
Dysfunctional mTORC1 Signaling: A Convergent Mechanism between Syndromic and Nonsyndromic Forms of Autism Spectrum Disorder?


Abstract
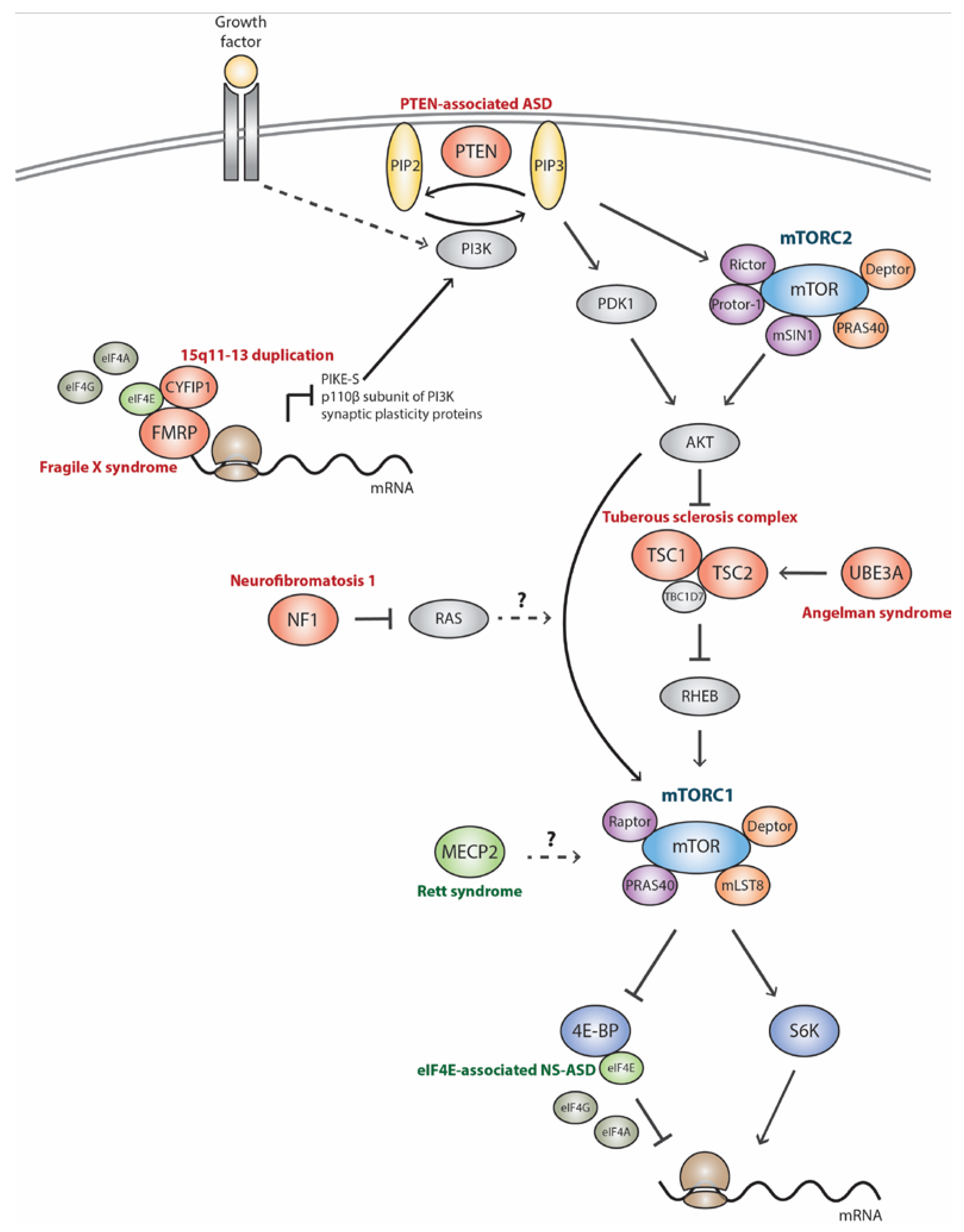
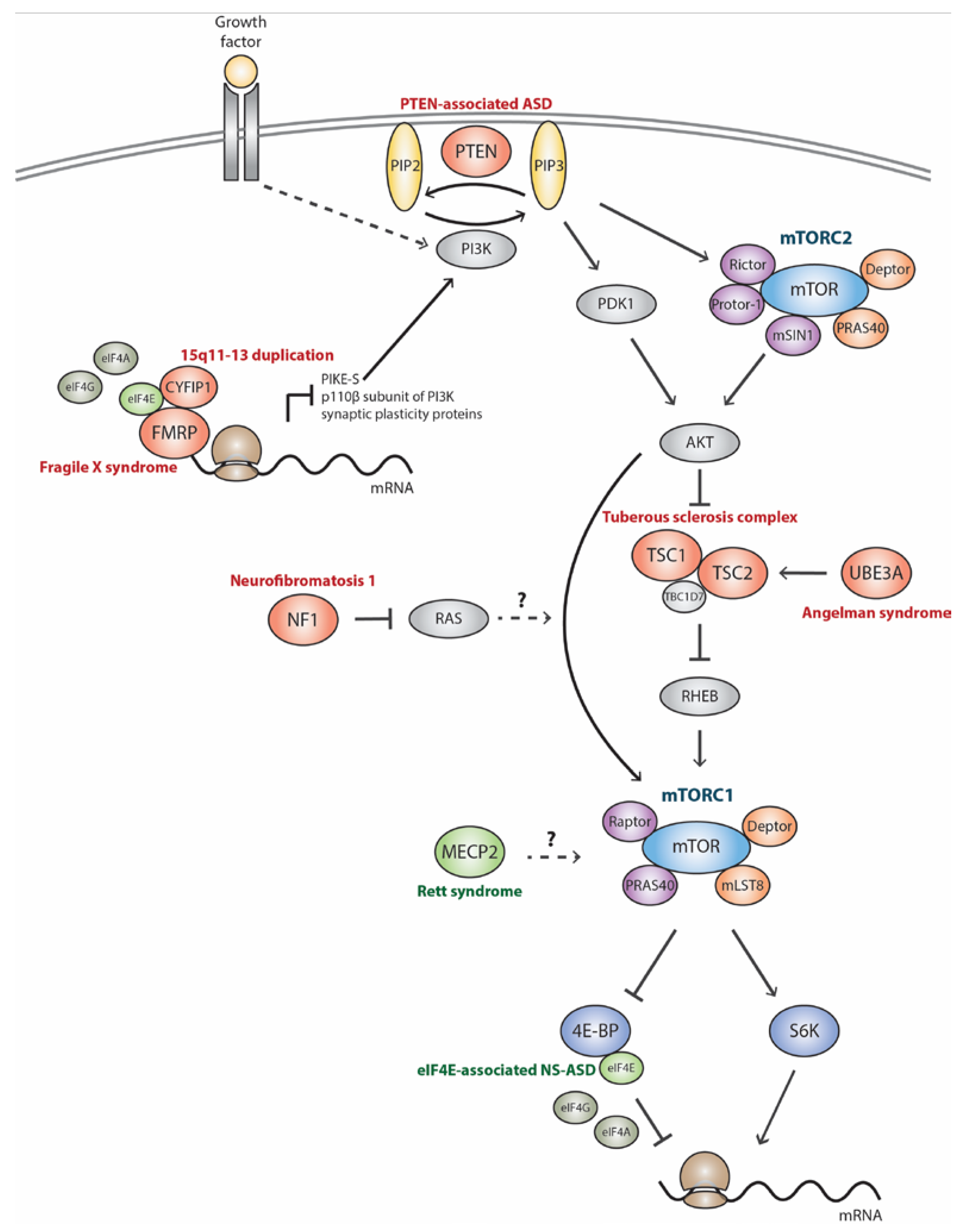
:1. Introduction
2. mTORC1 Signaling Pathway in Monogenic Autism Spectrum Disorder-Related Syndromes
2.1. Tuberous Sclerosis Complex (TSC)
2.2. Phosphatase and Tensin Homolog-Associated ASD (PTEN-ASD)
2.3. Neurofibromatosis Type I (NF1)
2.4. Fragile X Syndrome (FXS)
2.5. Angelman Syndrome (AS)
2.6. Rett Syndrome (RTT)
3. mTORC1 Signaling Pathway in Nonsyndromic/Idiopathic Autism Spectrum Disorder
3.1. 15q11-13 Duplication (Dup15q)
3.2. eIF4E-Associated NS-ASD (eIF4E-NS-ASD)
3.3. Idiopathic Autism Spectrum Disorder
4. Discussion
5. Conclusions and Future Directions
Author Contributions
Conflicts of Interest
References
- Christensen, D.L.; Baio, J.; van Naarden Braun, K.; Bilder, D.; Charles, J.; Constantino, J.; Daniels, J.; Durkin, M.; Fitzgerald, R.T.; Kurzius-Spencer, M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill. Summ. 2016, 65, 1–23. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (5th ed.; DSM–5); American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Newschaffer, C.; Croen, L.A.; Daniels, J.; Giarelli, E.; Grether, J.K.; Levy, S.E.; Mandell, D.S.; Miller, L.A.; Pinto-Martin, J.; Reaven, J.; et al. The Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2007, 28, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 2009, 65, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Jeste, S.S.; Sahin, M.; Bolton, P. Characterization of Autism in Young Children with Tuberous Sclerosis Complex. J. Child Neurol. 2008, 23, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Budimirovic, D.B.; Kaufmann, W.E. What Can We Learn about Autism from Studying Fragile X Syndrome? Dev. Neurosci. 2011, 33, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 2012, 14, 253–262. [Google Scholar] [PubMed]
- Williams, C.A.; Beaudet, A.L.; Clayton-Smith, J.; Knoll, J.H.; Kyllerman, M.; Laan, L.A.; Magenis, R.E.; Moncla, A.; Schinzel, A.A.; Summers, J.A.; et al. Angelman Syndrome 2005: Updated Consensus for Diagnostic Criteria. Am. J. Med. Genet. 2006, 140, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K. Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 Calcium Channel Dysfunction Causes a Multisystem Disorder Including Arrhythmia and Autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.; McDermid, H.E. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2011, 2, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Bear, M.F. The Autistic Neuron: Troubled Translation? Cell 2008, 135, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Sahin, M. Autism and the synapse: Emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol. 2015, 28, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Ronald, A.; Hoekstra, R.A. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Larsson, H.; Cm, H.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- State, M.W.; Levitt, P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci. 2011, 14, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Klei, L.; Sanders, S.J.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; Yu, T.W.; Fombonne, E.; Geschwind, D.; et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong Association of De Novo Copy Number Mutations with Autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.; Wang, K.; Cai, G.; Korvatska, O. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Brett, S.; et al. Functional Impact of Global Rare Copy Number Variation in Autism Spectrum Disorder. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380, 42–77. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Yamrom, B.; Lee, Y.; Narzisi, G.; Leotta, A.; Grabowska, E.; et al. De novo Gene Disruptions in children on the Autistic Spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.; Kou, Y.; Liu, L.; Ma’ayan, A. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2013, 485, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Ronemus, M.; Iossifov, I.; Levy, D.; Wigler, M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 2014, 15, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Simons Foundation Autism Research Initiative (SFARI). Available online: https://gene.sfari.org (accessed on 16 March 2017).
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare De Novo Variants Associated with Autism Implicate a Large Functional Network of Genes Involved in Formation and Function of Synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef]
- Baudouin, S.J.; Gaudias, J.; Gerharz, S.; Hatstatt, L.; Zhou, K.; Punnakkal, P.; Tanaka, K.F.; Spooren, W.; Hen, R.; De Zeeuw, C.I.; et al. Shared Synaptic Pathophysiology in Syndromic and Nonsyndromic Rodent Models of Autism. Science 2012, 338, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Mattioli, M.; Monteggia, L.M. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci 2013, 16, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.O.; Sahin, M. The Neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Silva, A.J. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 2011, 17, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Klann, E.; Costa-Mattioli, M.; Zukin, R.S. Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J. Neurosci. 2015, 35, 13836–13842. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Boggio, E.M.; Grosso, S.; Lonetti, G.; Forlani, G.; Stefanelli, G.; Calcagno, E.; Morello, N.; Landsberger, N.; Biffo, S.; et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 2011, 20, 1182–1196. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Moreno, S.; Baudry, M.; Bi, X. Imbalanced Mechanistic Target of Rapamycin C1 and C2 Activity in the Cerebellum of Angelman Syndrome Mice Impairs Motor Function. J. Neurosci. 2015, 35, 4706–4718. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Tran, J.; O’Neal, P.; Baudry, M.; Bi, X. mTORC1-S6K1 inhibition or mTORC2 activation improves hippocampal synaptic plasticity and learning in Angelman syndrome mice. Cell. Mol. Life Sci. 2016, 73, 4303–4314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Ash, R.T.; Baker, S.A.; Suter, B.; Ferguson, A.; Park, J.; Rudy, J.; Torsky, S.P.; Chao, H.-T.; Zoghbi, H.Y.; et al. Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J. Neurosci. 