
Molecular Basis for Modulation of Metabotropic Glutamate Receptors and Their Drug Actions by Extracellular Ca2+

Abstract
:1. Introduction
2. Extracellular Ca2+ and Dynamics in Nervous System
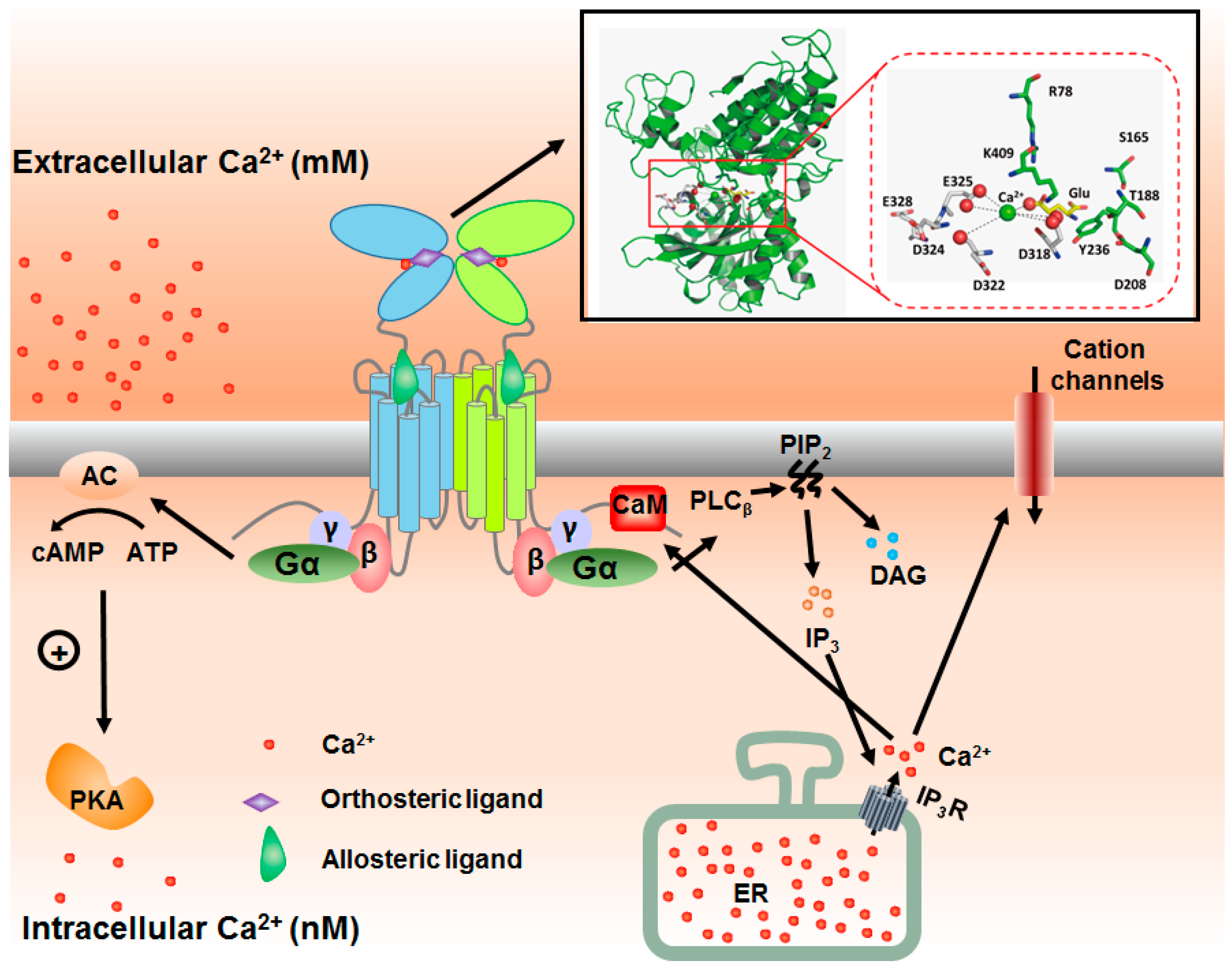
3. Integration of Extracellular and Intracellular Ca2+ Signaling via mGluRs
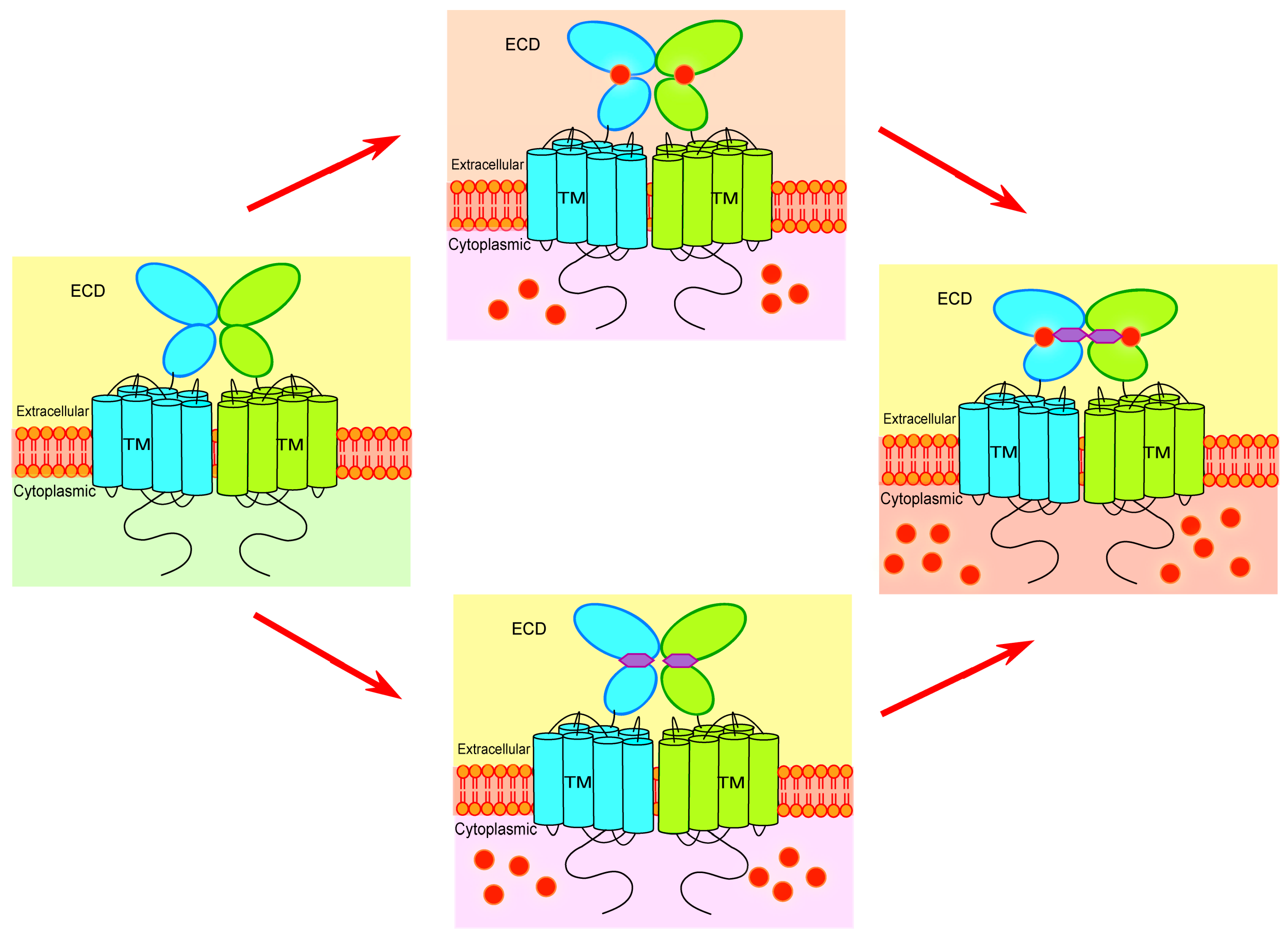
4. Key Determinants for Ligand Binding and Activation
5. Seeking for the Molecular Basis of Ca2+-Mediated Regulation of mGluRs
6. Overcoming the Challenges in Revealing Ca2+ Binding Site of mGluRs
7. Extracellular Ca2+ Modulates Actions of Orthosteric and Allosteric Drugs
8. Conclusions and Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Glusker, J.P. Structural aspects of metal liganding to functional groups in proteins. Adv. Protein Chem. 1991, 42, 1–76. [Google Scholar] [PubMed]
- Ikura, M.; Osawa, M.; Ames, J.B. The role of calcium-binding proteins in the control of transcription: Structure to function. Bioessays 2002, 24, 625–636. [Google Scholar] [PubMed]
- Falke, J.J.; Drake, S.K.; Hazard, A.L.; Peersen, O.B. Molecular tuning of ion binding to calcium signaling proteins. Q. Rev. Biophys. 1994, 27, 219–290. [Google Scholar] [PubMed]
- Teplyakov, A.V.; Kuranova, I.P.; Harutyunyan, E.H.; Vainshtein, B.K.; Frommel, C.; Hohne, W.E.; Wilson, K.S. Crystal structure of thermitase at 1.4 a resolution. J. Mol. Biol. 1990, 214, 261–279. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Brown, E.M.; MacLeod, R.J. Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [PubMed]
- Jingami, H.; Nakanishi, S.; Morikawa, K. Structure of the metabotropic glutamate receptor. Curr. Opin. Neurobiol. 2003, 13, 271–278. [Google Scholar] [CrossRef]
- Bai, M. Structure-function relationship of the extracellular calcium-sensing receptor. Cell Calcium 2004, 35, 197–207. [Google Scholar] [CrossRef]
- Bai, M.; Trivedi, S.; Kifor, O.; Quinn, S.J.; Brown, E.M. Intermolecular interactions between dimeric calcium-sensing receptor monomers are important for its normal function. Proc. Natl. Acad. Sci. USA 1999, 96, 2834–2839. [Google Scholar] [PubMed]
- Suzuki, Y.; Moriyoshi, E.; Tsuchiya, D.; Jingami, H. Negative cooperativity of glutamate binding in the dimeric metabotropic glutamate receptor subtype 1. J. Biol. Chem. 2004, 279, 35526–35534. [Google Scholar] [PubMed]
- Bai, M.; Trivedi, S.; Brown, E.M. Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J. Biol. Chem. 1998, 273, 23605–23610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, S.; Quinn, S.J.; Brown, E.M.; Bai, M. The extracellular calcium-sensing receptor dimerizes through multiple types of intermolecular interactions. J. Biol. Chem. 2001, 276, 5316–5322. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W.; Wisnoski, D.D.; Leister, W.H.; O’Brien, J.A.; Lemaire, W.; Williams, D.L., Jr.; Burno, M.; Sur, C.; Kinney, G.G.; Pettibone, D.J.; et al. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides that potentiate receptor function in vivo. J. Med. Chem. 2004, 47, 5825–5828. [Google Scholar] [CrossRef]
- Kubo, Y.; Miyashita, T.; Murata, Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science 1998, 279, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Kato, M. Quisqualate receptors are specifically involved in cerebellar synaptic plasticity. Nature 1987, 325, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Houamed, K.M.; Kuijper, J.L.; Gilbert, T.L.; Haldeman, B.A.; O’Hara, P.J.; Mulvihill, E.R.; Almers, W.; Hagen, F.S. Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science 1991, 252, 1318–1321. [Google Scholar] [CrossRef]
- Masu, M.; Tanabe, Y.; Tsuchida, K.; Shigemoto, R.; Nakanishi, S. Sequence and expression of a metabotropic glutamate receptor. Nature 1991, 349, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, Y.; Masu, M.; Ishii, T.; Shigemoto, R.; Nakanishi, S. A family of metabotropic glutamate receptors. Neuron 1992, 8, 169–179. [Google Scholar] [CrossRef]
- Abe, T.; Sugihara, H.; Nawa, H.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J. Biol. Chem. 1992, 267, 13361–13368. [Google Scholar]
- Nakajima, Y.; Iwakabe, H.; Akazawa, C.; Nawa, H.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for l-2-amino-4-phosphonobutyrate. J. Biol. Chem. 1993, 268, 11868–11873. [Google Scholar] [PubMed]
- Ohishi, H.; Shigemoto, R.; Nakanishi, S.; Mizuno, N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience 1993, 53, 1009–1018. [Google Scholar] [CrossRef]
