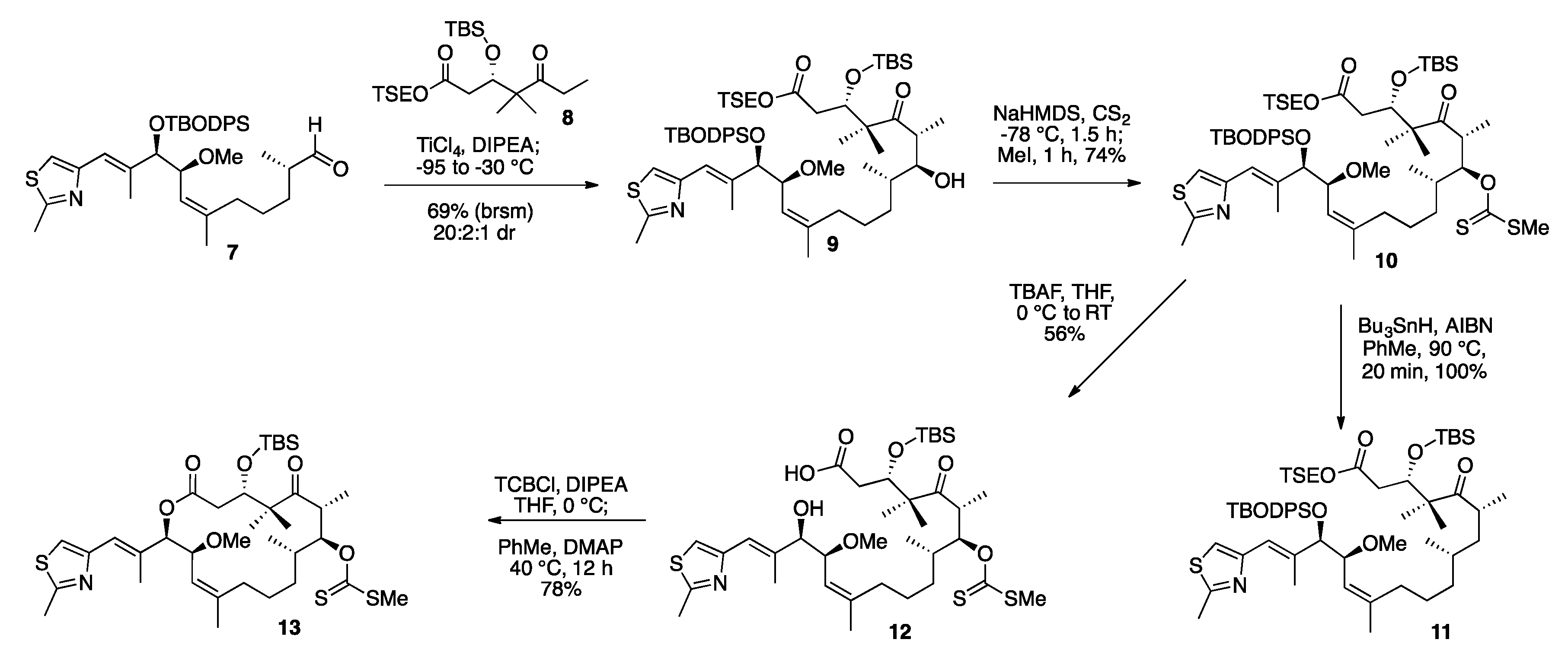
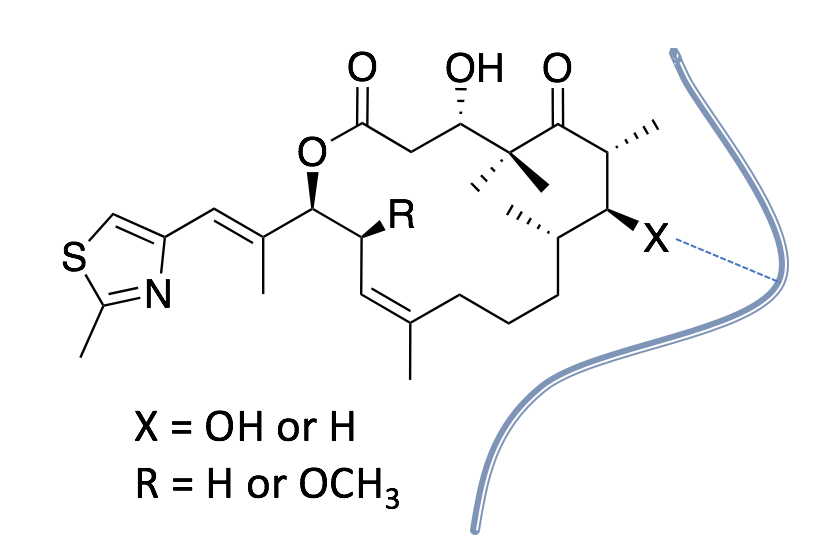
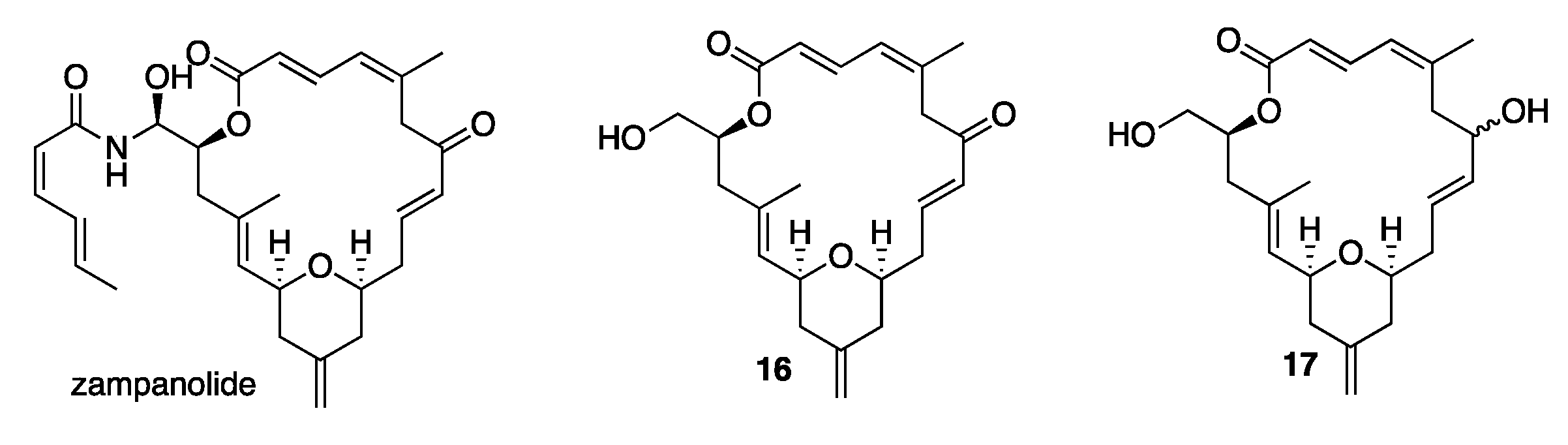
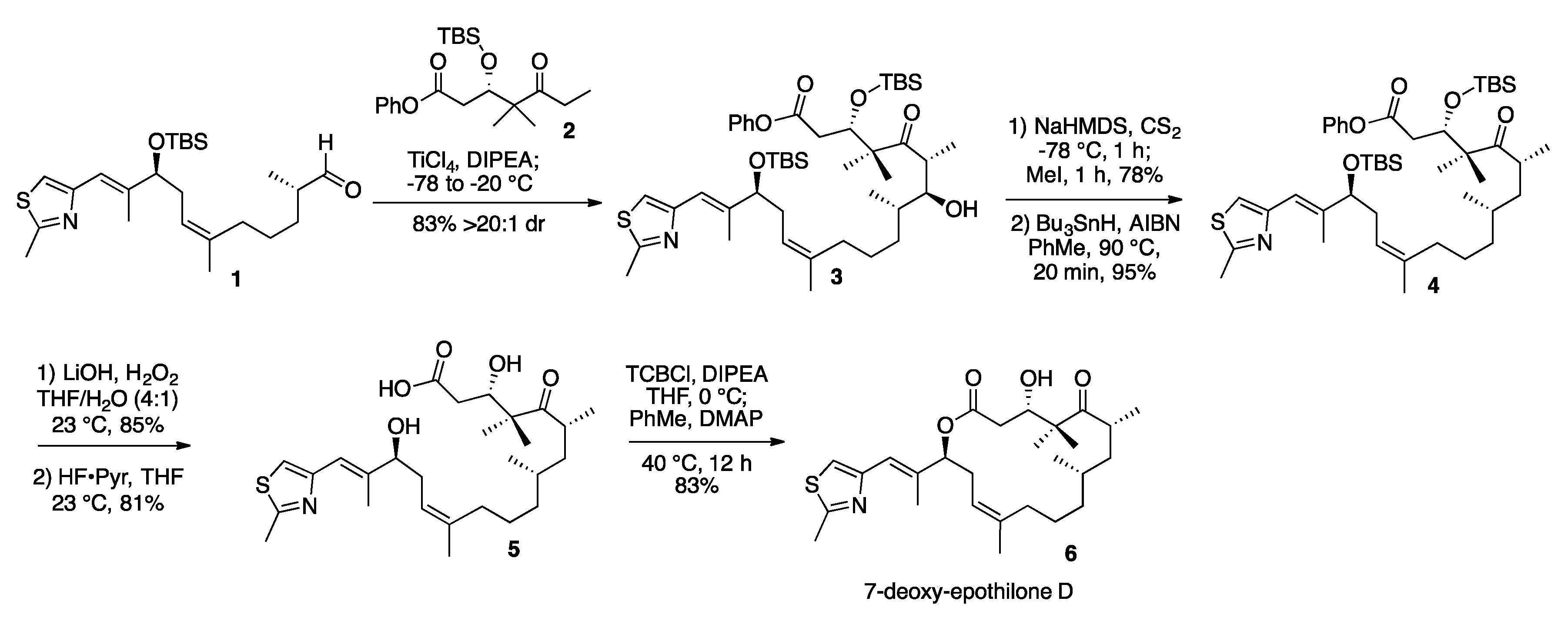
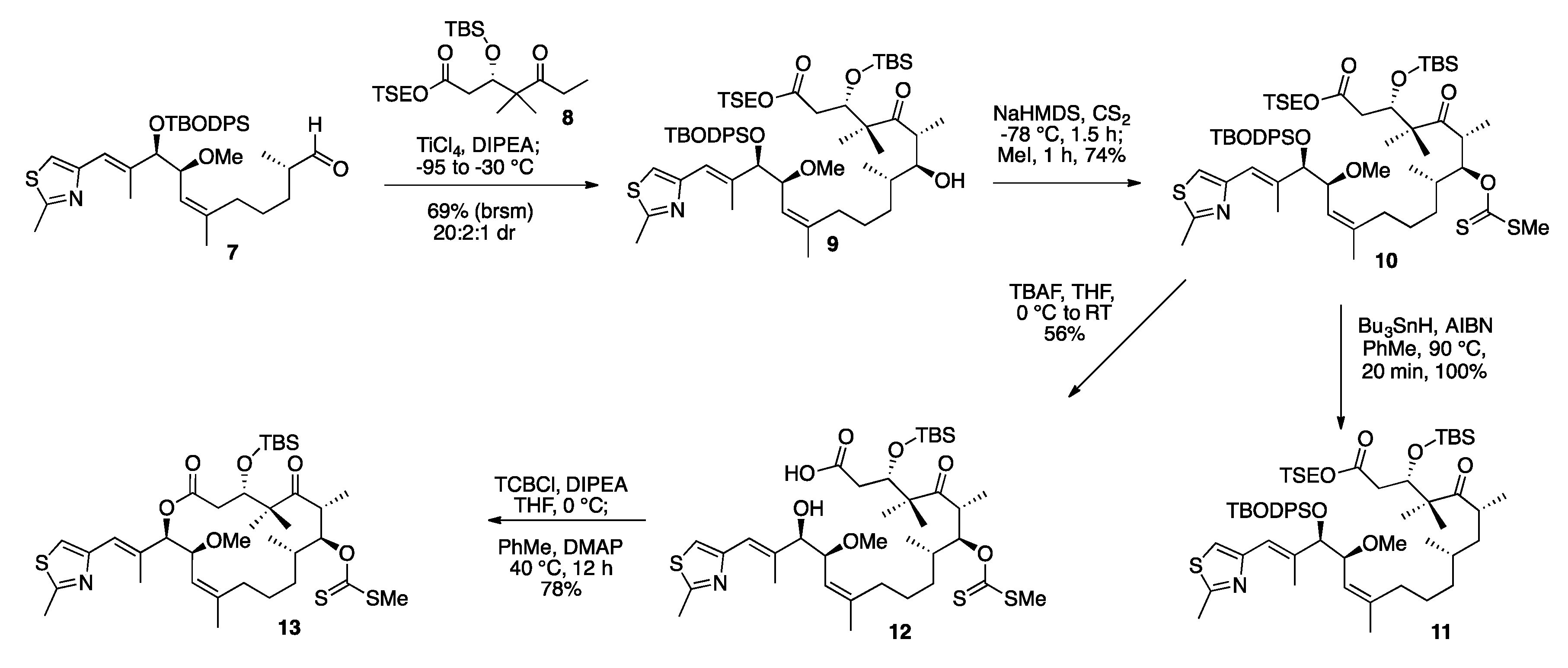
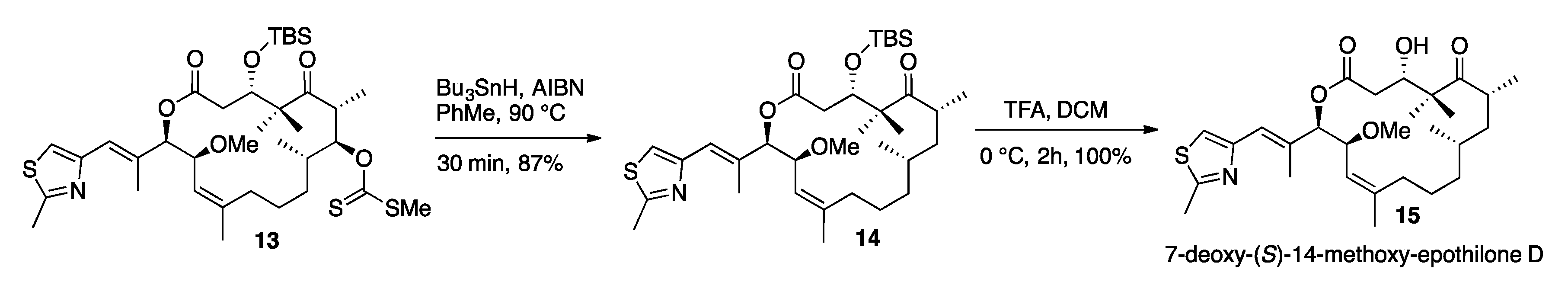
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. 7-Hydroxy-Epothilone D Phenyl Ester (3)
3.3. 7-O-Methylxanthate-Epothilone D Phenyl Ester (S1)
3.4. 7-Deoxy-Epothilone D Phenyl Ester (4)
3.5. 7-Deoxy-Epothilone D (6)
3.6. TSE Ester Ketone Fragment (8)
3.7. TSE Ester 14-(S)-Methoxy-Epothilone D (9)
3.8. TSE Ester 7-O-Methylxanthate-14-(S)-Methoxy-Epothilone D (10)
3.9. Deprotected Seco Acid 7-O-Methylxanthate-14-(S)-Methoxy-Epothilone D (12)
3.10. TBS Protected-7-O-Methylxanthate-14-(S)-Methoxy-Epothilone D (13)