2013, 33, 19518–19533. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, G.; Putignano, E.; Chelini, G.; Melani, R.; Calcagno, E.; Michele Ratto, G.; Amendola, E.; Gross, C.T.; Giustetto, M.; Pizzorusso, T. Dendritic Spine Instability in a Mouse Model of CDKL5 Disorder Is Rescued by Insulin-like Growth Factor 1. Biol. Psychiatry 2016, 80, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Neves-Pereira, M.; Müller, B.; Massie, D.; Williams, J.H.G.; O’Brien, P.C.M.; Hughes, A.; Shen, S.-B.; Clair, D.S.; Miedzybrodzka, Z. Deregulation of EIF4E: A novel mechanism for autism. J. Med. Genet. 2009, 46, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Huynh, T.N.; Macaskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; Kaphzan, H.; Klann, E. Exaggerated Translation Causes Synaptic and Behavioural Aberrations Associated with Autism. Nature 2013, 493, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Oguro-Ando, A.; Rosensweig, C.; Herman, E.; Nishimura, Y.; Werling, D.; Bill, B.R.; Berg, J.M.; Gao, F.; Coppola, G.; Abrahams, B.S.; et al. Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol. Psychiatry 2015, 20, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.M.; Griesi-Oliveira, K.; de Oliveira Freitas Machado, C.; Vadasz, E.; Zachi, E.C.; Passos-Bueno, M.R.; Sertie, A.L. Altered mTORC1 signaling in multipotent stem cells from nearly 25% of patients with nonsyndromic autism spectrum disorders. Mol. Psychiatry 2015, 20, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, C.; Ahn, Y.; Michalski, B.; Rho, J.M.; Fahnestock, M. Decreased mTOR signaling pathway in human idiopathic autism and in rats exposed to valproic acid. Acta Neuropathol. Commun. 2015, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Poopal, A.C.; Schroeder, L.M.; Horn, P.S.; Bassell, G.J.; Gross, C. Increased expression of the PI3K catalytic subunit p110δ underlies elevated S6 phosphorylation and protein synthesis in an individual with autism from a multiplex family. Mol. Autism 2016, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and Characterization of the Tuberous Sclerosis Gene on Chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar]
- Slegtenhorst, V.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, AH.D.; Young, J.; Burley, M.; et al. Identification of the Tuberous Sclerosis Gene TSC1 on Chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin Activation of Rheb, a Mediator of mTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis1, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Curatolo, P.; Moavero, R. mTOR Inhibitors in Tuberous Sclerosis Complex. Curr. Neuropharmacol. 2012, 10, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Jones, C.; Groves, L.; Moss, J.; Oliver, C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: A systematic review and meta-analysis. Lancet Psychiatry 2015, 2, 909–916. [Google Scholar] [CrossRef]
- Chu-Shore, C.J.; Major, P.; Camposano, S.; Muzykewicz, D.; Thiele, E.A. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010, 51, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Greenwood, J.S.F.; Calcagnotto, M.E.; Kirsch, H.E.; Barbaro, N.M.; Baraban, S.C. Neocortical Hyperexcitability in a Human Case of Tuberous Sclerosis Complex and Mice Lacking Neuronal Expression of TSC1. Ann. Neurol. 2007, 61, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Talos, D.M.; Kwiatkowski, D.J.; Cordero, K.; Black, P.M.; Jensen, F.E. Cell-Specific Alterations of Glutamate Receptor Expression in Tuberous Sclerosis Complex Cortical Tubers. Ann. Neurol. 2008, 63, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Ruppe, V.; Dilsiz, P.; Reiss, C.S.; Carlson, C.; Devinsky, O.; Zagzag, D.; Weiner, H.L.; Talos, D.M. Developmental brain abnormalities in tuberous sclerosis complex: A comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia 2014, 55, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, E.J.; Wong, M.; Baldwin, R.L.; Bajenaru, M.L.; Onda, H.; Kwiatkowski, D.J.; Yamada, K.; Gutmann, D.H. Astrocyte-Specific TSC1 Conditional Knockout Mice Exhibit Abnormal Neuronal Organization and Seizures. Ann. Neurol. 2002, 52, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Xu, L.; Gutmann, D.H.; Wong, M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 2008, 63, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Rensing, N.R.; Zhang, B.; Gutmann, D.H.; Gambello, M.J.; Wong, M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of Tuberous Sclerosis Complex. Hum. Mol. Genet. 2011, 20, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Ess, K.C. Conditional and domain-specific inactivation of the Tsc2 gene in neural progenitor cells. Genesis 2013, 51, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Magri, L.; Cambiaghi, M.; Cominelli, M.; Alfaro-Cervello, C.; Cursi, M.; Pala, M.; Bulfone, A.; Garca-Verdugo, J.M.; Leocani, L.; Minicucci, F.; et al. Sustained Activation of mTOR Pathway in Embryonic Neural Stem Cells Leads to Development of Tuberous Sclerosis Complex-Associated Lesions. Cell Stem Cell 2011, 9, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Crowell, B.; Hwa Lee, G.; Nikolaeva, I.; Dal Pozzo, V.; D’Arcangelo, G. Complex Neurological Phenotype in Mutant Mice Lacking Tsc2 in Excitatory Neurons of the Developing Forebrain. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Moon, U.Y.; Park, J.Y.; Park, R.; Cho, J.Y.; Hughes, L.J.; McKenna, J.; Goetzl, L.; Cho, S.-H.; Crino, P.B.; Gambello, M.J.; et al. Impaired Reelin-Dab1 Signaling Contributes to Neuronal Migration Deficits of Tuberous Sclerosis Complex. Cell Rep. 2015, 12, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.F.; Alvarez, V.A.; Ridenour, D.A.; Kwiatkowski, D.J.; Sabatini, B.L. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 2005, 8, 1727–1734. [Google Scholar] [CrossRef] [PubMed]
