
Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis

 and
and
Abstract
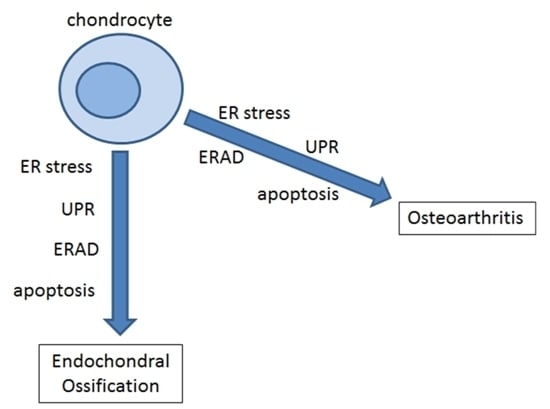
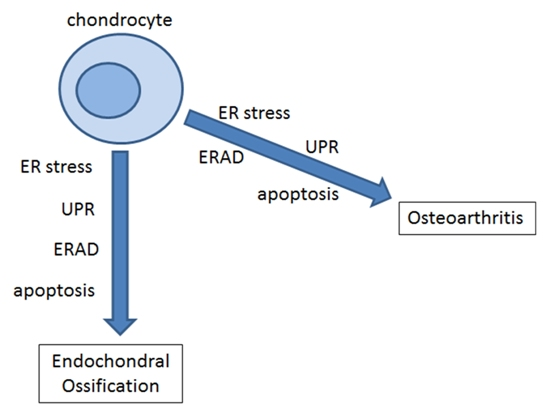
:
1. Introduction
2. Aetiology of Chondrodystrophies with Higher Risk of Osteoarthritis
3. Individuals with Chondrodystrophies Frequently Develop Early-Onset Osteoarthritis
4. A Role for the Unfolded Protein Response in Chondrodystrophies and Early Onset OA
5. Collagen Biosynthesis and the Unfolded Protein Response in Chondrodysplasia
6. The Unfolded Protein Response Has an Essential Role in Chondrogenesis
7. Osteoblast Physiology and Altered Bone Mineralization in Chondrodystrophies and Early Onset OA
8. Stickler Syndrome
9. Therapeutic Targets for the Modulation of UPR in Cartilage Pathophysiology
10. Discussion
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end-products |
| ALP | Alkaline phosphatase |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| BBF2H7 | BBF2 human homolog on chromosome 7 |
| BMP2 | Bone morphogenetic protein 2 |
| BSP | Bone sialoprotein |
| cho | Chondrodysplasia |
| CHOP | C/EBP homologous protein |
| COMP | Cartilage oligomeric matrix protein |
| CypB | Cyclophilin B |
| DMM | Disproportionate micromelia mouse |
| DDR2 | Discoidin domain receptor-2 |
| ECM | Extracellular matrix |
| ER | Endoplasmic reticulum |
| FACIT | Fibril-associated collagen with interrupted triple helices |
| GRP78 | Glucose-regulated protein, 78 kDa (also known as BiP/HSPA5) |
| HtrA1 | High temperature requirement protease A-1 |
| IHH | Indian hedgehog |
| IRE1 | Inositol-requiring endonuclease 1 |
| LH | Lysyl hydroxylases |
| MCDS | Metaphyseal chondrodysplasia, type Schmid |
| MED | Multiple epiphyseal dysplasia |
| MMP-13 | Matrix metalloproteinase-13 |
| OA | Osteoarthritis |
| OSX | Osterix (Osx/Sp7) |
| P3Hs | Prolyl 3-hydroxylases |
| P4Hs | Prolyl 4-hydroxylases |
| PERK | PKR-like endoplasmic reticulum kinase |
| PDI | Protein disulfide isomerase |
| PSACH | Pseudoachondroplasia |
| PTHrP | Parathyroid hormone related peptide |
| RAGE | Receptor for AGEs |
| RER | Rough endoplasmic reticulum |
| UPR | Unfolded protein response |
| VR | Variable region |
| XBP1 | X-box binding protein 1 |
| XBP1S | Spliced X-box binding protein 1 |
| XBP1U | Unspliced X-box binding protein 1 |
References
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.N.; Oxford, J.T. A lipid-rich gestational diet predisposes offspring to nonalcoholic fatty liver disease: A potential sequence of events. Hepat. Med. 2014, 6, 15–23. [Google Scholar] [PubMed]
- Koch, M.; Laub, F.; Zhou, P.; Hahn, R.A.; Tanaka, S.; Burgeson, R.E.; Gerecke, D.R.; Ramirez, F.; Gordon, M.K. Collagen XXIV, a vertebrate fibrillar collagen with structural features of invertebrate collagens: Selective expression in developing cornea and bone. J. Biol. Chem. 2003, 278, 43236–43244. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.M.; Corrado, M.; Missero, C.; Byers, P.H. Identification, characterization and expression analysis of a new fibrillar collagen gene, COL27A1. Matrix Biol. 2003, 22, 3–14. [Google Scholar] [CrossRef]
- Jenkins, E.; Moss, J.B.; Pace, J.M.; Bridgewater, L.C. The new collagen gene COL27A1 contains SOX9-responsive enhancer elements. Matrix Biol. 2005, 24, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Jacob, R.; McDougal, O.; Oxford, J.T. Minor fibrillar collagens, variable regions alternative splicing, intrinsic disorder, and tyrosine sulfation. Protein Cell 2012, 3, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Hamanaka, R.; Nakamura-Ota, M.; Adachi, S.; Zhang, J.J.; Matsuo, N.; Yoshioka, H. Sp7/Osterix induces the mouse pro-α2(I) collagen gene (Col1a2) expression via the proximal promoter in osteoblastic cells. Biochem. Biophys. Res. Commun. 