- Okamoto, N.; Hori, S.; Akazawa, C.; Hayashi, Y.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J. Biol. Chem. 1994, 269, 1231–1236. [Google Scholar]
- Tanabe, Y.; Nomura, A.; Masu, M.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J. Neurosci. 1993, 13, 1372–1378. [Google Scholar] [PubMed]
- Saugstad, J.A.; Kinzie, J.M.; Mulvihill, E.R.; Segerson, T.P.; Westbrook, G.L. Cloning and expression of a new member of the l-2-amino-4-phosphonobutyric acid-sensitive class of metabotropic glutamate receptors. Mol. Pharmacol. 1994, 45, 367–372. [Google Scholar] [PubMed]
- Duvoisin, R.M.; Zhang, C.; Ramonell, K. A novel metabotropic glutamate receptor expressed in the retina and olfactory bulb. J. Neurosci. 1995, 15, 3075–3083. [Google Scholar] [PubMed]
- Conn, P.J.; Pin, J.P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar] [PubMed]
- Knopfel, T.; Kuhn, R.; Allgeier, H. Metabotropic glutamate receptors: Novel targets for drug development. J. Med. Chem. 1995, 38, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [PubMed]
- Dhingra, A.; Vardi, N. “mGlu receptors in the retina”—Wires membrane transport and signaling. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 641–653. [Google Scholar] [PubMed]
- O’Connor, E.; Allen, L.E.; Bradshaw, K.; Boylan, J.; Moore, A.T.; Trump, D. Congenital stationary night blindness associated with mutations in GRM6 encoding glutamate receptor mGluR6. Br. J. Ophthalmol. 2006, 90, 653–654. [Google Scholar] [CrossRef] [PubMed]
- Brody, S.A.; Conquet, F.; Geyer, M.A. Disruption of prepulse inhibition in mice lacking mGluR1. Eur. J. Neurosci. 2003, 18, 3361–3366. [Google Scholar] [PubMed]
- Dolen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [PubMed]
- Marino, M.J.; Williams, D.L., Jr.; O’Brien, J.A.; Valenti, O.; McDonald, T.P.; Clements, M.K.; Wang, R.; DiLella, A.G.; Hess, J.F.; Kinney, G.G.; et al. Allosteric modulation of group III metabotropic glutamate receptor 4: A potential approach to Parkinson’s disease treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 13668–13673. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.T.; Zhang, L.; Martenyi, F.; Lowe, S.L.; Jackson, K.A.; Andreev, B.V.; Avedisova, A.S.; Bardenstein, L.M.; Gurovich, I.Y.; Morozova, M.A.; et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized phase 2 clinical trial. Nat. Med. 2007, 13, 1102–1107. [Google Scholar] [PubMed]
- Wu, Y.; Ding, H. Role of metabotropic glutamate receptors in cancer development and progression. Chin. J. Hepatol. 2014, 22, 558–560. [Google Scholar]
- Yu, L.J.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Metabotropic glutamate receptors in cancer. Neuropharmacology 2016. [Google Scholar] [CrossRef]
- Banda, M.; Speyer, C.L.; Semma, S.N.; Osuala, K.O.; Kounalakis, N.; Torres Torres, K.E.; Barnard, N.J.; Kim, H.J.; Sloane, B.F.; Miller, F.R.; et al. Metabotropic glutamate receptor-1 contributes to progression in triple negative breast cancer. PLoS ONE 2014, 9, e81126. [Google Scholar] [CrossRef]
- Speyer, C.L.; Hachem, A.H.; Assi, A.A.; Johnson, J.S.; DeVries, J.A.; Gorski, D.H. Metabotropic glutamate receptor-1 as a novel target for the antiangiogenic treatment of breast cancer. PLoS ONE 2014, 9, e88830. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.L.; Smith, J.S.; Banda, M.; DeVries, J.A.; Mekani, T.; Gorski, D.H. Metabotropic glutamate receptor-1: A potential therapeutic target for the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 132, 565–573. [Google Scholar] [PubMed]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.S.; et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar]
- Lee, H.J.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Regulation of mGluR1 expression in human melanocytes and melanoma cells. Biochim. Biophys. Acta 2012, 1819, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Takahashi, N.; Tokumaru, H. Distinct initial snare configurations underlying the diversity of exocytosis. Physiol. Rev. 2012, 92, 1915–1964. [Google Scholar] [PubMed]
- De Jong, A.P.; Fioravante, D. Translating neuronal activity at the synapse: Presynaptic calcium sensors in short-term plasticity. Front. Cell. Neurosci. 2014, 8, 356. [Google Scholar] [PubMed]
- Budde, T.; Meuth, S.; Pape, H.C. Calcium-dependent inactivation of neuronal calcium channels. Nat. Rev. Neurosci. 2002, 3, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, P.M.; Mitchel, J.; Vassilev, M.; Kanazirska, M.; Brown, E.M. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys. J. 1997, 72, 2103–2116. [Google Scholar]