- Meikle, L.; Pollizzi, K.; Egnor, A.; Kramvis, I.; Lane, H.; Sahin, M.; Kwiatkowski, D.J. Response of a Neuronal Model of Tuberous Sclerosis to Mammalian Target of Rapamycin (mTOR) Inhibitors: Effects on mTORC1 and Akt Signaling Lead to Improved Survival and Function. J. Neurosci. 2008, 28, 5422–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Di Nardo, A.; Kramvis, I.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M.; He, X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008, 22, 2485–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi-fakhari, D.; Saffari, A.; Wahlster, L.; Di Nardo, A.; Turner, D.; Lewis, T.L., Jr.; Conrad, C.; Rothberg, J.M.; Jonathan, O.; Kölker, S.; et al. Impaired Mitochondrial Dynamics and Mitophagy in Neuronal Models of Tuberous Sclerosis Complex. Cell Rep. 2016, 17, 1053–1070. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 In Vivo Impairs Hippocampal mGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. 2011, 31, 8862–8869. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/Inhibitory Synaptic Imbalance Leads to Hippocampal Hyperexcitability in Mouse Models of Tuberous Sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Von Der Brelie, C.; Waltereit, R.; Zhang, L.; Beck, H.; Kirschstein, T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur. J. Neurosci. 2006, 23, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Ouyang, Y.; Gazit, V.; Cirrito, J.R.; Jansen, L.A.; Ess, K.C.; Yamada, K.A.; Wozniak, D.F.; Holtzman, D.M.; Gutmann, D.H.; et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 2007, 28, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, S.; Sugiura, H.; Katsurabayashi, S.; Shimada, T.; Tanaka, H.; Takasaki, K.; Iwasaki, K.; Kobayashi, T.; Hino, O.; Yamagata, K. Activation of Rheb, but not of mTORC1, impairs spine synapse morphogenesis in tuberous sclerosis complex. Sci. Rep. 2014, 4, 5155. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Chen, Z.; Ebrahimi-Fakhari, D.; Di Nardo, A.; Julich, K.; Robson, V.K.; Cheng, Y.-C.; Woolf, C.J.; Heiman, M.; Sahin, M. The Stress-Induced Atf3-Gelsolin Cascade Underlies Dendritic Spine Deficits in Neuronal Models of Tuberous Sclerosis Complex. J. Neurosci. 2015, 35, 10762–10772. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; David, J. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 2012, 3, 1292. [Google Scholar] [CrossRef] [PubMed]
- Ercan, E.; Han, J.M.; Di Nardo, A.; Winden, K.; Han, M.-J.; Hoyo, L.; Saffari, A.; Leask, A.; Geschwind, D.H.; Sahin, M. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J. Exp. Med. 2017, 214, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J. T.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Cai, Q.; Lu, W.; Sheng, Z.-H.; Mochida, S. KIF5B Motor Adaptor Syntabulin Maintains Synaptic Transmission in Sympathetic Neurons. J. Neurosci. 2009, 29, 13019–13029. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Wertz, M.H.; Kwiatkowski, E.; Tsai, P.T.; Leech, J.D.; Greene-Colozzi, E.; Goto, J.; Dilsiz, P.; Talos, D.M.; Clish, C.B.; et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 2014, 23, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Cai, G.; Chaste, P.; Nygren, G.; Goldsmith, J.; Reichert, J.; Anckarsäter, H.; Rastam, M.; Smith, C.J.; Silverman, J.M.; et al. Mutation Screening of the PTEN Gene in Patients With Autism Spectrum Disorders and Macrocephaly. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2007, 144, 484–491. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Varga, E.A.; Pastore, M.T.; Prior, T.W.; Manickam, K.; Atkin, J.F.; Herman, G.E. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010, 3, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.A.; Pastore, M.; Prior, T.; Herman, G.E.; McBride, K.L. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 2009, 11, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hobert, J.A.; Embacher, R.; Mester, J.L.; Frazier, T.W.; Eng, C. Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur. J. Hum. Genet. 2014, 22, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Maehama, A.J.; Dixon, J.E. The Tumor Suppressor PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.-H.; Zhang, W.; Sinton, C.; Powell, C.M.; Parada, L.F. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 2009, 29, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Vanderver, A.; Tonduti, D.; Kahn, I.; Schmidt, J.; Medne, L.; Vento, J.; Chapman, K.A.; Lanpher, B.; Pearl, P.; Gropman, A.; et al. Characteristic brain magnetic resonance imaging pattern in patients with macrocephaly and PTEN mutations. Am. J. Med. Genet. Part A 2014, 164, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L.A.; Mirzaa, G.M.; Ishak, G.E.; O’Roak, B.J.; Hiatt, J.B.; Roden, W.H.; Gunter, S.A.; Christian, S.L.; Collins, S.; Adams, C.; et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015, 138, 1613–1628. [Google Scholar] [CrossRef] [PubMed]
- Conti, S.; Condo, M.; Posar, A.; Mari, F.; Resta, N.; Renieri, A.; Neri, I.; Patrizi, A.; Parmeggiani, A. Phosphatase and Tensin Homolog (PTEN) Gene Mutations and Autism: Literature Review and a Case Report of a Patient With Cowden Syndrome, Autistic Disorder, and Epilepsy. J. Child Neurol. 2012, 27, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Marchese, M.; Conti, V.; Valvo, G.; Moro, F.; Muratori, F.; Tancredi, R.; Santorelli, F.M.; Guerrini, R.; Sicca, F. Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med. Genet. 2014, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.M.; Zhu, X.; Kwon, C.H.; Uhlmann, E.J.; Gutmann, D.H.; Baker, S.J. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004, 64, 7773–7779. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.M.; Bayazitov, I.T.; Zakharenko, S.S.; Baker, S.J. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 2008, 151, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Luikart, B.W.; Schnell, E.; Washburn, E.K.; Bensen, A.L.; Tovar, K.R.; Westbrook, G.L. Pten Knockdown In Vivo Increases Excitatory Drive onto Dentate Granule Cells. J. Neurosci. 2011, 31, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; DeSpenza, T.; Li, M.; Gulledge, A.T.; Luikart, B.W. Hyperactivity of Newborn Pten Knock-out Neurons Results from Increased Excitatory Synaptic Drive. J. Neurosci. 2015, 35, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Chen, Y.; Page, D.T. Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun. 2016, 7, 13421. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Krimpenfort, P.; Leung, C.; van der Korput, H.A.G.M.; Trapman, J.; Camenisch, I.; Berns, A.; Brandner, S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 2002, 129, 3513–3522. [Google Scholar] [PubMed]