2014, 452, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Plumb, D.A.; Dhir, V.; Mironov, A.; Ferrara, L.; Poulsom, R.; Kadler, K.E.; Thornton, D.J.; Briggs, M.D.; Boot-Handford, R.P. Collagen XXVII is developmentally regulated and forms thin fibrillar structures distinct from those of classical vertebrate fibrillar collagens. J. Biol. Chem. 2007, 282, 12791–12795. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga-Jauregui, C.; Gamble, C.N.; Yuan, B.; Penney, S.; Jhangiani, S.; Muzny, D.M.; Gibbs, R.A.; Lupski, J.R.; Hecht, J.T. Mutations in COL27A1 cause Steel syndrome and suggest a founder mutation effect in the Puerto Rican population. Eur. J. Hum. Genet. 2015, 23, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Mendler, M. Cartilage contains mixed fibrils of collagen types II, IX, and XI. J. Cell Biol. 1989, 108, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D. Collagen of articular cartilage. Arthritis Res. 2002, 4, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imagawa, K.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Otero, M.; Roach, H.I.; Goldring, M.B.; Oreffo, R.O.C. Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis Rheumatol. 2014, 66, 3040–3051. [Google Scholar] [CrossRef] [PubMed]
- Jakkula, E.; Melkoniemi, M.; Kiviranta, I.; Lohiniva, J.; Räinä, S.S.; Perälä, M.; Warman, M.L.; Ahonen, K.; Kröger, H.; Göring, H.H.H.; et al. The role of sequence variations within the genes encoding collagen II, IX and XI in non-syndromic, early-onset osteoarthritis. Osteoarthr. Cartil. 2005, 13, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, G.; Snoeckx, R.L.; Hilgert, N.; van den Ende, J.; Fukuoka, H.; Wagatsuma, M.; Suzuki, H.; Smets, R.M.E.; Vanhoenacker, F.; Declau, F.; et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am. J. Hum. Genet. 2006, 79, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Couchouron, T.; Masson, C. Early-onset progressive osteoarthritis with hereditary progressive ophtalmopathy or Stickler syndrome. Jt. Bone Spine 2011, 78, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Steenvoorden, M.M.C.; Huizinga, T.W.J.; Verzijl, N.; Bank, R.A.; Ronday, H.K.; Luning, H.A.F.; Lafeber, F.P.J.G.; Toes, R.E.M.; DeGroot, J. Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 2006, 54, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Nah, S.-S.; Choi, I.-Y.; Yoo, B.; Kim, Y.G.; Moon, H.-B.; Lee, C.-K. Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13, and TNF-α in human osteoarthritic chondrocytes. FEBS Lett. 2007, 581, 1928–1932. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Haqqi, T.M. Endoplasmic reticulum stress induces the expression of COX-2 through activation of eIF2α, p38-MAPK and NF-κB in advanced glycation end products stimulated human chondrocytes. Biochim. Biophys. Acta 2012, 1823, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, C.; Farmaki, E.; Spilioti, E.; Kiaris, H.; Piperi, C.; Papavassiliou, A.G. Advanced glycation end-products induce endoplasmic reticulum stress in human aortic endothelial cells. Clin. Chem. Lab. Med. 2014, 52, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamabe, S.; Hirose, J.; Uehara, Y.; Okada, T.; Okamoto, N.; Oka, K.; Taniwaki, T.; Mizuta, H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013, 280, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, D.; Xiao, Y.; Zhou, X.; Wang, L.; Chen, B.; Li, Q.; Guo, X.; Huang, Q. Endoplasmic reticulum stress plays a role in the advanced glycation end product-induced inflammatory response in endothelial cells. Life Sci. 2014, 110, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Larkin, D.J.; Kartchner, J.Z.; Doxey, A.S.; Hollis, W.R.; Rees, J.L.; Wilhelm, S.K.; Draper, C.S.; Peterson, D.M.; Jackson, G.G.; Ingersoll, C.; et al. Inflammatory markers associated with osteoarthritis after destabilization surgery in young mice with and without Receptor for Advanced Glycation End-products (RAGE). Front. Physiol. 