- Jones, B.L.; Smith, S.M. Calcium-sensing receptor: A key target for extracellular calcium signaling in neurons. Front. Physiol. 2016, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Trinidad, B.J.; Shi, J. Hypocalcemia-induced seizure: Demystifying the calcium paradox. ASN Neuro 2015, 7, 1759091415578050. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar]
- Hardingham, N.R.; Bannister, N.J.; Read, J.C.; Fox, K.D.; Hardingham, G.E.; Jack, J.J. Extracellular calcium regulates postsynaptic efficacy through group 1 metabotropic glutamate receptors. J. Neurosci. 2006, 26, 6337–6345. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Reddish, F.; Zhuo, Y.; Yang, J.J. Fast kinetics of calcium signaling and sensor design. Curr. Opin. Chem. Biol. 2015, 27, 90–97. [Google Scholar] [PubMed]
- Tang, S.; Wong, H.C.; Wang, Z.M.; Huang, Y.; Zou, J.; Zhuo, Y.; Pennati, A.; Gadda, G.; Delbono, O.; Yang, J.J. Design and application of a class of sensors to monitor Ca2+ dynamics in high Ca2+ concentration cellular compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 16265–16270. [Google Scholar] [CrossRef]
- Wang, Z.M.; Tang, S.; Messi, M.L.; Yang, J.J.; Delbono, O. Residual sarcoplasmic reticulum Ca2+ concentration after Ca2+ release in skeletal myofibers from young adult and old mice. Pflugers Arch. 2012, 463, 615–624. [Google Scholar] [PubMed]
- Tabata, T.; Kano, M. Calcium dependence of native metabotropic glutamate receptor signaling in central neurons. Mol. Neurobiol. 2004, 29, 261–270. [Google Scholar] [CrossRef]
- Tabata, T.; Aiba, A.; Kano, M. Extracellular calcium controls the dynamic range of neuronal metabotropic glutamate receptor responses. Mol. Cell. Neurosci. 2002, 20, 56–68. [Google Scholar]
- Saunders, R.; Nahorski, S.R.; Challiss, R.A. A modulatory effect of extracellular Ca2+ on type 1α metabotropic glutamate receptor-mediated signalling. Neuropharmacology 1998, 37, 273–276. [Google Scholar] [PubMed]
- Kawabata, S.; Kohara, A.; Tsutsumi, R.; Itahana, H.; Hayashibe, S.; Yamaguchi, T.; Okada, M. Diversity of calcium signaling by metabotropic glutamate receptors. J. Biol. Chem. 1998, 273, 17381–17385. [Google Scholar] [PubMed]
- Nash, M.S.; Saunders, R.; Young, K.W.; Challiss, R.A.; Nahorski, S.R. Reassessment of the Ca2+ sensing property of a type I metabotropic glutamate receptor by simultaneous measurement of inositol 1,4,5-trisphosphate and Ca2+ in single cells. J. Biol. Chem. 2001, 276, 19286–19293. [Google Scholar] [PubMed]
- Lee, J.H.; Lee, J.; Choi, K.Y.; Hepp, R.; Lee, J.Y.; Lim, M.K.; Chatani-Hinze, M.; Roche, P.A.; Kim, D.G.; Ahn, Y.S.; et al. Calmodulin dynamically regulates the trafficking of the metabotropic glutamate receptor mglur5. Proc. Natl. Acad. Sci. USA 2008, 105, 12575–12580. [Google Scholar]
- El Far, O.; Bofill-Cardona, E.; Airas, J.M.; O’Connor, V.; Boehm, S.; Freissmuth, M.; Nanoff, C.; Betz, H. Mapping of calmodulin and Gβγ binding domains within the C-terminal region of the metabotropic glutamate receptor 7A. J. Biol. Chem. 2001, 276, 30662–30669. [Google Scholar] [PubMed]
- Nakajima, Y.; Yamamoto, T.; Nakayama, T.; Nakanishi, S. A relationship between protein kinase c phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J. Biol. Chem. 1999, 274, 27573–27577. [Google Scholar] [CrossRef] [PubMed]
- Pelkey, K.A.; Lavezzari, G.; Racca, C.; Roche, K.W.; McBain, C.J. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 2005, 46, 89–102. [Google Scholar] [PubMed]
- Fourgeaud, L.; Mato, S.; Bouchet, D.; Hemar, A.; Worley, P.F.; Manzoni, O.J. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J. Neurosci. 2004, 24, 6939–6945. [Google Scholar] [PubMed]
- Francesconi, A.; Kumari, R.; Zukin, R.S. Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J. Neurosci. 2009, 29, 3590–3602. [Google Scholar]
- Kubokawa, K.; Miyashita, T.; Nagasawa, H.; Kubo, Y. Cloning and characterization of a bifunctional metabotropic receptor activated by both extracellular calcium and glutamate. FEBS Lett. 1996, 392, 71–76. [Google Scholar] [PubMed]
- Vafabakhsh, R.; Levitz, J.; Isacoff, E.Y. Conformational dynamics of a class C G-protein-coupled receptor. Nature 2015, 524, 497–501. [Google Scholar] [PubMed]
- Takahashi, K.; Tsuchida, K.; Tanabe, Y.; Masu, M.; Nakanishi, S. Role of the large extracellular domain of metabotropic glutamate receptors in agonist selectivity determination. J. Biol. Chem. 1993, 268, 19341–19345. [Google Scholar] [PubMed]