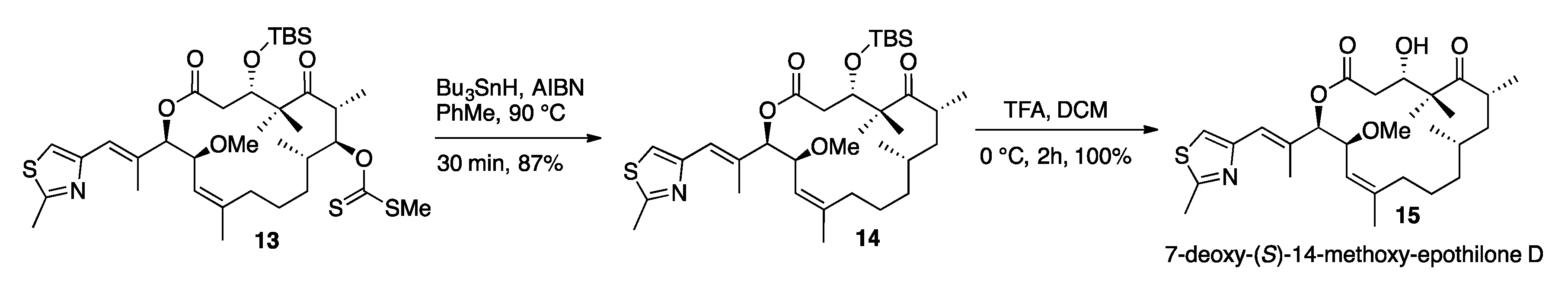
3.11. 7-Deoxy-14-(S)-Methoxy-TBS-Protected-Epothilone D (14)
3.12. 7-Deoxy-(S)-14-Methoxy-Epothilone D (15)
3.13. Cell Growth Inhibition by Compounds
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TLC | Thin layer chromatography |
| NMR | Nuclear magnetic resonance |
| AIBN | Azobisisobutyronitrile |
| TSE | 2-Trimethylsilylethoxy |
| NaHMDS | Sodium bis(trimethylsilyl)amide |
| FDA | Food and drug administration |
| TBODPS | Tert-butoxydiphenylsilyl |
| TBAF | Tetra-n-butylammonium fluoride |
| CS2 | Carbon disulfide |
| HF | Hydrogen fluoride |
| Bu3SnH | Tributyltinhydride |
| TBS | Tert-butyldimethylsilyl |
References
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Kinetic analysis of tubulin exchange at microtubule ends of low vinblastine