2013, 4, 121. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M.; Plumb, D.A.; Dragomir, C.; Favero, M.; El Hachem, K.; Hashimoto, K.; Roach, H.I.; Olivotto, E.; Borzì, R.M.; et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: Signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cell Mater. 2011, 21, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lacerda, D.; Warman, M.; Beier, D.; Yoshioka, H.; Ninomiya, Y.; Oxford, J.; Morris, N.; Andrikopoulos, K.; Ramirez, F.; et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell 1995, 80, 423–430. [Google Scholar] [CrossRef]
- Xu, L.; Peng, H.; Wu, D.; Hu, K.; Goldring, M.B.; Olsen, B.R.; Li, Y. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J. Biol. Chem. 2005, 280, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Raducanu, A.; Aszódi, A. Knock-Out Mice in Osteoarthritis Research. Curr. Rheumatol. Rev. 2008, 4, 183–192. [Google Scholar] [CrossRef]
- Ricks, M.L.; Farrell, J.T.; Falk, D.J.; Holt, D.W.; Rees, M.; Carr, J.; Williams, T.; Nichols, B.A.; Bridgewater, L.C.; Reynolds, P.R.; et al. Osteoarthritis in temporomandibular joint of Col2a1 mutant mice. Arch. Oral Biol. 2013, 58, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.N. Stickler Syndrome. Arch. Ophthalmol. 1995, 113, 1454. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.M.; Li, Y.; Seegmiller, R.E.; Teuscher, C.; Taylor, B.A.; Olsen, B.R. Disproportionate micromelia (Dmm) in mice caused by a mutation in the C-propeptide coding region of COL2A1. Dev. Dyn. 1997, 208, 25–33. [Google Scholar] [CrossRef]
- Fernandes, R.J.; Seegmiller, R.E.; Nelson, W.R.; Eyre, D.R. Protein consequences of the COL2A1 C-propeptide mutation in the chondrodysplastic Dmm mouse. Matrix Biol. 2003, 22, 449–453. [Google Scholar] [CrossRef]
- Uehara, Y.; Hirose, J.; Yamabe, S.; Okamoto, N.; Okada, T.; Oyadomari, S.; Mizuta, H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthr. Cartil. 2014, 22, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.V.; Fertala, A. Skeletal diseases caused by mutations that affect collagen structure and function. Int. J. Biochem. Cell Biol. 2013, 45, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, J.J.; May, K.L.; Thorpe, C.M.; Jandhyala, D.M.; Paton, J.C.; Paton, A.W. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell. Microbiol. 2008, 10, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Kung, L.H.W.; Rajpar, M.H.; Preziosi, R.; Briggs, M.D.; Boot-Handford, R.P. Increased Classical Endoplasmic Reticulum Stress Is Sufficient to Reduce Chondrocyte Proliferation Rate in the Growth Plate and Decrease Bone Growth. PLoS ONE 2015, 10, e0117016. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.E.; Dealy, C.N. Mechanisms and models of endoplasmic reticulum stress in chondrodysplasia. Dev. Dyn. 2014, 243, 875–893. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [PubMed]
- Fuest, M.; Willim, K.; MacNelly, S.; Fellner, N.; Resch, G.P.; Blum, H.E.; Hasselblatt, P. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology 2012, 55, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Singha, P.K.; Venkatachalam, M.A.; Saikumar, P. A novel role for MAP1 LC3 in nonautophagic cytoplasmic vacuolation death of cancer cells. Oncogene 2009, 28, 2556–2568. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-H.; Lim, S.-C. Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung ep. J. Pharmacol. Exp. Ther. 2009, 329, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Thiele, F.; Fuchs, H.; Hans, W.; Przemeck, G.K.H.; Abe, K.; Rathkolb, B.; Quintanilla-Martinez, L.; Hoelzlwimmer, G.; Helfrich, M.; et al. ER stress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet. 2008, 4, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Lian, C.; Huang, D.; Gao, W.; Liang, A.; Peng, Y.; Ye, W.; Wu, Z.; Su, P.; Huang, D. Endoplasmic reticulum stress-unfolding protein response-apoptosis cascade causes chondrodysplasia in a COL2A1 p.Gly1170Ser mutated mouse model. PLoS ONE 2014, 9, e86894. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.T.; Schwarze, U.; Goldstein, J.; Byers, P.H. Mutations in the COL3A1 gene result in the Ehlers-Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. J. Investig. Dermatol. 1997, 108, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; de Backer, J.; Vanakker, O.; Coucke, P.; de Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.Y.; Chan, D.; Cheslett, D.; Chan, W.C.W.; So, C.L.; Melhado, I.G.; Chan, T.W.Y.; Kwan, K.M.; Hunziker, E.B.; Yamada, Y.; et al. Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 2007, 5, e44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Bächinger, H.P. A molecular ensemble in the rER for procollagen maturation. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; Skach, W.R. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell Biol. 2011, 23, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Makareeva, E.; Aviles, N.A.; Leikin, S. Chaperoning osteogenesis: New protein-folding disease paradigms. Trends Cell Biol. 2011, 21, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Boudko, S.; Bächinger, H.P. Ziploc-ing the structure: Triple helix formation is coordinated by rough endoplasmic reticulum resident PPIases. Biochim. Biophys. Acta 2015, 1850, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Townley, A.K.; Feng, Y.; Schmidt, K.; Carter, D.A.; Porter, R.; Verkade, P.; Stephens, D.J. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J. Cell Sci. 2008, 121, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Katada, T. Mechanisms for exporting large-sized cargoes from the endoplasmic reticulum. Cell. Mol. Life Sci. 2015, 72, 3709–3720. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K. HSP47 as a collagen-specific molecular chaperone: Function and expression in normal mouse development. Semin. Cell Dev. Biol. 2003, 14, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.-G.; Kim, E.-J.; Kim, D.-K.; Ryoo, H.-M.; Lee, K.-B.; Kim, S.-H.; Choi, H.-S.; Koh, J.-T. BMP2 protein regulates osteocalcin expression via Runx2-mediated Atf6 gene transcription. J. Biol. Chem. 2012, 287, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Zhang, M.; Xie, R.; Wang, M.; Jin, H.; Hou, W.; Tang, D.; Harris, S.E.; Mishina, Y.; O’Keefe, R.J.; et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 2011, 124, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lian, N.; Li, L.; Moss, H.E.; Wang, W.; Perrien, D.S.; Elefteriou, F.; Yang, X. Atf4 regulates chondrocyte proliferation and differentiation during endochondral ossification by activating Ihh transcription. Development 2009, 136, 4143–4153. [Google Scholar] [CrossRef] [PubMed]
- Cameron, T.L.; Gresshoff, I.L.; Bell, K.M.; Piróg, K.A.; Sampurno, L.; Hartley, C.L.; Sanford, E.M.; Wilson, R.; Ermann, J.; Boot-Handford, R.P.; et al. Cartilage-Specific Ablation of XBP1 Signaling in Mouse Results in a Chondrodysplasia Characterized by Reduced Chondrocyte Proliferation and Delayed Cartilage Maturation and Mineralization. Osteoarthr. Cartil. 2015, 23, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Hino, S.; Murakami, T.; Kanemoto, S.; Kondo, S.; Saitoh, M.; Nishimura, R.; Yoneda, T.; Furuichi, T.; Ikegawa, S.; et al. Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat. Cell Biol. 2009, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-J.; Xiong, Z.; Han, X.; Liu, C.; Liu, Y.; Jiang, R.; Zhang, P. XBP1S, a BMP2-inducible transcription factor, accelerates endochondral bone growth by activating GEP growth factor. J. Cell. Mol. Med. 2014, 18, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534. [Google Scholar] [PubMed]