- Kunishima, N.; Shimada, Y.; Tsuji, Y.; Sato, T.; Yamamoto, M.; Kumasaka, T.; Nakanishi, S.; Jingami, H.; Morikawa, K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000, 407, 971–977. [Google Scholar] [PubMed]
- Tsuchiya, D.; Kunishima, N.; Kamiya, N.; Jingami, H.; Morikawa, K. Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc. Natl. Acad. Sci. USA 2002, 99, 2660–2665. [Google Scholar]
- Muto, T.; Tsuchiya, D.; Morikawa, K.; Jingami, H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 3759–3764. [Google Scholar] [PubMed]
- Tsuji, Y.; Shimada, Y.; Takeshita, T.; Kajimura, N.; Nomura, S.; Sekiyama, N.; Otomo, J.; Usukura, J.; Nakanishi, S.; Jingami, H. Cryptic dimer interface and domain organization of the extracellular region of metabotropic glutamate receptor subtype 1. J. Biol. Chem. 2000, 275, 28144–28151. [Google Scholar] [PubMed]
- Ray, K.; Hauschild, B.C.; Steinbach, P.J.; Goldsmith, P.K.; Hauache, O.; Spiegel, A.M. Identification of the cysteine residues in the amino-terminal extracellular domain of the human Ca2+ receptor critical for dimerization. Implications for function of monomeric Ca2+ receptor. J. Biol. Chem. 1999, 274, 27642–27650. [Google Scholar] [PubMed]
- Ray, K.; Hauschild, B.C. Cys-140 is critical for metabotropic glutamate receptor-1 dimerization. J. Biol. Chem. 2000, 275, 34245–34251. [Google Scholar] [PubMed]
- O’Hara, P.J.; Sheppard, P.O.; Thogersen, H.; Venezia, D.; Haldeman, B.A.; McGrane, V.; Houamed, K.M.; Thomsen, C.; Gilbert, T.L.; Mulvihill, E.R. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 1993, 11, 41–52. [Google Scholar]
- Pin, J.P.; Galvez, T.; Prezeau, L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol. Ther. 2003, 98, 325–354. [Google Scholar] [PubMed]
- Abe, H.; Tateyama, M.; Kubo, Y. Functional identification of Gd3+ binding site of metabotropic glutamate receptor 1α. FEBS Lett. 2003, 545, 233–238. [Google Scholar] [PubMed]
- Yang, W.; Wilkins, A.L.; Li, S.; Ye, Y.; Yang, J.J. The effects of Ca2+ binding on the dynamic properties of a designed Ca2+-binding protein. Biochemistry 2005, 44, 8267–8273. [Google Scholar]
- Nagar, B.; Overduin, M.; Ikura, M.; Rini, J.M. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature 1996, 380, 360–364. [Google Scholar] [PubMed]
- Hu, J.; Spiegel, A.M. Naturally occurring mutations of the extracellular Ca2+-sensing receptor: Implications for its structure and function. Trends Endocrinol. Metab. 2003, 14, 282–288. [Google Scholar] [CrossRef]
- Yang, J.J.; Gawthrop, A.; Ye, Y. Obtaining site-specific calcium-binding affinities of calmodulin. Protein Pept. Lett. 2003, 10, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Linse, S.; Forsen, S. Determinants that govern high-affinity calcium binding. Adv. Second Messenger Phosphoprotein Res. 1995, 30, 89–151. [Google Scholar] [PubMed]
- Ye, Y.; Lee, H.W.; Yang, W.; Shealy, S.; Yang, J.J. Probing site-specific calmodulin calcium and lanthanide affinity by grafting. J. Am. Chem. Soc. 2005, 127, 3743–3750. [Google Scholar]
- Ye, Y.; Lee, H.W.; Yang, W.; Shealy, S.J.; Wilkins, A.L.; Liu, Z.R.; Torshin, I.; Harrison, R.; Wohlhueter, R.; Yang, J.J. Metal binding affinity and structural properties of an isolated EF-loop in a scaffold protein. Protein Eng. 2001, 14, 1001–1013. [Google Scholar] [CrossRef]
- Ye, Y.; Lee, H.W.; Yang, W.; Yang, J.J. Calcium and lanthanide affinity of the EF-loops from the C-terminal domain of calmodulin. J. Inorg. Biochem. 2005, 99, 1376–1383. [Google Scholar] [PubMed]
- Ye, Y.; Shealy, S.; Lee, H.W.; Torshin, I.; Harrison, R.; Yang, J.J. A grafting approach to obtain site-specific metal-binding properties of EF-hand proteins. Protein Eng. 2003, 16, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, K.; Kirberger, M.; Wong, H.; Chen, G.; Yang, J.J. Analysis and prediction of calcium-binding pockets from APO-protein structures exhibiting calcium-induced localized conformational changes. Protein Sci. 2010, 19, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kirberger, M.; Qiu, F.; Chen, G.; Yang, J.J. Towards predicting Ca2+-binding sites with different coordination numbers in proteins with atomic resolution. Proteins 2009, 75, 787–798. [Google Scholar] [PubMed]
- Kirberger, M.; Wang, X.; Deng, H.; Yang, W.; Chen, G.; Yang, J.J. Statistical analysis of structural characteristics of protein Ca2+-binding sites. J. Biol. Inorg. Chem. 2008, 13, 1169–1181. [Google Scholar]
