
ROS Production and ERK Activity Are Involved in the Effects of d-β-Hydroxybutyrate and Metformin in a Glucose Deficient Condition

 ,
,
Abstract
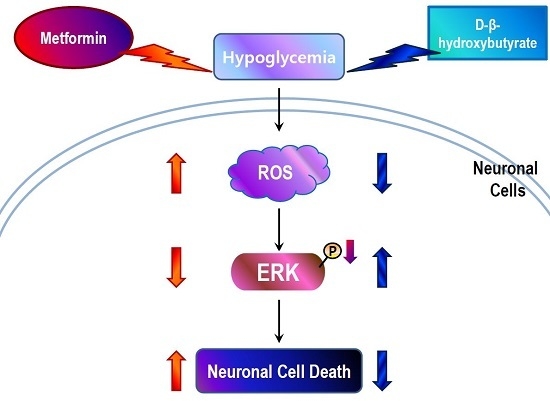
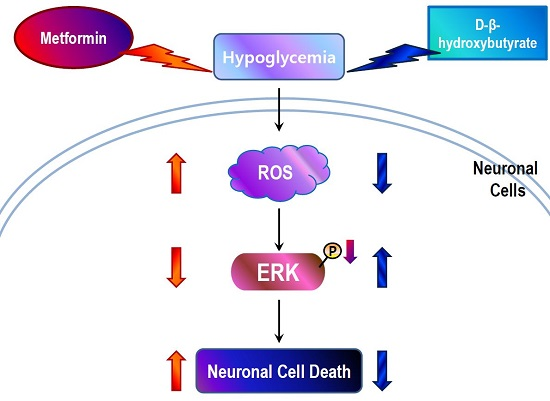
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
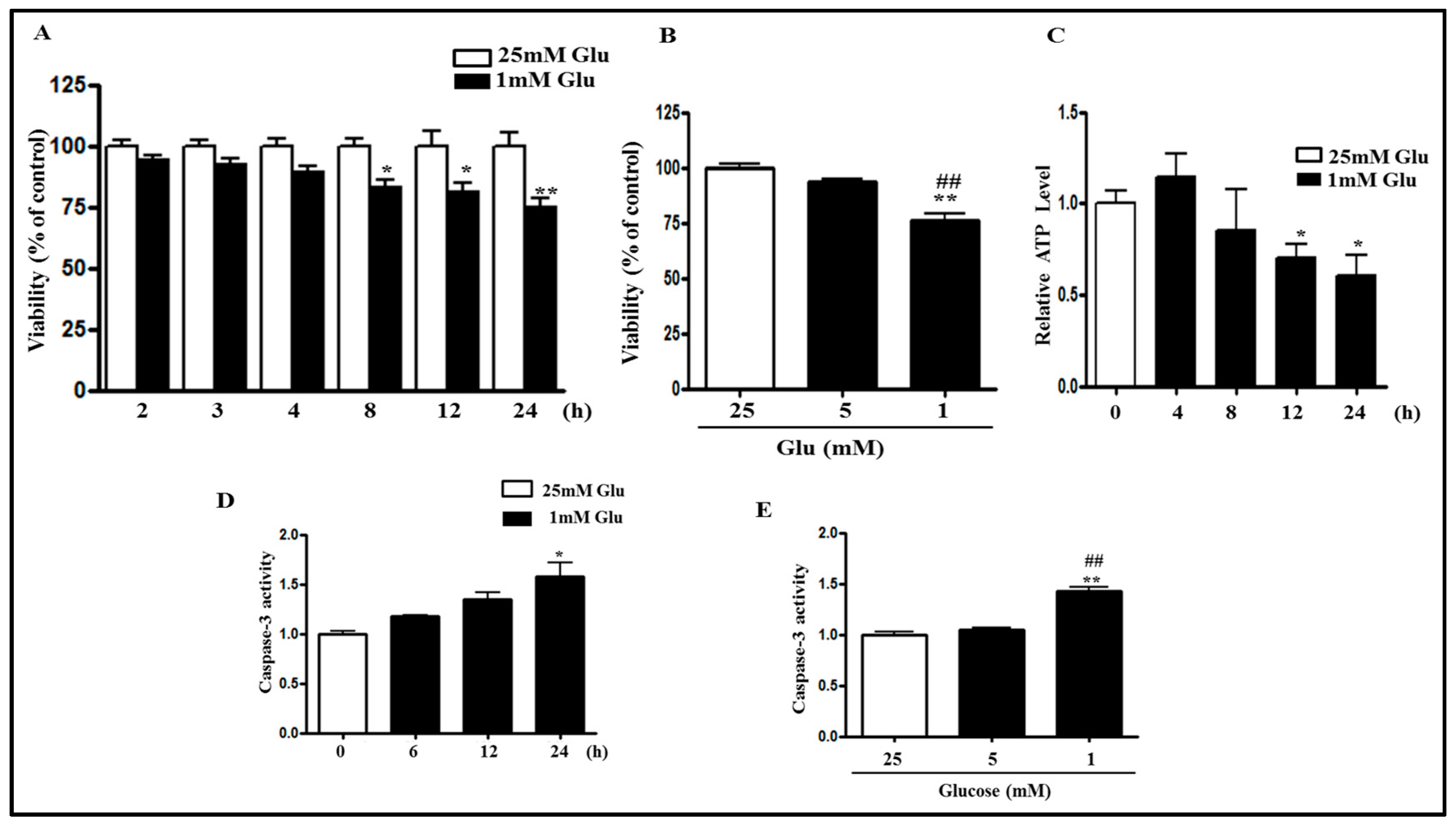
2.1. Glucose Deficiency Decreases Cell Viability and Intracellular ATP Level in SH-SY5Y Cells
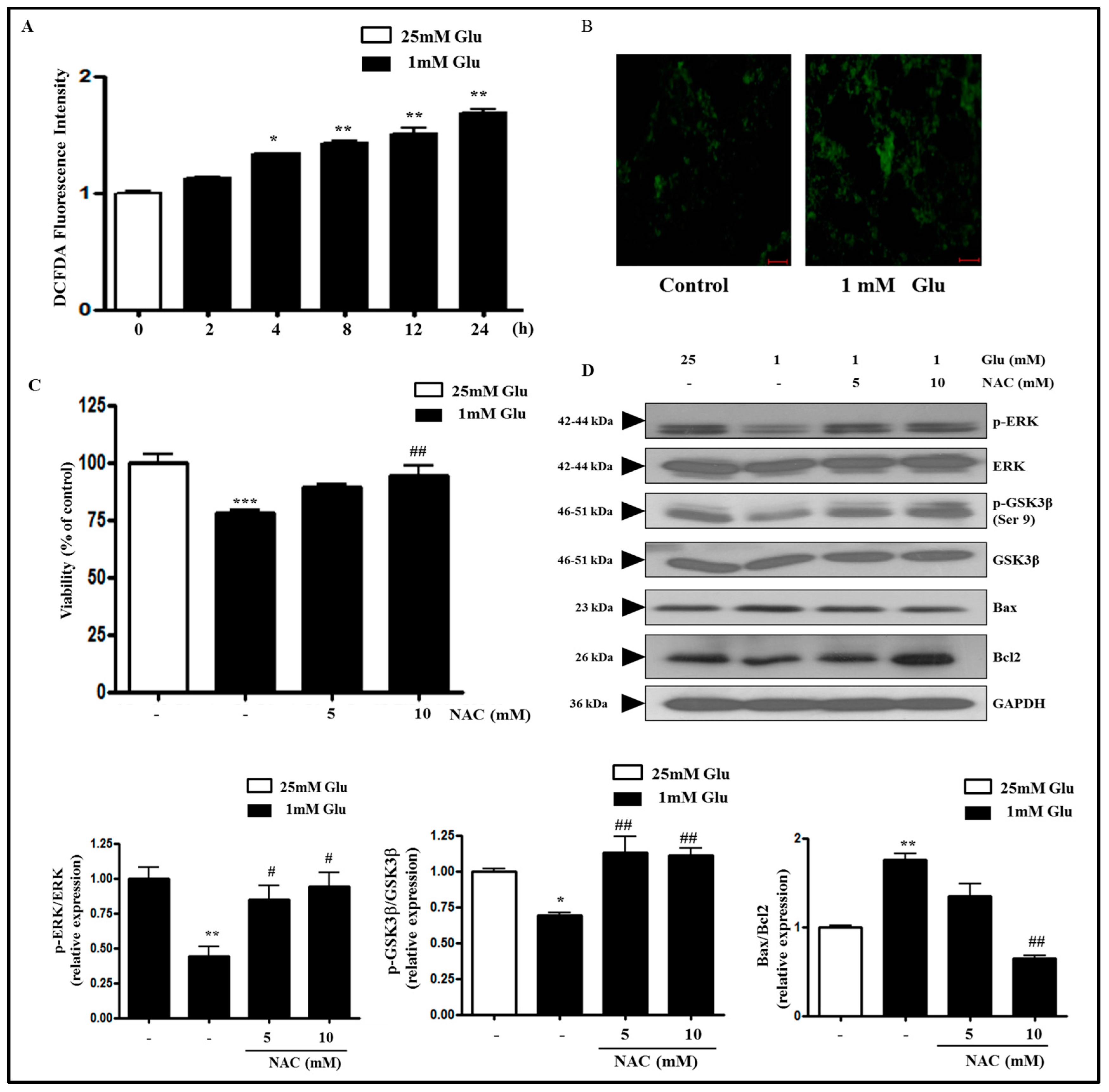
2.2. Glucose Deficiency Induces Cell Death through ROS Accumulation in SH-SY5Y Cells
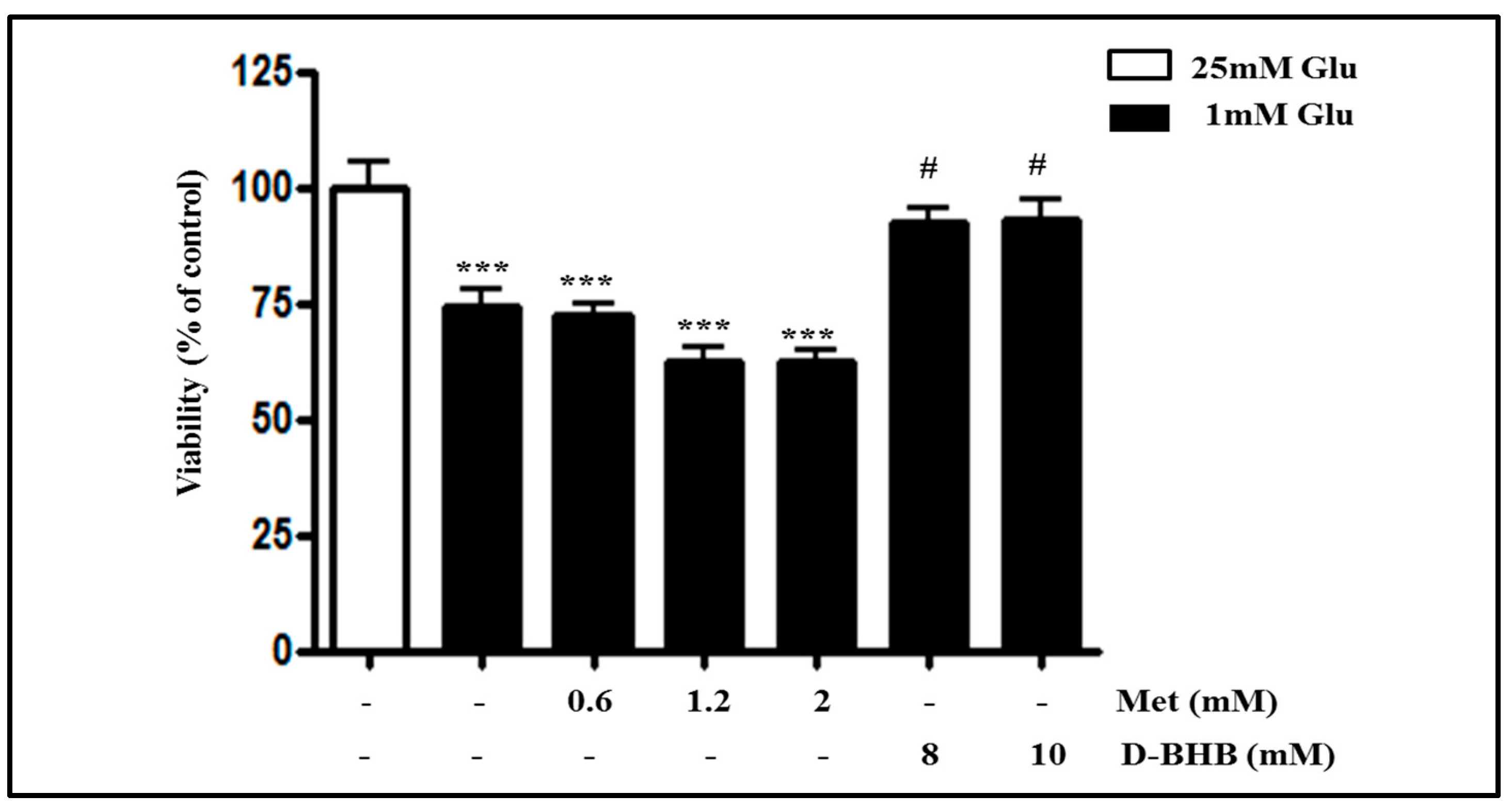
2.3. Metformin Aggravates Glucose Deficiency-Induced Cell Death in SH-SY5Y Cells
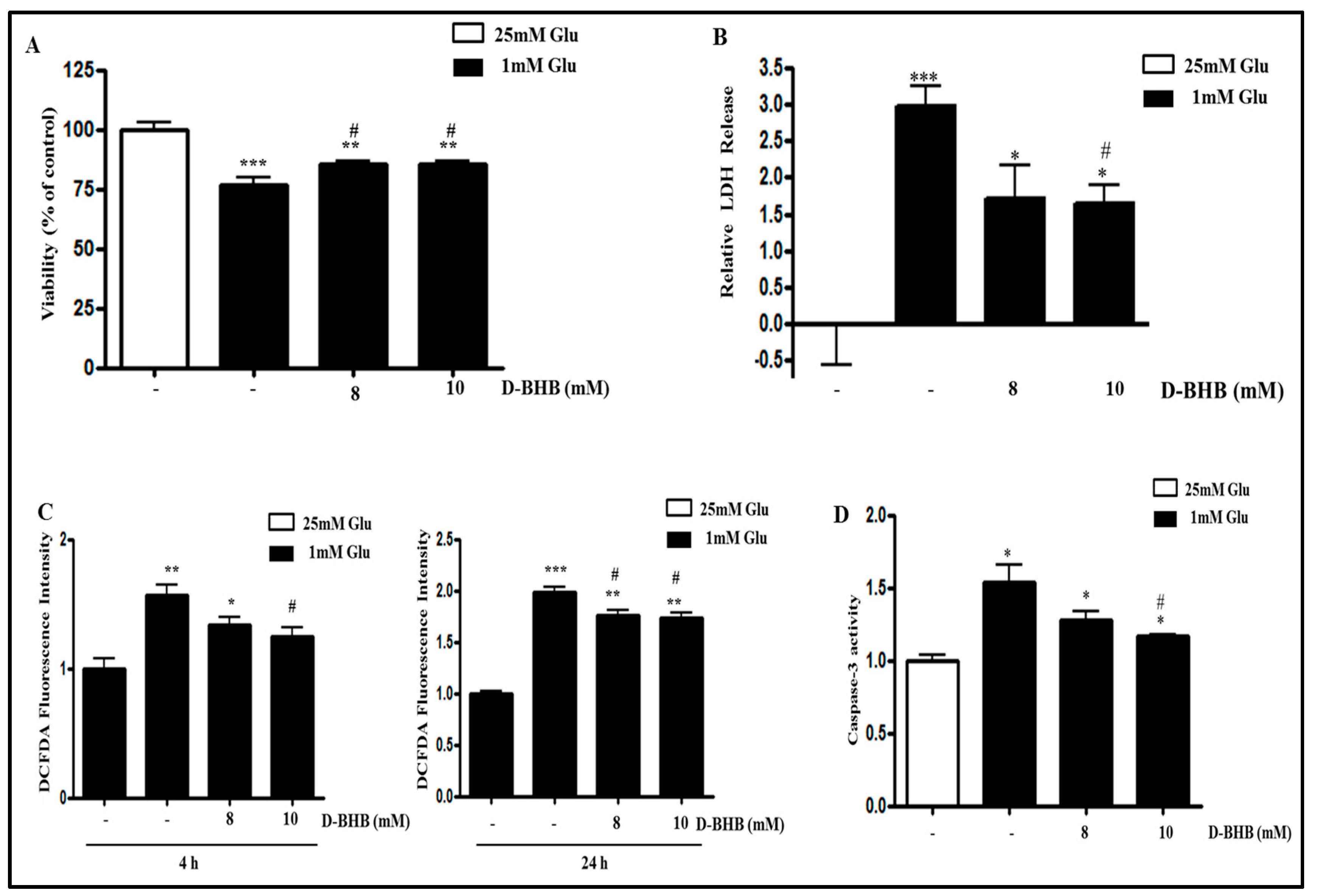
2.4. D-BHB Attenuates Glucose Deficiency-Induced Cytotoxicity and ROS Production in SH-SY5Y Cells
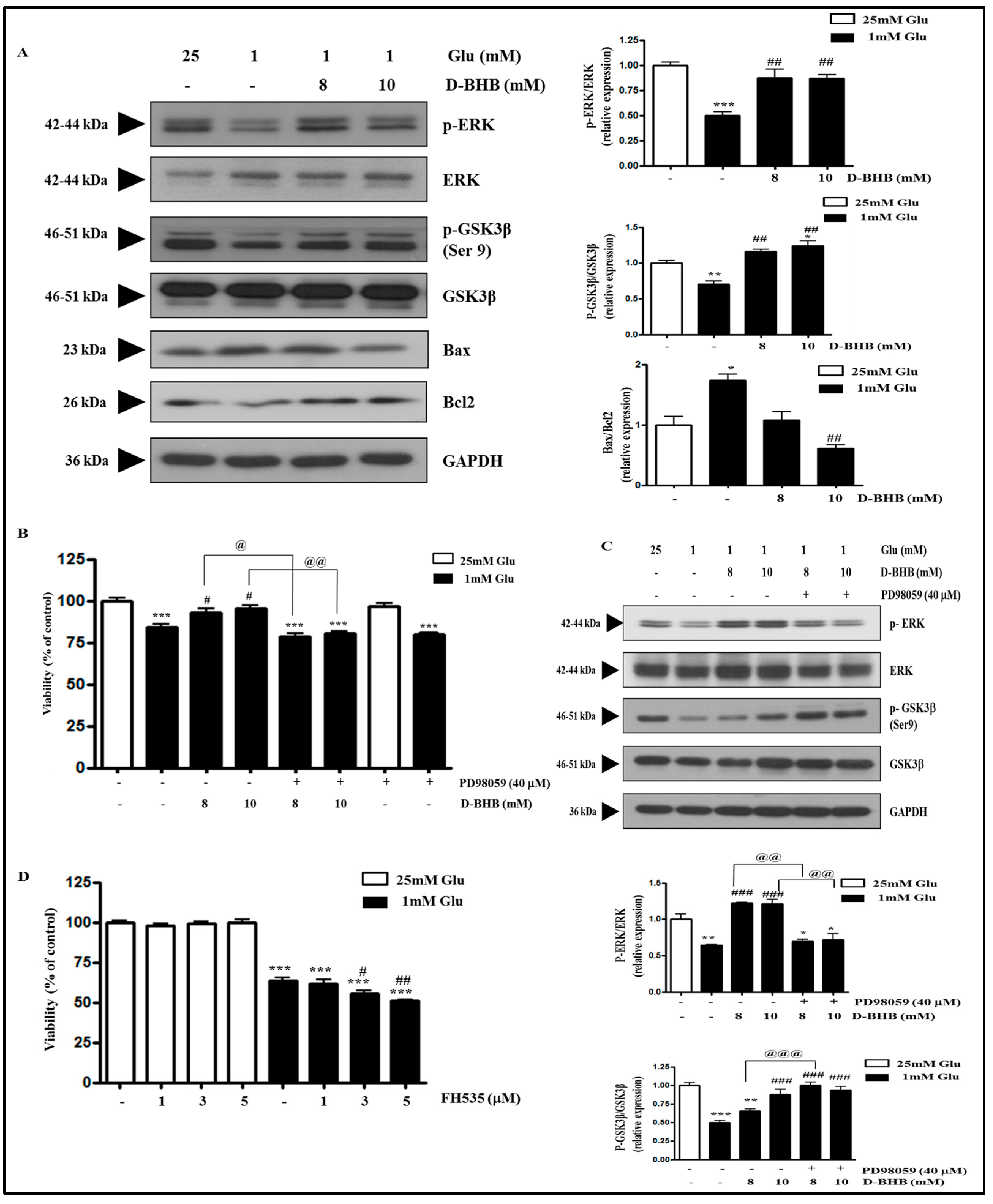
2.5. D-BHB Reverses Glucose Deficiency-Induced Reduction in the Phosphorylation of ERK and GSK3β in SH-SY5Y Cells