- Li, T.-F.; Yukata, K.; Yin, G.; Sheu, T.; Maruyama, T.; Jonason, J.H.; Hsu, W.; Zhang, X.; Xiao, G.; Konttinen, Y.T.; et al. BMP-2 induces ATF4 phosphorylation in chondrocytes through a COX-2/PGE2 dependent signaling pathway. Osteoarthr. Cartil. 2014, 22, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Kamekura, S.; Mabuchi, A.; Kou, I.; Seki, S.; Takato, T.; Nakamura, K.; Kawaguchi, H.; Ikegawa, S.; Chung, U. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004, 50, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Zehentner, B.K.; Dony, C.; Burtscher, H. The transcription factor Sox9 is involved in BMP-2 signaling. J. Bone Miner. Res. 1999, 14, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.-C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef] [PubMed]
- Hida, M.; Hamanaka, R.; Okamoto, O.; Yamashita, K.; Sasaki, T.; Yoshioka, H.; Matsuo, N. Nuclear factor Y (NF-Y) regulates the proximal promoter activity of the mouse collagen α1(XI) gene (Col11a1) in chondrocytes. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Saito, A.; Kido, M.; Kanemoto, S.; Asada, R.; Takai, T.; Cui, M.; Cui, X.; Imaizumi, K. Master regulator for chondrogenesis, Sox9, regulates transcriptional activation of the endoplasmic reticulum stress transducer BBF2H7/CREB3L2 in chondrocytes. J. Biol. Chem. 2014, 289, 13810–13820. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Kanemoto, S.; Zhang, Y.; Asada, R.; Hino, K.; Imaizumi, K. Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol. Cell 2014, 53, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.R.; Lapierre, L.A.; Frotscher, M.; Goldenring, J.R.; Knapik, E.W. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat. Genet. 2006, 38, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Izumi, S.; Saito, A.; Kanemoto, S.; Kawasaki, N.; Asada, R.; Iwamoto, H.; Oki, M.; Miyagi, H.; Ochi, M.; Imaizumi, K. The endoplasmic reticulum stress transducer BBF2H7 suppresses apoptosis by activating the ATF5-MCL1 pathway in growth plate cartilage. J. Biol. Chem. 2012, 287, 36190–36200. [Google Scholar] [CrossRef] [PubMed]
- Cameron, T.L.; Bell, K.M.; Tatarczuch, L.; Mackie, E.J.; Rajpar, M.H.; McDermott, B.T.; Boot-Handford, R.P.; Bateman, J.F. Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS ONE 2011, 6, e24600. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Soegiarto, D.W.; Yang, Y.; Lanske, B.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Investig. 2005, 115, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, Q.; Lanske, B.; Fleming, B.C.; Terek, R.; Wei, X.; Zhang, G.; Wang, S.; Li, K.; Wei, L. Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1-CreERT2; Ihhfl/fl mice. Arthritis Res. Ther. 2014, 16, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Ali, A.S.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhou, J.; Wei, X.; Zhang, J.; Fleming, B.C.; Terek, R.; Pei, M.; Chen, Q.; Liu, T.; Wei, L. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthr. Cartil. 2012, 20, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Acke, F.R.; Dhooge, I.J.; Malfait, F.; de Leenheer, E.M. Hearing impairment in Stickler syndrome: A systematic review. Orphanet J. Rare Dis. 2012, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, A.J.; Baguley, D.M.; Yates, J.R.; Lane, C.; Nicol, M.; Harper, P.S.; Scott, J.D.; Snead, M.P. Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen Gly-X-Y triple helix. Am. J. Hum. Genet. 2000, 67, 1083–1094. [Google Scholar] [PubMed]