- Jiang, Y.; Huang, Y.; Wong, H.C.; Zhou, Y.; Wang, X.; Yang, J.; Hall, R.A.; Brown, E.M.; Yang, J.J. Elucidation of a novel extracellular calcium-binding site on metabotropic glutamate receptor 1{α} (mGluR1{α}) that controls receptor activation. J. Biol. Chem. 2010, 285, 33463–33474. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, Y.; Castiblanco, A.; Yang, W.; Brown, E.M.; Yang, J.J. Multiple Ca2+-binding sites in the extracellular domain of the Ca2+-sensing receptor corresponding to cooperative Ca2+ response. Biochemistry 2009, 48, 388–398. [Google Scholar] [PubMed]
- Huang, Y.; Zhou, Y.; Yang, W.; Butters, R.; Lee, H.W.; Li, S.; Castiblanco, A.; Brown, E.M.; Yang, J.J. Identification and dissection of Ca2+-binding sites in the extracellular domain of Ca2+-sensing receptor. J. Biol. Chem. 2007, 282, 19000–19010. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, Y.; Wong, H.C.; Chen, Y.; Chen, Y.; Wang, S.; Castiblanco, A.; Liu, A.; Yang, J.J. A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+. FEBS J. 2009, 276, 5589–5597. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zou, J.; Wang, S.; Ye, Y.; Huang, Y.; Gadda, G.; Yang, J.J. Designing protease sensors for real-time imaging of trypsin activation in pancreatic cancer cells. Biochemistry 2009, 48, 3519–3526. [Google Scholar]
- Zou, J.; Hofer, A.M.; Lurtz, M.M.; Gadda, G.; Ellis, A.L.; Chen, N.; Huang, Y.; Holder, A.; Ye, Y.; Louis, C.F.; et al. Developing sensors for real-time measurement of high Ca2+ concentrations. Biochemistry 2007, 46, 12275–12288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tzeng, W.P.; Yang, W.; Zhou, Y.; Ye, Y.; Lee, H.W.; Frey, T.K.; Yang, J. Identification of a Ca2+-binding domain in the rubella virus nonstructural protease. J. Virol. 2007, 81, 7517–7528. [Google Scholar] [PubMed]
- Abe, H.; Misaka, T.; Tateyama, M.; Kubo, Y. Effects of coexpression with homer isoforms on the function of metabotropic glutamate receptor 1α. Mol. Cell. Neurosci. 2003, 23, 157–168. [Google Scholar] [CrossRef]
- Hemstapat, K.; de Paulis, T.; Chen, Y.; Brady, A.E.; Grover, V.K.; Alagille, D.; Tamagnan, G.D.; Conn, P.J. A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol. Pharmacol. 2006, 70, 616–626. [Google Scholar] [PubMed]
- Sheffler, D.J.; Conn, P.J. Allosteric potentiators of metabotropic glutamate receptor subtype 1a differentially modulate independent signaling pathways in baby hamster kidney cells. Neuropharmacology 2008, 55, 419–427. [Google Scholar]
- Litschig, S.; Gasparini, F.; Rueegg, D.; Stoehr, N.; Flor, P.J.; Vranesic, I.; Prezeau, L.; Pin, J.P.; Thomsen, C.; Kuhn, R. Cpccoet, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol. Pharmacol. 1999, 55, 453–461. [Google Scholar]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [PubMed]
- Thomsen, C.; Hansen, L.; Suzdak, P.D. l-glutamate uptake inhibitors may stimulate phosphoinositide hydrolysis in baby hamster kidney cells expressing mGluR1a via heteroexchange with l-glutamate without direct activation of mGluR1a. J. Neurochem. 1994, 63, 2038–2047. [Google Scholar] [PubMed]
- Carruthers, A.M.; Challiss, R.A.; Mistry, R.; Saunders, R.; Thomsen, C.; Nahorski, S.R. Enhanced type 1α metabotropic glutamate receptor-stimulated phosphoinositide signaling after pertussis toxin treatment. Mol. Pharmacol. 1997, 52, 406–414. [Google Scholar]
- Desai, M.A.; Burnett, J.P.; Mayne, N.G.; Schoepp, D.D. Cloning and expression of a human metabotropic glutamate receptor 1α: Enhanced coupling on co-transfection with a glutamate transporter. Mol. Pharmacol. 1995, 48, 648–657. [Google Scholar] [PubMed]
- Pickering, D.S.; Thomsen, C.; Suzdak, P.D.; Fletcher, E.J.; Robitaille, R.; Salter, M.W.; MacDonald, J.F.; Huang, X.P.; Hampson, D.R. A comparison of two alternatively spliced forms of a metabotropic glutamate receptor coupled to phosphoinositide turnover. J. Neurochem. 1993, 61, 85–92. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Steensma, D.; Araldi, G.L.; Tuckmantel, W.; Wang, S.; Pshenichkin, S.; Surina, E.; Wroblewski, J.T. Synthesis and biology of the conformationally restricted acpd analogue, 2-aminobicyclo[2.1.1]hexane-2,5-dicarboxylic acid-I, a potent mglur agonist. J. Med. Chem. 1998, 41, 1641–1650. [Google Scholar] [CrossRef]
- Parmentier, M.L.; Joly, C.; Restituito, S.; Bockaert, J.; Grau, Y.; Pin, J.P. The G protein-coupling profile of metabotropic glutamate receptors, as determined with exogenous G proteins, is independent of their ligand recognition domain. Mol. Pharmacol. 1998, 53, 778–786. [Google Scholar] [PubMed]