- Getz, S.A.; DeSpenza, T.; Li, M.; Luikart, B.W. Rapamycin prevents, but does not reverse, aberrant migration in Pten knockout neurons. Neurobiol. Dis. 2016, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Gertner, M.J.; Zhou, J.; Parada, L.F.; Bennett, M.V.L.; Zukin, R.S. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc. Natl. Acad. Sci. USA 2013, 110, 4738–4743. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Green, J.; Leadbitter, K.; Emsley, R.; Lehtonen, A.; Evans, G.; Huson, S.M. Neurofibromatosis type 1 and autism spectrum disorder. Pediatrics 2013, 132, e1642–e1648. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Weiss, R.; Xu, G.; Viskochil, D.; Culver, M.; Stevens, J.; Robertson, M.; Dunn, D.; Gesteland, R.; O’Connell, P.; et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990, 62, 193–201. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Yi, Y.; Chen, D.Y.; Weber, J.D.; Gutmann, D.H. Proteomic Analysis Reveals Hyperactivation of the Mammalian Target of Rapamycin Pathway in Neurofibromatosis 1—Associated Human and Mouse Brain Tumors. Cancer Res. 2005, 65, 2755–2760. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Crouse, N.R.; Emnett, R.J.; Gianino, S.M.; Gutmann, D.H. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc. Natl. Acad. Sci. USA 2011, 108, 15996–16001. [Google Scholar] [CrossRef] [PubMed]
- Kulkantrakorn, K.; Geller, T.J. Seizures in neurofibromatosis 1. Pediatr. Neurol. 1998, 19, 347–350. [Google Scholar] [CrossRef]
- Vivarelli, R.; Grosso, S.; Calabrese, F.; Farnetani, M.; Di, B.R.; Morgese, G.; Balestri, P. Epilepsy in neurofibromatosis 1. J. Child Neurol. 2003, 18, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Ostendorf, A.P.; Gutmann, D.H.; Weisenberg, J.L.Z. Epilepsy in individuals with neurofibromatosis type 1. Epilepsia 2013, 54, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.D.; Slopis, J.M.; Jackson, E.F.; De Winter, A.E.; Leeds, N.E. Brain volume in children with neurofibromatosis type 1. Neurology 2000, 54, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Cutting, L.E.; Koth, C.W.; Burnette, C.P.; Abrams, M.T.; Kaufmann, W.E.; Denckla, M.B. Relationship of Cognitive Functioning, Whole Brain Volumes, and T2-Weighted Hyperintensities in Neurofibromatosis-1. J. Child Neurol. 2000, 15, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Margariti, P.N.; Blekas, K.; Katzioti, F.G.; Zikou, A.K.; Tzoufi, M.; Argyropoulou, M.I. Magnetization transfer ratio and volumetric analysis of the brain in macrocephalic patients with neurofibromatosis type 1. Eur. Radiol. 2007, 17, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Karlsgodt, K.H.; Rosser, T.; Lutkenhoff, E.S.; Cannon, T.D.; Silva, A.; Bearden, C.E. Alterations in White Matter Microstructure in Neurofibromatosis-1. PLoS ONE 2012, 7, e47854. [Google Scholar] [CrossRef] [PubMed]
- Petrella, L.I.; Cai, Y.; Sereno, J.V.; Gon??alves, S.I.; Silva, A.J.; Castelo-Branco, M. Brain and behaviour phenotyping of a mouse model of neurofibromatosis type-1: An MRI/DTI study on social cognition. Genes Brain Behav. 2016, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Korf, B.R.; Schneider, G.; Poussaint, T.Y. Structural anomalies revealed by neuroimaging studies in the brains of patients with neurofibromatosis type 1 and large deletions. Genet. Med. 1999, 1, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Balestri, P.; Vivarelli, R.; Grosso, S.; Santori, L.; Farnetani, M.A.; Galluzzi, P.; Vatti, G.P.; Calabrese, F.; Morgese, G. Malformations of cortical development in neurofibromatosis type 1. Neurology 2003, 61, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Huijbregts, S.C.; Loitfelder, M.; Rombouts, S.A.; Swaab, H.; Verbist, B.M.; Arkink, E.B.; van Buchem, M.A.; Veer, I.M. Cerebral volumetric abnormalities in Neurofibromatosis type 1: Associations with parent ratings of social and attention problems, executive dysfunction, and autistic mannerisms. J. Neurodev. Disord. 2015, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Wang, Y.; Kim, S.-J.; Bornhorst, M.; Jecrois, E.S.; Anthony, T.E.; Wang, C.; Li, Y.E.; Guan, J.-L.; Murphy, G.G.; et al. Transient inhibition of the ERK pathway prevents cerebellar developmental defects and improves long-term motor functions in murine models of neurofibromatosis type 1. Elife 2014, 3, e05151. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ortiz, E.; Cho, W.; Nazarenko, I.; Mo, W.; Chen, J.; Parada, L.F. NF1 regulation of RAS/ERK signaling is required for appropriate granule neuron progenitor expansion and migration in cerebellar development. Genes Dev. 2014, 28, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Bajenaru, M.L.; Zhu, Y.; Hedrick, N.M.; Donahoe, J.; Parada, L.F.; Gutmann, D.H. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol. Cell. Biol. 2002, 22, 5100–5113. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, B.; Dasgupta, B.; Shin, J.E.; Emnett, R.J.; Hart-Mahon, E.K.; Elghazi, L.; Bernal-Mizrachi, E.; Gutmann, D.H. Neurofibromatosis-1 Regulates Neuronal and Glial Cell Differentiation from Neuroglial Progenitors In Vivo by Both cAMP- and Ras-Dependent Mechanisms. Cell Stem Cell 2007, 1, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Gianino, S.M.; Gutmann, D.H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 2010, 30, 5579–5589. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Lei, Y.T.; Hong, C.J.; Hsueh, Y.P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Shih, Y.T.; Chen, C.Y.; Chao, H.W.; Lee, M.J.; Hsueh, Y.P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Investig. 2011, 121, 4820–4837. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.M.; Federov, N.B.; Kogan, J.H.; Murphy, G.G.; Stern, J.; Ohno, M.; Kucherlapati, R.; Jacks, T.; Silva, A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002, 415, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Guilding, C.; McNair, K.; Stone, T.W.; Morris, B.J. Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur. J. Neurosci. 2007, 25, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A neurofibromatosis clinical trials consortium phase II study. Neurol. Oncol. 2015, 17, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Zehou, O.; Ducassou, S.; Minard-Colin, V.; Hamel-Teillac, D.; Wolkenstein, P.; Valeyrie-Allanore, L. Sirolimus Improves Pain in NF1 Patients with Severe Plexiform Neurofibromas. Pediatrics 2014, 133, e1792–e1797. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, B.W.; Williams, S.M.G.; Chan, A.W.; Reczek, E.E.; Lynch, R.C.; Rioth, M.J.; McClatchey, A.; Ryeom, S.; Cichowski, K. TORC1 Is Essential for NF1-Associated Malignancies. Curr. Biol. 2008, 18, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Bhola, P.; Banerjee, S.; Mukherjee, J.; Balasubramanium, A.; Arun, V.; Karim, Z.; Burrell, K.; Croul, S.; Gutmann, D.H.; Guha, A. Preclinical in vivo evaluation of rapamycin in human malignant peripheral nerve sheath explant xenograft. Int. J. Cancer 2010, 126, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romero, M.I.; Ghosh, P.; Ye, Z.; Charnay, P.; Rushing, E.J.; Marth, J.D.; Parada, L.F. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001, 15, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hoem, G.; Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol. Autism 2010, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Bagni, C.; Greenough, W.T. From mRNP trafficking to spine dysmorphogenesis: The roots of fragile X syndrome. Nat. Rev. Neurosci. 2005, 6, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Klann, E. The translation of translational control by FMRP: Therapeutic targets for FXS. Nat. Neurosci. 2013, 16, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Incorpora, G.; Sorge, G.; Sorge, A.; Pavone, L. Epilepsy in fragile X syndrome. Brain Dev. 2002, 24, 766–769. [Google Scholar] [CrossRef]
- Hagerman, P.J.; Stafstrom, C.E. Origins of Epilepsy in Fragile X Syndrome. Epilepsy Curr. 2009, 9, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Brunschwig, A.; Miller, L.K.; Hagerman, R.J. Standards for Selected Anthropometric Measurements in Males With the Fragile X Syndrome. Pediatrics 1992, 89, 1059–1062. [Google Scholar] [PubMed]
- Chiu, S.; Wegelin, J.A.; Blank, J.; Jenkins, M.; Day, J.; Hessl, D.; Tassone, F.; Hagerman, R. Early acceleration of head circumference in children with fragile x syndrome and autism. J. Dev. Behav. Pediatr. 2007, 28, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Hinton, V.J.; Brown, W.T.; Wisniewski, K.; Rudelli, R.D. Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 1991, 41, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Patel, B.; Idupulapati, M.; Harris, J.B.; Crisostomo, R.A.; Larsen, B.P.; Kooy, F.; Willems, P.J.; Cras, P.; Kozlowski, P.B.; et al. Abnormal Dendritic Spine Characteristics in the Temporal and Visual Cortices of Patients With Fragile-X Syndrome: A Quantitative Examination. Am. J. Med. Genet. 2001, 98, 161–167. [Google Scholar] [CrossRef]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic Characterization of the FMR1 Gene and Aberrant Neurodevelopment in Human Induced Pluripotent Stem Cell Models of Fragile X Syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.-C.; Abbeduto, L.; Bhattacharyya, A. iPSC-Derived Forebrain Neurons from FXS Individuals Show Defects in Initial Neurite Outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Kuznitsov-Yanovsky, L.; Segal, M.; Ben-Yosef, D. Functional Deficiencies in Fragile X Neurons Derived from Human Embryonic Stem Cells. J. Neurosci. 2015, 35, 15295–15306. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, S.A.; Calabrese, G.; Bonaccorso, C.M.; D’Antoni, S.; Brouwer, J.R.; Bakker, C.E.; Elia, M.; Ferri, R.; Nelson, D.L.; Oostra, B.A.; et al. Audiogenic seizure susceptibility is reduced in fragile X knockout mice after introduction of FMR1 transgenes. Exp. Neurol. 2007, 203, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic Removal of p70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Uutela, M.; Lindholm, J.; Louhivuori, V.; Wei, H.; Louhivuori, L.M.; Pertovaara, A.; Åkerman, K.; Castrén, E.; Castrén, M.L. Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensorimotor deficits. Genes Brain Behav. 2012, 11, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef] [PubMed]
- Galvez, R.; Greenough, W.T. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am. J. Med. Genet. 2005, 135A, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.W.; Elisseou, N.M.; McKinney, B.C.; Greenough, W.T. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006, 1084, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Chuang, D.-M.; Smith, C.B. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 2011, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Cao, R.; Jafarnejad, S.M.; Prager-Khoutorsky, M.; Giannakas, N.; Kaminari, A.; Fragkouli, A.; Nader, K.; Price, T.J.; et al. Pharmacogenetic Inhibition of eIF4E-Dependent Mmp9 mRNA Translation Reverses Fragile X Syndrome-like Phenotypes. Cell Rep. 2014, 9, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- La Fata, G.; Gärtner, A.; Domínguez-Iturza, N.; Dresselaers, T.; Dawitz, J.; Poorthuis, R.B.; Averna, M.; Himmelreich, U.; Meredith, R.M.; Achsel, T.; et al. FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat. Neurosci. 2014, 17, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Pacey, L.K.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400. [Google Scholar] [CrossRef] [PubMed]
- Pacey, L.K.K.; Xuan, I.C.Y.; Guan, S.; Sussman, D.; Henkelman, R.M.; Chen, Y.; Thomsen, C.; Hampson, D.R. Delayed myelination in a mouse model of fragile X syndrome. Hum. Mol. Genet. 2013, 22, 3920–3930. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed]
- Nosyreva, E.D.; Huber, K.M.; Elena, D.; Metabotropic, K.M. H. Metabotropic Receptor-Dependent Long-Term Depression Persists in the Absence of Protein Synthesis in the Mouse Model of Fragile X Syndrome. J. Neurophysiol. 2006, 95, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.T.; Anstey, J.E.; Golshani, P.; Portera-Cailliau, C. Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 2013, 16, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.Y.; Rotman, Z.; Blundon, J.A.; Cho, Y.; Cui, J.; Cavalli, V.; Zakharenko, S.S.; Klyachko, V.A. FMRP Regulates Neurotransmitter Release and Synaptic Information Transmission by Modulating Action Potential Duration via BK Channels. Neuron 2013, 77, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.; Segal, M. FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb. Cortex 2000, 10, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Selby, L.; Zhang, C.; Sun, Q. Major Defects in Neocortical GABAergic Inhibitory Circuits in Mice Lacking the Fragile X Mental Retardation Protein. Neurosci. Lett. 2007, 412, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.; Yim, S.Y.; Warren, S.T.; Bassell, G.J. Excess PI3K subunit synthesis and activity as a novel therapeutic target in Fragile X Syndrome. Neuroscience 2010, 30, 10624–10638. [Google Scholar] [PubMed]