- Lincoln, J.; Florer, J.B.; Deutsch, G.H.; Wenstrup, R.J.; Yutzey, K.E. ColVa1 and ColXIa1 are required for myocardial morphogenesis and heart valve development. Dev. Dyn. 2006, 235, 3295–3305. [Google Scholar] [CrossRef] [PubMed]
- Kahler, R.A.; Yingst, S.M.C.; Hoeppner, L.H.; Jensen, E.D.; Krawczak, D.; Oxford, J.T.; Westendorf, J.J. Collagen 11a1 is indirectly activated by lymphocyte enhancer-binding factor 1 (Lef1) and negatively regulates osteoblast maturation. Matrix Biol. 2008, 27, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Paic, F.; Igwe, J.C.; Nori, R.; Kronenberg, M.S.; Franceschetti, T.; Harrington, P.; Kuo, L.; Shin, D.-G.; Rowe, D.W.; Harris, S.E.; et al. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone 2009, 45, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Cocco, G.; Gasparyan, A.Y. A case report of a patient with Ribbing disease underlines the connections between the skeletal and cardiovascular complications. Clin. Pract. 2011, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Al Kaissi, A.; Roschger, P.; Nawrot-Wawrzyniak, K.; Krebs, A.; Grill, F.; Klaushofer, K. Evidence of reduced bone turnover and disturbed mineralization process in a boy with Stickler syndrome. Calcif. Tissue Int. 2010, 86, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Hafez, A.; Squires, R.; Pedracini, A.; Joshi, A.; Seegmiller, R.E.; Oxford, J.T. Col11a1 Regulates Bone Microarchitecture during Embryonic Development. J. Dev. Biol. 2015, 3, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Matsui, Y.; Fernandes, R.J.; Hanson, D.A.; Kubo, T.; Yukata, K.; Michigami, T.; Komori, T.; Fujita, T.; Yang, L.; et al. Sp1 family of transcription factors regulates the human α2 (XI) collagen gene (COL11A2) in Saos-2 osteoblastic cells. J. Bone Miner. Res. 2006, 21, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Furmanek, T.; Kryvi, H.; Krossøy, C.; Totland, G.K.; Grotmol, S.; Wargelius, A. Transcriptome sequencing of Atlantic salmon (Salmo salar L.) notochord prior to development of the vertebrae provides clues to regulation of positional fate, chordoblast lineage and mineralisation. BMC Genom. 2014, 15, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.J.; Iida, K.; Egusa, H.; Hokugo, A.; Jewett, A.; Nishimura, I. Trabecular bone deterioration in col9a1+/− mice associated with enlarged osteoclasts adhered to collagen IX-deficient bone. J. Bone Miner. Res. 2008, 23, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Jobling, R.; D’Souza, R.; Baker, N.; Lara-Corrales, I.; Mendoza-Londono, R.; Dupuis, L.; Savarirayan, R.; Ala-Kokko, L.; Kannu, P. The collagenopathies: Review of clinical phenotypes and molecular correlations. Curr. Rheumatol. Rep. 2014, 16, 394. [Google Scholar] [CrossRef] [PubMed]
- Al Kaissi, A.; Chehida, F.B.; Ganger, R.; Kenis, V.; Zandieh, S.; Hofstaetter, J.G.; Klaushofer, K.; Grill, F. Radiographic and tomographic analysis in patients with stickler syndrome type I. Int. J. Med. Sci. 2013, 10, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Sakao, K.; Takahashi, K.A.; Arai, Y.; Saito, M.; Honjo, K.; Hiraoka, N.; Asada, H.; Shin-Ya, M.; Imanishi, J.; Mazda, O.; et al. Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J. Bone Miner. Metab. 2009, 27, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The Novel Zinc Finger-Containing Transcription Factor Osterix Is Required for Osteoblast Differentiation and Bone Formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [PubMed]