- Hayashi, Y.; Tanabe, Y.; Aramori, I.; Masu, M.; Shimamoto, K.; Ohfune, Y.; Nakanishi, S. Agonist analysis of 2-(carboxycyclopropyl)glycine isomers for cloned metabotropic glutamate receptor subtypes expressed in chinese hamster ovary cells. Br. J. Pharmacol. 1992, 107, 539–543. [Google Scholar]
- Brabet, I.; Mary, S.; Bockaert, J.; Pin, J.P. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharmacology 1995, 34, 895–903. [Google Scholar] [CrossRef]
- Flor, P.J.; Gomeza, J.; Tones, M.A.; Kuhn, R.; Pin, J.P.; Knopfel, T. The c-terminal domain of the mGluR1 metabotropic glutamate receptor affects sensitivity to agonists. J. Neurochem. 1996, 67, 58–63. [Google Scholar] [CrossRef]
- Akam, E.C.; Carruthers, A.M.; Nahorski, S.R.; Challiss, R.A. Pharmacological characterization of type 1α metabotropic glutamate receptor-stimulated [35s]-GTPγs binding. Br. J. Pharmacol. 1997, 121, 1203–1209. [Google Scholar]
- Sortino, M.A.; Nicoletti, F.; Canonico, P.L. ‘Metabotropic’ glutamate receptors in rat hypothalamus: Characterization and developmental profile. Brain Res. Dev. Brain Res. 1991, 61, 169–172. [Google Scholar] [CrossRef]
- Littman, L.; Glatt, B.S.; Robinson, M.B. Multiple subtypes of excitatory amino acid receptors coupled to the hydrolysis of phosphoinositides in rat brain. J. Neurochem. 1993, 61, 586–593. [Google Scholar] [CrossRef]
- Godfrey, P.P.; Wilkins, C.J.; Tyler, W.; Watson, S.P. Stimulatory and inhibitory actions of excitatory amino acids on inositol phospholipid metabolism in rat cerebral cortex. Br. J. Pharmacol. 1988, 95, 131–138. [Google Scholar] [CrossRef]
- Manahan-Vaughan, D.; Reiser, M.; Pin, J.P.; Wilsch, V.; Bockaert, J.; Reymann, K.G.; Riedel, G. Physiological and pharmacological profile of trans-azetidine-2,4-dicarboxylic acid: Metabotropic glutamate receptor agonism and effects on long-term potentiation. Neuroscience 1996, 72, 999–1008. [Google Scholar]
- Moroni, F.; Lombardi, G.; Thomsen, C.; Leonardi, P.; Attucci, S.; Peruginelli, F.; Torregrossa, S.A.; Pellegrini-Giampietro, D.E.; Luneia, R.; Pellicciari, R. Pharmacological characterization of 1-aminoindan-1,5-dicarboxylic acid, a potent mGluR1 antagonist. J. Pharmacol. Exp. Ther. 1997, 281, 721–729. [Google Scholar] [PubMed]
- Kingston, A.E.; Ornstein, P.L.; Wright, R.A.; Johnson, B.G.; Mayne, N.G.; Burnett, J.P.; Belagaje, R.; Wu, S.; Schoepp, D.D. Ly341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 1998, 37, 1–12. [Google Scholar] [CrossRef]
- Thomsen, C.; Klitgaard, H.; Sheardown, M.; Jackson, H.C.; Eskesen, K.; Jacobsen, P.; Treppendahl, S.; Suzdak, P.D. (s)-4-carboxy-3-hydroxyphenylglycine, an antagonist of metabotropic glutamate receptor (mGluR) 1a and an agonist of mGluR2, protects against audiogenic seizures in DBA/2 mice. J. Neurochem. 1994, 62, 2492–2495. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Kingston, A.; O’Neill, M.J.; Catania, M.V.; Di Grezia, R.; Nicoletti, F. Neuroprotective activity of the potent and selective mglu1a metabotropic glutamate receptor antagonist, (+)-2-methyl-4 carboxyphenylglycine (ly367385): Comparison with ly357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology 1999, 38, 199–207. [Google Scholar]
- Kingston, A.E.; Burnett, J.P.; Mayne, N.G.; Lodge, D. Pharmacological analysis of 4-carboxyphenylglycine derivatives: Comparison of effects on mGluR1 alpha and mGluR5a subtypes. Neuropharmacology 1995, 34, 887–894. [Google Scholar]
- Pellicciari, R.; Luneia, R.; Costantino, G.; Marinozzi, M.; Natalini, B.; Jakobsen, P.; Kanstrup, A.; Lombardi, G.; Moroni, F.; Thomsen, C. 1-aminoindan-1,5-dicarboxylic acid: A novel antagonist at phospholipase C-linked metabotropic glutamate receptors. J. Med. Chem. 1995, 38, 3717–3719. [Google Scholar] [CrossRef]
- Hermans, E.; Challiss, R.A.; Nahorski, S.R. Effects of varying the expression level of recombinant human mGlu1α receptors on the pharmacological properties of agonists and antagonists. Br. J. Pharmacol. 1999, 126, 873–882. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Cozzi, A.; Peruginelli, F.; Leonardi, P.; Meli, E.; Pellicciari, R.; Moroni, F. 1-aminoindan-1,5-dicarboxylic acid and (s)-(+)-2-(3′-carboxybicyclo[1.1.1] pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischaemia. Eur. J. Neurosci. 1999, 11, 3637–3647. [Google Scholar]
- Costantino, G.; Maltoni, K.; Marinozzi, M.; Camaioni, E.; Prezeau, L.; Pin, J.P.; Pellicciari, R. Synthesis and biological evaluation of 2-(3′-(1h-tetrazol-5-yl) bicyclo[1.1.1]pent-1-yl)glycine (S-TBPG), a novel mGlu1 receptor antagonist. Bioorg. Med. Chem. 2001, 9, 221–227. [Google Scholar]