concentrations. Biochemistry 1990, 29, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Hastie, S.B. Interactions of colchicine with tubulin. Pharmacol. Ther. 1991, 51, 377. [Google Scholar] [CrossRef]
- Fanale, D.; Bronte, G.; Passiglia, F.; Calo, V.; Castiglia, M.; di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.; Wikerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef] [PubMed]
- TAXOL® (Paclitaxel) for Injection. Available online: www.rxlist.com/taxol-drug.html (accessed on 1 July 2016).
- Höfle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reinchenbach, H. Epothilone A and B-Novel 16-membered macrolides with cytotoxic activity: Isolation, crystal structure, and conformation in solution. Angew. Chem. Int. Ed. 1996, 35, 1567–1569. [Google Scholar] [CrossRef]
- Gerth, K.; Bedorf, N.; Höfle, G.; Irschik, H.; Reinchenbach, H. Antibiotics from gliding bacteria. 74. Epothilons A and B: Antifungal and cytotoxic compounds from Sorangium cellulosum (myxobacteria): Production, physic-chemical and biological properties. J. Antibiot. 1996, 49. [Google Scholar] [CrossRef]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E. Woods, Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar] [PubMed]
- Forli, S.; Manetti, F.; Altmann, K.H.; Botta, M. Evaluation of novel epothilone analogues bymeans of a common pharmacophore and a QSAR pseudoreceptor model for taxanes and epothilones. Chem. Med. Chem. 2010, 5, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Heise, H.; Blommers, M.J.; Krastel, P.; Schmitt, E.; Petersen, F.; Jeganathan, S.; Mandelkow, E.M.; Carlomagno, T.; Griesinger, C.; et al. Interaction of epothilone B (patupilone) with microtubules as detected by two-dimensional solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2010, 49, 7504–7507. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.; Gaugaz, F.Z.; Schiess, R. Diversity through semisynthesis: The chemistry and biological activity of semisynthetic epothilone derivatives. Mol. Divers. 2011, 15, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Ritzen, A.; Namoto, K. Recent developments in the chemistry, biology and medicine of the epothilones. Chem. Commun. 2001, 1523–1535. [Google Scholar] [CrossRef]
- IXEMPRA® Kit (Ixabepilone) for Injection [Package Insert]. Available online: www.ixempra.com (accessed on 1 July 2016).