- Hayashi, M.L.; Rao, B.S.S.; Seo, J.-S.; Choi, H.-S.; Dolan, B.M.; Choi, S.-Y.; Chattarji, S.; Tonegawa, S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11489–11494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-G.; Toyoda, H.; Ko, S.W.; Ding, H.-K.; Wu, L.-J.; Zhou, M. Deficits in Trace Fear Memory and Long-Term Potentiation in a Mouse Model for Fragile X Syndrome. J. Neurosci. 2005, 25, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Pilpel, Y.; Kolleker, A.; Berberich, S.; Ginger, M.; Frick, A.; Mientjes, E.; Oostra, B.A.; Seeburg, P.H. Synaptic ionotropic glutamate receptors and plasticity are developmentally altered in the CA1 field of Fmr1 knockout mice. J. Physiol. 2009, 587, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Seese, R.R.; Babayan, A.H.; Katz, A.M.; Cox, C.D.; Lauterborn, J.C.; Lynch, G.; Gall, C.M. LTP induction translocates cortactin at distant synapses in wild-type but not Fmr1 knock-out mice. J. Neurosci. 2012, 32, 7403–7413. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, U.; Nalavadi, V.; Nakamoto, M.; Thomas, G.; Ceman, S.; Bassell, G.J.; Warren, S.T. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 2008, 283, 18478–18482. [Google Scholar] [CrossRef] [PubMed]
- Bartley, C.M.; O’Keefe, R.A.; Bordey, A. FMRP S499 is phosphorylated independent of mTORC1-S6K1 activity. PLoS ONE 2014, 9, e96956. [Google Scholar]
- Vu, T.H.; Hoffman, A.R. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 1997, 17, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Grier, M.D.; Carson, R.P.; Lagrange, A.H. Toward a broader view of Ube3a in a mouse model of Angelman Syndrome: Expression in brain, spinal cord, sciatic nerve, glial cells. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Bonati, M.T.; Russo, S.; Finelli, P.; Valsecchi, M.R.; Cogliati, F.; Cavalleri, F.; Roberts, W.; Elia, M.; Larizza, L. Evaluation of autism traits in Angelman syndrome: A resource to unfold autism genes. Neurogenetics 2007, 8, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Kumagai, T.; Miura, K.; Miyazaki, S.; Hayakawa, C.; Yamanaka, T. Epilepsy in Angelman Syndrome Associated with Chromosome 15q Deletion. Epilepsia 1992, 33, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Clayton-Smith, J. Clinical research on Angelman syndrome in the United Kingdom: Observations on 82 affected individuals. Am. J. Med. Genet. 1993, 46, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.H.; Bacino, C.A.; Skinner, S.A.; Anselm, I.; Barbieri-Welge, R.; Bauer-Carlin, A.; Beaudet, A.L.; Bichell, T.J.; Gentile, J.K.; Glaze, D.G.; et al. Angelman syndrome: Mutations influence features in early childhood. Am. J. Med. Genet. Part A 2011, 155, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Harting, I.; Seitz, A.; Rating, D.; Sartor, K.; Zschocke, J.; Janssen, B.; Ebinger, F.; Wolf, N.I. Abnormal myelination in Angelman syndrome. Eur. J. Paediatr. Neurol. 2009, 13, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gago, M.; Gómez-Lado, C.; Eirís-Puñal, J. Abnormal myelination in Angelman syndrome. Eur. J. Paediatr. Neurol. 2010, 14, 292. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef]
- Miura, K.; Kishino, T.; Li, E.; Webber, H.; Dikkes, P.; Holmes, G.L.; Wagstaff, J. Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol. Dis. 2002, 9, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Grier, M.D.; Carson, R.P.; Lagrange, A.H. Of mothers and myelin: Aberrant myelination phenotypes in mouse model of Angelman syndrome are dependent on maternal and dietary influences. Behav. Brain Res. 2015, 291, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Dindot, S.V.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008, 17, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.L.; Burette, A.C.; Weinberg, R.J.; Philpot, B.D. Maternal Loss of Ube3a Produces an Excitatory/Inhibitory Imbalance through Neuron Type-Specific Synaptic Defects. Neuron 2012, 74, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Chen, R.; Ye, J.; Tan, G.-H.; Li, S.; Zhang, J.; Jiang, Y.-H.; Xiong, Z.-Q. The Angelman Syndrome Protein Ube3a Is Required for Polarized Dendrite Morphogenesis in Pyramidal Neurons. J. Neurosci. 2013, 33, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Tonazzini, I.; Meucci, S.; van Woerden, G.M.; Elgersma, Y.; Cecchini, M. Impaired Neurite Contact Guidance in Ubiquitin Ligase E3a (Ube3a)-Deficient Hippocampal Neurons on Nanostructured Substrates. Adv. Healthc. Mater. 2016, 5, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Riday, T.T.; Condon, K.H.; Roberts, A.C.; Bernardo, D.R.; Prakash, R.; Weinberg, R.J.; Ehlers, M.D.; Philpot, B.D. Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 2009, 12, 777–783. [Google Scholar] [CrossRef]
- Pignatelli, M.; Piccinin, S.; Molinaro, G.; Di Menna, L.; Riozzi, B.; Cannella, M.; Motolese, M.; Vetere, G.; Catania, M.V.; Battaglia, G.; et al. Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling angelman syndrome. J. Neurosci. 2014, 34, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome Protein Ube3A Regulates Synapse Development by Ubiquitinating Arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Bienvenu, T.; Chelly, J. Molecular genetics of Rett syndrome: When DNA methylation goes unrecognized. Nat. Rev. Genet. 2006, 7, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Cardoza, B.; Clarke, A.; Wilcox, J.; Gibbon, F.; Smith, P.E.M.; Archer, H.; Hryniewiecka-Jaworska, A.; Kerr, M. Epilepsy in Rett syndrome: Association between phenotype and genotype, and implications for practice. Seizure 2011, 20, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Dolce, A.; Ben-Zeev, B.; Naidu, S.; Kossoff, E.H. Rett syndrome and epilepsy: An update for child neurologists. Pediatr. Neurol. 2013, 48, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.; Armstrong, D.; Zoghbi, H.Y.; Percy, A.K.; Boltzmann, L.; Wien, A. Neuropathology of Rett syndrome. Acta Neuropathol. 1988, 76, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, G.; Stenbom, Y.; Witt Engerström, I. Head growth in Rett syndrome. Brain Dev. 2001, 23, 227–229. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L.; Arin, D.M. Microscopic observations of the brain in Rett syndrome. Neuropediatrics 1995, 26, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L.; Kemper, T.L.; Arin, D.M. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology 1995, 45, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Lipani, J.D.; Bhattacharjee, M.B.; Corey, D.M.; Lee, D.A. Reduced nerve growth factor in Rett syndrome postmortem brain tissue. J. Neuropathol. Exp. Neurol. 2000, 59, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; Dunn, J.K.; Antalffy, B.T.R. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 1995, 54, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.D. Neuropathology of Rett syndrome. J. Child Neurol. 2005, 20, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, P.V.; Oldfors, A.; Hagberg, B.; Dahlström, A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 1994, 5, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Taylor, C.; Hohmann, C.; Sanwal, I.; Naidu, S. Abnormalities in neuronal maturation in Rett syndrome neocortex: Preliminary molecular correlates. Eur. Child Adolesc. Psychiatry 1997, 6, 75–77. [Google Scholar]
- Chapleau, C.A.; Larimore, J.L.; Theibert, A.; Pozzo-Miller, L. Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: Fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J. Neurodev. Disord. 2009, 1, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Young, J.I.; Yuva-Paylor, L.A.; Spencer, C.M.; Antalffy, B.A.; Noebels, J.L.; Armstrong, D.L.; Paylor, R.; Zoghbi, H.Y. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002, 35, 243–254. [Google Scholar] [CrossRef]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Yamashita, Y.; Nagamitsu, S.; Miyamoto, K.; Jin, J.J.; Ohmori, I.; Ohtsuka, Y.; Kuwajima, K.; Endo, S.; Iwai, T.; et al. Methyl-CpG binding protein 2 gene (MECP2) variations in Japanese patients with Rett syndrome: Pathological mutations and polymorphisms. Brain Dev. 2005, 27, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; McCurry, C.; Fu, D.D.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.V.C.; Du, F.; Felice, C.A.; Shan, X.; Nigam, A.; Mandel, G.; Robinson, J.K.; Ballas, N. MeCP2 is critical for maintainng mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 2012, 32, 10021–10034. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Garcia, R.I.; Kwok, S.; Banerjee, A.; Petravicz, J.; Woodson, J.; Mellios, N.; Tropea, D.; Sur, M. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9941–9946. [Google Scholar] [CrossRef] [PubMed]
- Baj, G.; Patrizio, A.; Montalbano, A.; Sciancalepore, M.; Tongiorgi, E. Developmental and maintenance defects in Rett syndrome neurons identified by a new mouse staging system in vitro. Front. Cell. Neurosci. 2014, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Asaka, Y.; Jugloff, D.G.M.; Zhang, L.; Eubanks, J.H.; Fitzsimonds, R.M. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 2006, 21, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P. Learning and Memory and Synaptic Plasticity Are Impaired in a Mouse Model of Rett Syndrome. J. Neurosci. 2006, 26, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 Controls Excitatory Synaptic Strength by Regulating Glutamatergic Synapse Number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Ballas, N.; Lioy, D.T.; Grunseich, C.; Mandel, G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 2009, 12, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Swanberg, S.; Harvey, D.; LaSalle, J.M.; Jin, L.-W. Rett Syndrome Astrocytes Are Abnormal and Spread MeCP2 Deficiency through Gap Junctions. J. Neurosci. 2009, 29, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Kishi, N.; Macklis, J.D. MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp. Neurol. 2010, 222, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic- stem-cell-derived rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.C.; Zhong, X.; Mohamed, A.; Li, R.; Liu, Y.; Dong, Q.; Ananiev, G.E.; Choongmok, J.C.; Lin, B.R.; Lu, J.; et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wildtype neurons. Hum. Mol. Genet. 2014, 23, 2968–2980. [Google Scholar] [CrossRef] [PubMed]
- Djuric, U.; Cheung, A.Y.L.; Zhang, W.; Mok, R.S.; Lai, W.; Piekna, A.; Hendry, J.A.; Ross, P.J.; Pasceri, P.; Kim, D.S.; et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 2015, 76, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-N.; Freitas, B.C.; Qian, H.; Lux, J.; Acab, A.; Trujillo, C.A.; Herai, R.H.; Nguyen Huu, V.A.; Wen, J.H.; Joshi-Barr, S.; et al. Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, 3185–3190. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or Atypical Autism in Maternally but Not Paternally Derived Proximal 15q Duplication. Am. J. Hum. Genet. 1997, 60, 928–934. [Google Scholar] [PubMed]
- Cook, E.H.; Scherer, S.W. Copy-number variations associated with neuropsychiatric conditions. Nature 2008, 455, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.; Waters, M.; Williams, C.; Zori, R.; Glenn, C.; Avidano, K.; Nicholls, R. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics 1992, 13, 917–924. [Google Scholar] [CrossRef]
- Smith, S.E. P.; Zhou, Y.-D.; Zhang, G.; Jin, Z.; Stoppel, D.C.; Anderson, M.P. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 2011, 3, 103ra97. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, J.; Tamada, K.; Hatanaka, F.; Ise, S.; Ohta, H.; Inoue, K.; Tomonaga, S.; Watanabe, Y.; Chung, Y.J.; Banerjee, R.; et al. Abnormal Behavior in a Chromosome- Engineered Mouse Model for Human 15q11-13 Duplication Seen in Autism. Cell 2009, 137, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, M.; Tanaka, S.; Kuriu, T.; Tabuchi, K.; Takumi, T.; Okabe, S. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun. 2014, 5, 4742. [Google Scholar] [CrossRef] [PubMed]