- Vieira, E.; Huwyler, J.; Jolidon, S.; Knoflach, F.; Mutel, V.; Wichmann, J. Fluorinated 9H-xanthene-9-carboxylic acid oxazol-2-yl-amides as potent, orally available mGlu1 receptor enhancers. Bioorg. Med. Chem. Lett. 2009, 19, 1666–1669. [Google Scholar] [CrossRef]
- Lavreysen, H.; Janssen, C.; Bischoff, F.; Langlois, X.; Leysen, J.E.; Lesage, A.S. [3h]r214127: A novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol. Pharmacol. 2003, 63, 1082–1093. [Google Scholar]
- Lavreysen, H.; Wouters, R.; Bischoff, F.; Nobrega Pereira, S.; Langlois, X.; Blokland, S.; Somers, M.; Dillen, L.; Lesage, A.S. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology 2004, 47, 961–972. [Google Scholar]
- Knoflach, F.; Mutel, V.; Jolidon, S.; Kew, J.N.; Malherbe, P.; Vieira, E.; Wichmann, J.; Kemp, J.A. Positive allosteric modulators of metabotropic glutamate 1 receptor: Characterization, mechanism of action, and binding site. Proc. Natl. Acad. Sci. USA 2001, 98, 13402–13407. [Google Scholar]
- Suzuki, G.; Kimura, T.; Satow, A.; Kaneko, N.; Fukuda, J.; Hikichi, H.; Sakai, N.; Maehara, S.; Kawagoe-Takaki, H.; Hata, M.; et al. Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl- 3,6-dihydropyridine-1(2H)-carboxamide (FTIDC). J. Pharmacol. Exp. Ther. 2007, 321, 1144–1153. [Google Scholar]
- EL-Kouhen, O.; Lehto, S.G.; Pan, J.B.; Chang, R.; Baker, S.J.; Zhong, C.; Hollingsworth, P.R.; Mikusa, J.P.; Cronin, E.A.; Chu, K.L.; et al. Blockade of mGluR1 receptor results in analgesia and disruption of motor and cognitive performances: Effects of A-841720, a novel non-competitive mGluR1 receptor antagonist. Br. J. Pharmacol. 2006, 149, 761–774. [Google Scholar]
- Micheli, F.; di Fabio, R.; Bordi, F.; Cavallini, P.; Cavanni, P.; Donati, D.; Faedo, S.; Maffeis, M.; Sabbatini, F.M.; Tarzia, G.; et al. 2,4-Dicarboxy-pyrroles as selective non-competitive mGluR1 antagonists: Further characterization of 3,5-dimethyl pyrrole-2,4-dicarboxylic acid 2-propyl ester 4-(1,2,2-trimethyl-propyl) ester and structure-activity relationships. Bioorg. Med. Chem. Lett. 2003, 13, 2113–2118. [Google Scholar] [CrossRef]
- Micheli, F.; Fabio, R.D.; Cavanni, P.; Rimland, J.M.; Capelli, A.M.; Chiamulera, C.; Corsi, M.; Corti, C.; Donati, D.; Feriani, A.; et al. Synthesis and pharmacological characterisation of 2,4-dicarboxy-pyrroles as selective non-competitive mGluR1 antagonists. Bioorg. Med. Chem. 2003, 11, 171–183. [Google Scholar] [CrossRef]
- Kohara, A.; Toya, T.; Tamura, S.; Watabiki, T.; Nagakura, Y.; Shitaka, Y.; Hayashibe, S.; Kawabata, S.; Okada, M. Radioligand binding properties and pharmacological characterization of 6-amino-N-cyclohexyl-n,3-dimethylthiazolo[3,2-a]benzimidazole-2-carboxamide (ym-298198), a high-affinity, selective, and noncompetitive antagonist of metabotropic glutamate receptor type 1. J. Pharmacol. Exp. Ther. 2005, 315, 163–169. [Google Scholar]
- Carroll, F.Y.; Stolle, A.; Beart, P.M.; Voerste, A.; Brabet, I.; Mauler, F.; Joly, C.; Antonicek, H.; Bockaert, J.; Muller, T.; et al. Bay36-7620: A potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol. Pharmacol. 2001, 59, 965–973. [Google Scholar]
- Malherbe, P.; Kratochwil, N.; Knoflach, F.; Zenner, M.T.; Kew, J.N.; Kratzeisen, C.; Maerki, H.P.; Adam, G.; Mutel, V. Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J. Biol. Chem. 2003, 278, 8340–8347. [Google Scholar] [CrossRef]
- Fukuda, J.; Suzuki, G.; Kimura, T.; Nagatomi, Y.; Ito, S.; Kawamoto, H.; Ozaki, S.; Ohta, H. Identification of a novel transmembrane domain involved in the negative modulation of mglur1 using a newly discovered allosteric mGluR1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4h-chromen-4-one. Neuropharmacology 2009, 57, 438–445. [Google Scholar] [CrossRef]
- Kohara, A.; Nagakura, Y.; Kiso, T.; Toya, T.; Watabiki, T.; Tamura, S.; Shitaka, Y.; Itahana, H.; Okada, M. Antinociceptive profile of a selective metabotropic glutamate receptor 1 antagonist ym-230888 in chronic pain rodent models. Eur. J. Pharmacol. 2007, 571, 8–16. [Google Scholar] [CrossRef]
- Jiang, J.Y.; Nagaraju, M.; Meyer, R.C.; Zhang, L.; Hamelberg, D.; Hall, R.A.; Brown, E.M.; Conn, P.J.; Yang, J.J. Extracellular calcium modulates actions of orthosteric and allosteric ligands on metabotropic glutamate receptor 1α. J. Biol. Chem. 2014, 289, 1649–1661. [Google Scholar] [CrossRef]