- Nicolaou, K.C.; Vourloumis, D.; Li, T.; Pastor, J.; Winssinger, N.; He, Y.; Ninkovic, S.; Sarabia, F.; Vallberg, H.; Roschangar, F.; et al. Designed epothilones: Combinatorial synthesis, tubulin assembly properties, and cytotoxic action against taxol-resistant tumor cells. Angew. Chem. Int. Ed. 1997, 36, 2097–2103. [Google Scholar] [CrossRef]
- Su, D.; Balog, A.; Meng, D.; Bertinato, P.; Danishefsky, S.J.; Zheng, Y.; Chou, T.; He, L.; Horwitz, S.B. Structure-activity relationship of the epothilones and the first in vivo comparison with paclitaxel. Angew. Chem. Int. Ed. 1997, 36, 2093–2096. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Roschangar, F.; Vourloumis, D. Chemical biology of epothilone. Angew. Chem. Int. Ed. 1998, 37, 2014–2045. [Google Scholar] [CrossRef]
- Larsen, E.M.; Wilson, M.R.; Taylor, R.E. Conformation-activity relationships of polyketide natural products. Nat. Prod. Rep. 2015, 32, 1183–1206. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.E.; Zajicek, J. Conformational properties of epothilone. J. Org. Chem. 1999, 64, 7224–7228. [Google Scholar] [CrossRef]
- Yoshimura, F.; Rivkin, A.; Gabarda, A.E.; Chou, T.-C.; Dong, H.; Sukenick, G.; Morel, F.F.; Taylor, R.E.; Danishefsky, S.J. Synthesis and Conformational Analysis of (E)-9,10-Dehydro Epothilone B. A Suggestive Linkage between Observations in the Chemistry and Biology of Epothilones. Angew. Chem. Int. Ed. 2003, 42, 2518–2521. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.E.; Chen, Y.; Beatty, A.; Myles, D.C.; Zhou, Y. Conformational-activity relationships in polyketide natural products: A new perspective on the rational design of epothilone analogues. J. Am. Chem. Soc. 2003, 125, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.E.; Chen, Y.; Galvin, G.M.; Pabba, P.K. Conformation-activity relationships in polyketide natural products. Towards the biologically active conformation of epothilone. Org. Biomol. Chem. 2004, 2, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Frein, J.D.; Taylor, R.E.; Sackett, D.L. New sources of chemical diversity inspired by biosynthesis: Rational design of a potent epothilone analogue. Org. Lett. 2009, 11, 3186–3189. [Google Scholar] [CrossRef] [PubMed]
- Giannakakou, P.; Gussio, R.; Nogales, E.; Downing, K.H.; Zaharevitz, D.; Bollbuck, B.; Poy, G.; Sachett, D.; Nicolaou, K.C. A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Manetti, F.; Forli, S.; Maccari, L.; Corelli, F.; Botta, M. 3D QSAR studies of the interaction between β-tubulin and microtubule stabilizing antimitotic agents (MSAA). A combined pharmacophore generation and pseudoreceptor modeling approach applied to taxanes and epothilones. Farmaco 2003, 58, 357–361. [Google Scholar] [CrossRef]
- Manetti, F.; Maccari, L.; Corelli, F.; Botta, M. 3D QSAR models of interactions between β-tubulin and microtubule stabilizing antimitotic agents (MSAA): A survey on taxanes and epothilones. Curr. Top. Med. Chem. 2004, 4, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, T.; Sánchez, V.M.; Blommers, M.J.J.; Griesinger, C. Derivation of dihedral angles from CH–CH dipolar-dipolar cross-correlated relaxation rates: A C–C torsion involving a quaternary carbon atom in epothilone A bound to tubulin. Angew. Chem. Int. Ed. 2003, 42, 2515–2517. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, T.; Blommers, M.J.J.; Meiler, J.; Jahnke, W.; Schupp, T.; Petersen, F.; Schinzer, D.; Altmann, K.; Griesinger, C. The high-resolution solution structure of epothilone A bound to tubulin: An understanding of the structure-activity relationships for a powerful class of antitumor agents. Angew. Chem. Int. Ed. 2003, 42, 2511–2515. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Schupp, T.; Petersen, F.; Carlomagno, T.; Baldus, M. High-resolution solid-state NMR structure of an anticancer agent. ChemMedChem 2007, 2, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Erdélyi, M; Pfeiffer, B.; Hauenstein, K.; Fohrer, J.; Gertsch, J.; Altmann, K.; Carlomagno, T. Conformational preferences of natural and C3-modified epothilones in aqueous solution. J. Med. Chem. 2008, 51, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Sefkow, M.; Kiffe, M.; Schummer, D.; Hofle, G. Oxidative and reductive transformations of epothilone A. Bioorg. Med. Chem. Lett. 1998, 8, 3025–3030. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ninkovic, S.; Sarabia, F.; Vourloumis, D.; He, Y.; Vallberg, H.; Finlay, M.R.V.; Yang, Z. Total syntheses of epothilone A and B via a macrolactonization-based strategy. J. Am. Chem. Soc. 1997, 119, 7974–7991. [Google Scholar] [CrossRef]