- Tohmonda, T.; Miyauchi, Y.; Ghosh, R.; Yoda, M.; Uchikawa, S.; Takito, J.; Morioka, H.; Nakamura, M.; Iwawaki, T.; Chiba, K.; et al. The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011, 12, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-H.; Park, S.-Y.; de Crombrugghe, B.; Kim, J.-E. Chondrocyte-specific ablation of Osterix leads to impaired endochondral ossification. Biochem. Biophys. Res. Commun. 2012, 418, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Saito, A.; Hino, S.; Kondo, S.; Kanemoto, S.; Chihara, K.; Sekiya, H.; Tsumagari, K.; Ochiai, K.; Yoshinaga, K.; et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 2009, 11, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Robin, N.H.; Moran, R.T.; Ala-Kokko, L. Stickler Syndrome; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hoornaert, K.P.; Vereecke, I.; Dewinter, C.; Rosenberg, T.; Beemer, F.A.; Leroy, J.G.; Bendix, L.; Björck, E.; Bonduelle, M.; Boute, O.; et al. Stickler syndrome caused by COL2A1 mutations: Genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. 2010, 18, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.J.; McNinch, A.; Martin, H.; Oakhill, K.; Rai, H.; Waller, S.; Treacy, B.; Whittaker, J.; Meredith, S.; Poulson, A.; et al. Stickler syndrome and the vitreous phenotype: Mutations in COL2A1 and COL11A1. Hum. Mutat. 2010, 31, E1461–E1471. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.J.; Fincham, G.S.; McNinch, A.; Hill, D.; Poulson, A.V.; Castle, B.; Lees, M.M.; Moore, A.T.; Scott, J.D.; Snead, M.P. Alternative splicing modifies the effect of mutations in COL11A1 and results in recessive type 2 Stickler syndrome with profound hearing loss. J. Med. Genet. 2013, 50, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Nikopoulos, K.; Schrauwen, I.; Simon, M.; Collin, R.W.J.; Veckeneer, M.; Keymolen, K.; van Camp, G.; Cremers, F.P.M.; van den Born, L.I. Autosomal recessive Stickler syndrome in two families is caused by mutations in the COL9A1 gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4774–4779. [Google Scholar] [CrossRef] [PubMed]
- Tran-Viet, K.-N.; Soler, V.; Quiette, V.; Powell, C.; Yanovitch, T.; Metlapally, R.; Luo, X.; Katsanis, N.; Nading, E.; Young, T.L. Mutation in collagen II α 1 isoforms delineates Stickler and Wagner syndrome phenotypes. Mol. Vis. 2013, 19, 759–766. [Google Scholar] [PubMed]
- Faletra, F.; D’Adamo, A.P.; Bruno, I.; Athanasakis, E.; Biskup, S.; Esposito, L.; Gasparini, P. Autosomal recessive Stickler syndrome due to a loss of function mutation in the COL9A3 gene. Am. J. Med. Genet. A 2014, 164A, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Stickler, G.B.; Belau, P.G.; Farrell, F.J.; Jones, J.D.; Pugh, D.G.; Steinberg, A.G.; Ward, L.E. Hereditary Progressive Arthro-Ophthalmopathy. Mayo Clin. Proc. 1965, 40, 433–455. [Google Scholar] [PubMed]
- Li, Y.-H.; Tardif, G.; Hum, D.; Kapoor, M.; Fahmi, H.; Pelletier, J.-P.; Martel-Pelletier, J. The unfolded protein response genes in human osteoarthritic chondrocytes: PERK emerges as a potential therapeutic target. Arthritis Res. Ther. 2016, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Clavarino, G.; Adriouach, S.; Quesada, J.-L.; Clay, M.; Chevreau, M.; Trocmé, C.; Grange, L.; Gaudin, P.; Gatti, E.; Pierre, P.; et al. Unfolded protein response gene GADD34 is overexpressed in rheumatoid arthritis and related to the presence of circulating anti-citrullinated protein antibodies. Autoimmunity 2016, 49, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.Y.; Chan, D.; Bateman, J.F.; Cheah, K.S.E. In vivo cellular adaptation to ER stress: Survival strategies with double-edged consequences. J. Cell Sci. 2010, 123, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, K.; Olsson, H.; Morgelin, M.; Heinegard, D. Cartilage Oligomeric Matrix Protein Shows High Affinity Zinc-dependent Interaction with Triple Helical Collagen. J. Biol. Chem. 1998, 273, 20397–20403. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Mallory, C.; McDougal, O.M.; Oxford, J.T. Proteomic analysis of Col11a1-associated protein complexes. Proteomics 2011, 11, 4660–4676. [Google Scholar] [CrossRef] [PubMed]