- Tateyama, M.; Abe, H.; Nakata, H.; Saito, O.; Kubo, Y. Ligand-induced rearrangement of the dimeric metabotropic glutamate receptor 1α. Nat. Struct. Mol. Biol. 2004, 11, 637–642. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, T.; Zou, J.; Miller, C.L.; Gorkhali, R.; Yang, J.Y.; Schilmiller, A.; Wang, S.; Huang, K.; Brown, E.M.; et al. Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci. Adv. 2016, 2, e1600241. [Google Scholar] [CrossRef]
- Geng, Y.; Mosyak, L.; Kurinov, I.; Zuo, H.; Sturchler, E.; Cheng, T.C.; Subramanyam, P.; Brown, A.P.; Brennan, S.C.; Mun, H.C.; et al. Structural mechanism of ligand activation in human calcium-sensing receptor. eLife 2016, 5, e13662. [Google Scholar]



{kind=link}
{kind=link}
| Group | Receptor | Coupled G Protein | Signaling Pathways | Associated Disease |
|---|---|---|---|---|
| Group I | mGluR1 | Predominantly Gαq/s | PLC stimulation, MAP kinase phosphorylation, AC stimulation (some cases) | Schizophrenia, breast cancer, depression, and bipolar disorder |
| mGluR5 | Schizophrenia, anxiety, chronic pain, Alzheimer’s disease, drug addiction, fragile X syndrome, gastroesophageal reflux disease | |||
| Group II | mGluR2 | Predominantly Gαi | AC inhibition, activation of K+ channel, inhibition of Ca2+ channel | Anxiety, epilepsy, Parkinson’s disease, depression, addictive disorders, schizophrenia |
| mGluR3 | ||||
| Group III | mGluR4 | Predominantly Gαi | AC inhibition, activation of K+ channel, inhibition of Ca2+ channel, stimulation of cGMP (some cases) | Parkinson’s disease |
| mGluR6 | congenital stationary night blindness | |||
| mGluR7 | Schizophrenia, anxiety | |||
| mGluR8 | Alzheimer’s disease, Parkinson’s disease |
| Agonists | ||||
| Ligand | Binding Site | Action | Potency | References |
| Quisqualate | ECD | Full agonist | EC50: 0.2–3.0 μM | [16,17,100,101,102,103] |
| ABHx D-I | ECD | Full agonist | EC50: 2.0 μM | [104] |
| 3,5-DHPG | ECD | Full agonist | EC50: 6.6 μM | [105] |
| l-glutamate | ECD | Full agonist | EC50: 9–13 μM | [16,17,106,107] |
| (1S,3R)-ACPD | ECD | Full agonist | EC50: 10–80 μM | [102,108,109] |
| Ibotenate | ECD | Full agonist | EC50: 10–100 μM | [110,111,112] |
| l-CCG-I | ECD | Full agonist | EC50: 50 μM | [106] |
| (S)-3HPG | ECD | Partial agonist | EC50: 97 μM | [107] |
| t-ADA | ECD | Full agonist | EC50: 190 μM | [113] |
| Antagonists | ||||
| Ligand | Binding Site | Action | Potency | References |
| AIDA | ECD | Antagonist | IC50: 214 μM | [114] |
| LY341495 | ECD | Antagonist | IC50: 7.8 μM | [115] |
| (S)-4C3HPG | ECD | Antagonist | IC50: 15 μM | [116] |
| LY367385 | ECD | Antagonist | IC50: 8.8 μM | [117] |
| (S)-4CPG | ECD | Antagonist | IC50: 44–72 μM | [118] |
| AIDC | ECD | Antagonist | IC50: 7.0 μM | [119] |
| (+)-MCPG | ECD | Antagonist | IC50: 3.8 μM | [120] |
| (S)-(+)-CBPG | ECD | Antagonist | IC50: 65 μM | [121] |
| (S)-TBPG | ECD | Antagonist | IC50: 69 μM | [122] |
| Allosteric Regulators | ||||
| Ligand | Possible Binding Site | Action | Potency | References |
| VU-71 | 7TMD | Positive | EC50: 2.4 μM | [96] |
| Ro 07-11401 | 7TMD | Positive | EC50: 56 nM | [123] |
| NPS2390 | 7TMD | Negative | Ki: 1.4 nM | [124] |
| R214127 | 7TMD | Negative | KD: 0.9 nM | [124] |
| JNJ16259685 | 7TMD | Negative | IC50: 3.2 nM | [125] |
| Ro 67-7476 | 7TMD | Positive | EC50: 174 nM | [126] |
| Ro 01-6128 | 7TMD | Positive | EC50: 200 nM | [126] |
| CPCCOEt | 7TMD | Negative | IC50: 6.6 nM | [98] |
| Ro 67-4853 | 7TMD | Positive | EC50: 69 nM | [126] |
| FTIDC | 7TMD | Negative | IC50: 6 nM | [127] |
| A841720 | 7TMD | Negative | IC50: 11 nM | [128] |
| DM-PPP | 7TMD | Negative | IC50: 15.8 nM | [129,130] |
| YM298198 | 7TMD | Negative | IC50: 16 nM | [131] |
| BAY 367620 | 7TMD | Negative | IC50: 160 nM | [132] |
| EM-TBPC | 7TMD | Negative | IC50: 15 nM | [133] |
| CFMMC | 7TMD | Negative | IC50: 50 nM | [134] |
| YM-230888 | 7TMD | Negative | IC50: 13 nM | [135] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, J.; Jiang, J.Y.; Yang, J.J. Molecular Basis for Modulation of Metabotropic Glutamate Receptors and Their Drug Actions by Extracellular Ca2+. Int. J. Mol. Sci. 2017, 18, 672. https://doi.org/10.3390/ijms18030672
Zou J, Jiang JY, Yang JJ. Molecular Basis for Modulation of Metabotropic Glutamate Receptors and Their Drug Actions by Extracellular Ca2+. International Journal of Molecular Sciences. 2017; 18(3):672. https://doi.org/10.3390/ijms18030672
Chicago/Turabian StyleZou, Juan, Jason Y. Jiang, and Jenny J. Yang. 2017. "Molecular Basis for Modulation of Metabotropic Glutamate Receptors and Their Drug Actions by Extracellular Ca2+" International Journal of Molecular Sciences 18, no. 3: 672. https://doi.org/10.3390/ijms18030672