- Schinzer, D.; Bauer, A.; Schieber, J. Synthesis of (−)-epothilone B. Chem. Eur. J. 1999, 5, 2492. [Google Scholar] [CrossRef]
- Mulzer, J.; Mantoulidis, A.; Ohler, E. Total syntheses of epothilones B and D. J. Org. Chem. 2000, 65, 7456–7467. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.E.; Chen, Y. Total synthesis of epothilones B and D. Org. Lett. 2001, 3, 2221–2224. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Loiseleur, O.; Fuentes, D.; Jantsch, A.; Altmann, K.-H. Diastereoselective titanium enolate aldol reaction for the total synthesis of epothilones. Org. Lett. 2002, 4, 3811–3814. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R.; McCombie, S.W. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc. Perkin Trans. I 1975, 1574–1585. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef]
- Hardt, I.H.; Steinmetz, H.; Gerth, K.; Sasse, F.; Reichenbach, H.; Höfle, G. New natural epothilones from Sorangium cellulosum, strains So ce90/B2 and So ce90/D13: Isolation, structure elucidation, and SAR studies. J. Nat. Prod. 2001, 64, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Diaz, J.F.; Altmann, K.H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canales, A.; Nieto, L.; Rodríguez-Salarichs, J.; Sánchez-Murcia, P.A.; Coderich, C.; Cortés-Cabrera, A.; Patterson, I.; Carlomagno, T.; Gago, F.; Andreu, J.M.; et al. Molecular recognition of epothilones by microtubules and tubulin dimers revealed by biochemical and NMR approaches. ACS Chem. Biol. 2014, 9, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Jennings, M.P. Total synthesis of (−)-dactylolide and formal synthesis of (−)-zampanolide via target oriented β-C-glycoside formation. J. Org. Chem. 2008, 73, 5965–5976. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R. Synthesis, Conformational Analysis, and Biological Evaluation of the Potent Microtubule-Stabilizing Agents (−)-Zampanolide and (−)-Dactylolide. Ph.D. Thesis, University of Notre Dame du Lac, South Bend, IN, USA, 2014. [Google Scholar]
- Daly, E.M. Probing Tubulin Interactions with 14-Methyl Epothilone D Analogues. Ph.D. Thesis, University of Notre Dame du Lac, South Bend, IN, USA, 2011. [Google Scholar]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–6084. [Google Scholar] [CrossRef] [PubMed]
- Begaye, A.; Trostel, S.; Zhao, Z.; Taylor, R.E.; Schriemer, D.C.; Sackett, D.L. Mutations in the β-tubulin binding site for peloruside A confer resistance by targeting a cleft significant in side chain binding. Cell Cycle 2011, 10, 3387–3396. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GI50 Value |
|---|---|
| Taxol® | 0.6 nM |
| epothilone A | 9.5 nM |
| epothilone B | 0.2 nM |
| epothilone D | 7.5 nM |
| 7-deoxy-epothilone D | 900 nM |
| 7-deoxy-(S)-14-methoxy epothilone D | 2200 nM |
| 14-methoxy-epothilone D | 29 nM |
| E-9,10-dehydro-epothilone D | <1.7 nM |
| zampanolide | 0.25 nM |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woods, L.M.; Arico, J.W.; Frein, J.D.; Sackett, D.L.; Taylor, R.E. Synthesis and Biological Evaluation of 7-Deoxy-Epothilone Analogues. Int. J. Mol. Sci. 2017, 18, 648. https://doi.org/10.3390/ijms18030648
Woods LM, Arico JW, Frein JD, Sackett DL, Taylor RE. Synthesis and Biological Evaluation of 7-Deoxy-Epothilone Analogues. International Journal of Molecular Sciences. 2017; 18(3):648. https://doi.org/10.3390/ijms18030648
Chicago/Turabian StyleWoods, Laura M., Joseph W. Arico, Jeffrey D. Frein, Dan L. Sackett, and Richard E. Taylor. 2017. "Synthesis and Biological Evaluation of 7-Deoxy-Epothilone Analogues" International Journal of Molecular Sciences 18, no. 3: 648. https://doi.org/10.3390/ijms18030648