- Holden, P.; Meadows, R.S.; Chapman, K.L.; Grant, M.E.; Kadler, K.E.; Briggs, M.D. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J. Biol. Chem. 2001, 276, 6046–6055. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.G.; Phamluong, K.; Li, L.; Sun, M.; Cao, T.C.; Liu, P.S.; Modrusan, Z.; Sandoval, W.N.; Rangell, L.; Carano, R.A.D.; et al. Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J. Cell Biol. 2011, 193, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Suleman, F.; Gualeni, B.; Gregson, H.J.; Leighton, M.P.; Piróg, K.A.; Edwards, S.; Holden, P.; Boot-Handford, R.P.; Briggs, M.D. A novel form of chondrocyte stress is triggered by a COMP mutation causing pseudoachondroplasia. Hum. Mutat. 2012, 33, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Coustry, F.; Posey, K.L.; Liu, P.; Alcorn, J.L.; Hecht, J.T. D469del-COMP Retention in Chondrocytes Stimulates Caspase-Independent Necroptosis. Am. J. Pathol. 2012, 180, 738–748. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
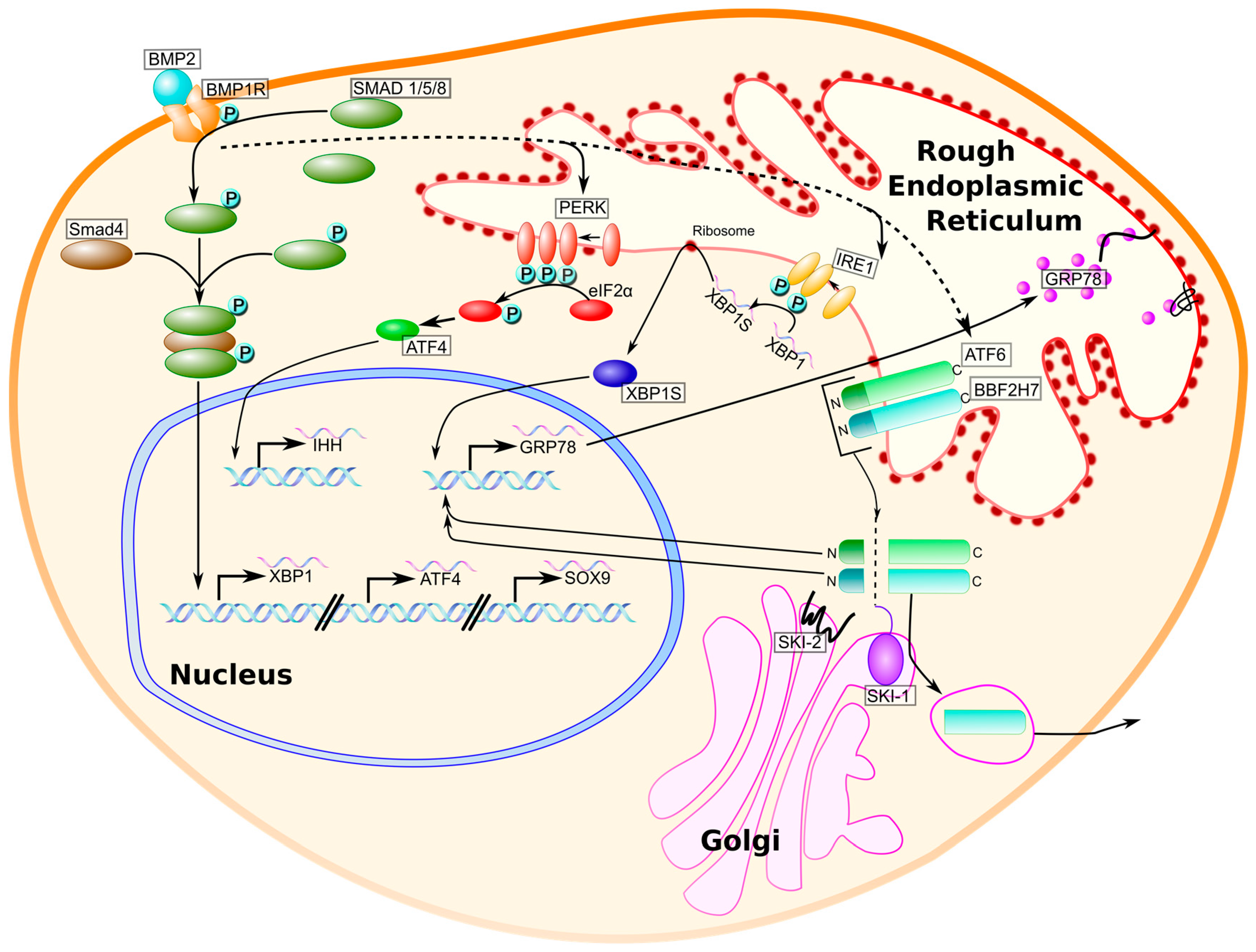{kind=link}
| Collagen Chain | Mutation Described | Dysplasia Resulting from Mutation(s) | Observed | References |
|---|---|---|---|---|
| COL1A1 | Aga2/+ mouse; C-propeptide mutation in col1a1 | Osteogenesis imperfecta | UPR leads to osteoblast apoptosis | [44] |
| COL2A1 | Col2a1 p.Gly1170Ser mouse | Chondrodysplasia | UPR leads to apoptosis of chondrocytes prior to hypertrophy; no hypertrophic zone formed | [45] |
| Dmm/+ mouse; C-propeptide mutation in col2a1 | Chondrodysplasia with early-onset OA | UPR leads to articular chondrocyte apoptosis, contributes to early OA | [28] | |
| COL3A1 | 15 human patients with unique col3a1 mutations | Ehlers-Danlos type IV | Retention of Col3a1 procollagen in ER; distension of rough ER | [46] |
| COL5A1 | 21 patients harboring unique mutations in Col5a1 or Col5a2 | Classic Ehlers-Danlos | Variability in collagen fibril diameter, collagen cauliflowers (aggregates), and dilated ER | [47] |
| COL5A2 | ||||
| COL10A1 | Transgenic mouse: 13 bp deletion within NC1 domain (13del) | Metaphyseal chondrodysplasia, type Schmid (MCDS) | UPR induction in hypertrophic chondrocytes and dedifferentiation to a pre-hypertrophic state | [48] |
| Gene Symbol | Gene Name | Function in Chondrocytes/Osteoblasts | Knockout Phenotype | Reference |
|---|---|---|---|---|
| ATF4 | Activating transcription factor 4 | Induces CHOP activity (pro-apoptotis) | Severe skeletal defects, delayed ossif., short stature/limbs, disorganization of growth plate chondrocyte columns | [58] |
| BMP2 | Bone morphogenetic protein 2 | Induces physiological UPR in chondrocytes and osteoblasts | Severe chondrodysplasia, disorganization of growth plate chondrocytes | [57] |
| XBP1 | X-box binding protein 1 | May promote chondrocyte proliferation | Delayed ossification that resolves by maturity | [59] |
| BBF2H7 | BBF2 human homolog on chromosome 7 | Sequence similarity to ATF6 (in CREB/ATF family); expressed in chondrocytes; targets Sec23a in ER-to-Golgi transport | Severe chondrodysplasia, proliferating chondrocytes show abnormal ER distension | [60] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, A.; Oxford, A.E.; Tawara, K.; Jorcyk, C.L.; Oxford, J.T. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 665. https://doi.org/10.3390/ijms18030665
Hughes A, Oxford AE, Tawara K, Jorcyk CL, Oxford JT. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. International Journal of Molecular Sciences. 2017; 18(3):665. https://doi.org/10.3390/ijms18030665
Chicago/Turabian StyleHughes, Alexandria, Alexandra E. Oxford, Ken Tawara, Cheryl L. Jorcyk, and Julia Thom Oxford. 2017. "Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis" International Journal of Molecular Sciences 18, no. 3: 665. https://doi.org/10.3390/ijms18030665