- Piochon, C.; Kloth, A.D.; Grasselli, G.; Titley, H.K.; Nakayama, H.; Hashimoto, K.; Wan, V.; Simmons, D.H.; Eissa, T.; Nakatani, J.; et al. Masanobu Kano8, Samuel S-H Wang2, 3, and C.H. Cerebellar Plasticity and Motor Learning Deficits in a Copy Number Variation Mouse Model of Autism. Nat. Commun. 2014, 5, 5586. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.; Sikkema-Raddatz, B.; Ruijvenkamp, C.A.L.; Dijkhuizen, T.; Bijlsma, E.K.; Gijsbers, A.C.J.; Hilhorst-Hofstee, Y.; Hordijk, R.; Verbruggen, K.T.; Kerstjens-Frederikse, W.S.; et al. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur. J. Med. Genet. 2009, 52, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zwaag, B.; Staal, W.G.; Hochstenbach, R.; Poot, M.; Spierenburg, H.; De Jonge, M.V.; Verbeek, N.E.; van ’t Slot, R.; van Es, M.A.; Staal, F.J.; et al. A co-segregating microduplication of chromosome 15q11.2 pinpoints two risk genes for autism spectrum disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 153, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I.; Mercaldo, V.; Boyl, P.P.; Eleuteri, B.; Zalfa, F.; De Rubeis, S.; Di Marino, D.; Mohr, E.; Massimi, M.; Falconi, M.; et al. The Fragile X Syndrome Protein Represses Activity-Dependent Translation through CYFIP1, a New 4E-BP. Cell 2008, 134, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kuroda, S.; Fukata, M.; Nakamura, T.; Nagase, T.; Nomura, N.; Matsuura, Y.; Yoshida-Kubomura, N.; Iwamatsu, A.; Kaibuchi, K. p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J. Biol. Chem. 1998, 273, 291–295. [Google Scholar] [CrossRef] [PubMed]
- DeRubeis, S.; Pasciuto, E.; Li, K.W.; Fernández, E.; DiMarino, D.; Buzzi, A.; Ostroff, L.E.; Klann, E.; Zwartkruis, F.J.T.; Komiyama, N.H.; Grant, S.G.N.; et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic Spine formation. Neuron 2013, 79, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Martin, C.L.; Vazquez-Lopez, A.; Spence, S.J.; Alvarez-Retuerto, A.I.; Sigman, M.; Steindler, C.; Pellegrini, S.; Schanen, N.C.; Warren, S.T.; et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum. Mol. Genet. 2007, 16, 1682–1698. [Google Scholar] [CrossRef] [PubMed]
- Pathania, M.; Davenport, E.C.; Muir, J.; Sheehan, D.F.; López-Doménech, G.; Kittler, J.T. The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl. Psychiatry 2014, 4, e374. [Google Scholar] [CrossRef] [PubMed]
- Spence, S.J.; Schneider, M.T. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr. Res. 2009, 65, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Alessandri, M.; Cuccaro, M. Autism spectrum disorders and epilepsy: Moving towards a comprehensive approach to treatment. Brain Dev. 2010, 32, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Pierce, K.; Schumann, C.M.; Redcay, E.; Buckwalter, J.A.; Kennedy, D.P.; Morgan, J.T. Mapping early brain development in autism. Neuron 2007, 56, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Campbell, K.; Solso, S. Brain growth across the life span in autism: Age-specific changes in anatomical pathology. Brain Res. 2011, 1380, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Pickett, J. The Neuropathology of Autism. In Imaging the Brain in Autism; Casanova, M., El-Baz, A., Suri, J., Eds.; Springer: New York, NY, USA, 2013; pp. 27–41. [Google Scholar]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, M.; Nielson, D.M.; Lenroot, R.K.; Ostuni, J.L.; Luckenbaugh, D.A.; Thurm, A.E.; Giedd, J.N.; Swedo, S.E. A magnetization transfer imaging study of corpus callosum myelination in young children with autism. Biol. Psychiatry 2012, 72, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Deoni, S.C. L.; Dean, D.C.; Remer, J.; Dirks, H.; O’Muircheartaigh, J. Cortical maturation and myelination in healthy toddlers and young children. Neuroimage 2015, 115, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.H.; Janzarik, W.G.; Delvendahl, I.; Münchau, A.; Biscaldi, M.; Mainberger, F.; Bäumer, T.; Rauh, R.; Mall, V. Impaired induction of long-term potentiation-like plasticity in patients with high-functioning autism and Asperger syndrome. Dev. Med. Child Neurol. 2012, 55, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Capal, J.K.; Franz, D.N. Profile of everolimus in the treatment of tuberous sclerosis complex: An evidence-based review of its place in therapy. Neuropsychiatr. Dis. Treat. 2016, 12, 2165–2172. [Google Scholar] [PubMed]
- Angliker, N.; Rüegg, M. In vivo evidence for mTORC2-mediated actin cytoskeleton rearrangement in Neurons. Bioarchitecture 2013, 3, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crino, P.B. The mTOR signalling cascade: Paving new roads to cure neurological disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Phenotypes | ASD-Related Syndromes | Nonsyndromic or Idiopathic ASD | |||||||
|---|---|---|---|---|---|---|---|---|---|
| TSC | PTEN | NF1 | FXS | AS | RTT | Dup15q | eIF4E-ASD | Idiopathic | |
| mTORC1 signaling | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | ↑ | ↑ | ↑/↓ |
| Seizures | Present *,1 | Present *,1 | Present | Present | Present | Present | Present | ND | Present |
| Brain size | ↑ *,1 | ↑ *,1 | ↑ | ↑ | ↓ | ↓ | ↓ | ND | ↑ |
| Neuron size | ↑ *,1 | ↑ *,1,3 | ND | ↑/↓/normal | ND | ↓ *,5,6 | ↑ *,1 (Cyfip1) | ND | ↓/normal |
| Neuronal migration | Abnormal *,1 | Abnormal *,1 | Abnormal | Abnormal | ND | Abnormal | Abnormal | ND | Abnormal |
| Neurite arborization | ↑ *,1 | ↑ *,3 | ND | ↓/normal | ↓ | ↓ *,5,6 | ↑ *,1 (Cyfip1) | ND | ND |
| Neurite length | ↑/normal | ↑ | ↓ | ↓ | ↓ | ↓ | ↑/↓ *,1 (Cyfip1) | ND | ND |
| Spine density | ↑ *,1/↓ *,1/normal | ↑ *,1 | ↓ | ↑/↓/normal | ↓ *,1 | ↓ | ↑ * (Cyfip1) | ↑ | ↑ |
| Spine length | ↑/↓ *,1/normal | ↑ | ND | ↑/↓/normal | ↓ | ND | ND | ND | ND |
| Immature spine morphology | ↓ *,1/↑ | ↑/↓ | ND | ↑ *,3,4 | Abnormal *,1 | ↑ | ↓ (Cyfip1) | ND | ND |
| LTP | ↓ | ↑/↓ | ↓ | ↑/↓ | ↓ *,1,3 | ↓ | ↓ (patDp/+) | ↑ *,7 | ↓ |
| mGluR-LTD | ↓ *,1 | ↓ | ND | ↑ *,3,4 | ↑ | ND | ND | ↑ *,7 | ND |
| Protein synthesis | ↓ *,1/↑ *,1 | ↑ | ↑ *,1 | ↑ *,1,2,3,4 | ND | ↓ *,5,6 | ND | ↑ *,7 | ↑ *,8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magdalon, J.; Sánchez-Sánchez, S.M.; Griesi-Oliveira, K.; Sertié, A.L. Dysfunctional mTORC1 Signaling: A Convergent Mechanism between Syndromic and Nonsyndromic Forms of Autism Spectrum Disorder? Int. J. Mol. Sci. 2017, 18, 659. https://doi.org/10.3390/ijms18030659
Magdalon J, Sánchez-Sánchez SM, Griesi-Oliveira K, Sertié AL. Dysfunctional mTORC1 Signaling: A Convergent Mechanism between Syndromic and Nonsyndromic Forms of Autism Spectrum Disorder? International Journal of Molecular Sciences. 2017; 18(3):659. https://doi.org/10.3390/ijms18030659
Chicago/Turabian StyleMagdalon, Juliana, Sandra M. Sánchez-Sánchez, Karina Griesi-Oliveira, and Andréa L. Sertié. 2017. "Dysfunctional mTORC1 Signaling: A Convergent Mechanism between Syndromic and Nonsyndromic Forms of Autism Spectrum Disorder?" International Journal of Molecular Sciences 18, no. 3: 659. https://doi.org/10.3390/ijms18030659