2.6. Metformin and D-BHB Affect Low Glucose-Induced Cell Death in Primary Neuronal Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. MTT Viability Assay
4.4. LDH Cytotoxicity Assay
4.5. ATP Measurement Assay
4.6. DCFDA Fluorescence Assay
4.7. Caspase-3 Activity Assay
4.8. Western Blot Analysis
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Russo, V.C.; Higgins, S.; Werther, G.A.; Cameron, F.J. Effects of fluctuating glucose levels on neuronal cells in vitro. Neurochem. Res. 2012, 37, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Isaev, N.K.; Stel Ashuk, E.V.; Zorov, D.B. Cellular mechanisms of brain hypoglycemia. Biochemistry (Moscow) 2007, 72, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, X.D.; Liu, W.; Zhang, T.Y.; Zuo, J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. J. Cell Mol. Med. 2003, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal nadph oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Takeshima, T.; Kashiwaya, Y.; Nakaso, K.; Nakashima, K. d-β-hydroxybutyrate protects dopaminergic SH-SY5Y cells in a rotenone model of Parkinson’s disease. J. Neurosci. Res. 2006, 84, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.S.; Hah, Y.S.; Nilufar, R.; Han, J.; Bong, J.H.; Kang, S.S.; Cho, G.J.; Choi, W.S. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res. 2006, 83, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Haces, M.L.; Hernandez-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kitamura, Y.; Mori, S.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn. J. Pharmacol. 2002, 89, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.K.; Nguyen, N.; Pham, L.; Truong, D.D. Ketogenic diet prevents cardiac arrest-induced cerebral ischemic neurodegeneration. J. Neural Transm. (Vienna) 2008, 115, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.A.; Rensing, N.; Thio, L.L. Ketogenic diet reduces hypoglycemia-induced neuronal death in young rats. Neurosci. Lett. 2005, 385, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Julio-Amilpas, A.; Montiel, T.; Soto-Tinoco, E.; Geronimo-Olvera, C.; Massieu, L. Protection of hypoglycemia-induced neuronal death by β-hydroxybutyrate involves the preservation of energy levels and decreased production of reactive oxygen species. J. Cereb. Blood Flow Metab. 2015, 35, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Qaseem, A.; Humphrey, L.L.; Sweet, D.E.; Starkey, M.; Shekelle, P. Oral pharmacologic treatment of type 2 diabetes mellitus: A clinical practice guideline from the american college of physicians. Annul. Int. Med. 2012, 156, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.M.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.B.; Matsuzaki, H.; Haorah, J.; Ulrich, A.; Standop, J.; Ding, X.Z.; Adrian, T.E.; Pour, P.M. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology 2001, 120, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin d1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Al-Sammarraie, N.; DiPette, D.J.; Singh, U.S. Metformin impairs RHO GTPase signaling to induce apoptosis in neuroblastoma cells and inhibits growth of tumors in the xenograft mouse model of neuroblastoma. Oncotarget 2014, 5, 11709–11722. [Google Scholar] [CrossRef] [PubMed]
- Mouhieddine, T.H.; Nokkari, A.; Itani, M.M.; Chamaa, F.; Bahmad, H.; Monzer, A.; El-Merahbi, R.; Daoud, G.; Eid, A.; Kobeissy, F.H.; et al. Metformin and ARA-A effectively suppress brain cancer by targeting cancer stem/progenitor cells. Front. Neurosci. 2015, 9, 442. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Gigoni, A.; Wurth, R.; Cancedda, R.; Florio, T.; Pagano, A. Metformin inhibition of neuroblastoma cell proliferation is differently modulated by cell differentiation induced by retinoic acid or overexpression of NDM29 non-coding RNA. Cancer Cell. Int. 2014, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin prevents dopaminergic neuron death in MPTP/p-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [PubMed]
- Dulovic, M.; Jovanovic, M.; Xilouri, M.; Stefanis, L.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Paunovic, V.; Ardah, M.T.; El-Agnaf, O.M.; Kostic, V.; et al. The protective role of AMP-activated protein kinase in α-synuclein neurotoxicity in vitro. Neurobiol. Dis. 2014, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Chan, D.K.; Haugrud, A.B.; Miskimins, W.K. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PLoS ONE 2014, 9, e108444. [Google Scholar] [CrossRef] [PubMed]
- Watcharasit, P.; Bijur, G.N.; Zmijewski, J.W.; Song, L.; Zmijewska, A.; Chen, X.; Johnson, G.V.W.; Jope, R.S. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc. Natl. Acad. Sci. USA 2002, 99, 7951–7955. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Barnard, D.; Diaz, B.; Clawson, D.; Marshall, M. Oncogenes, growth factors and phorbol esters regulate Raf-1 through common mechanisms. Oncogene 1998, 17, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.L.; Figueroa, C.; Brtva, T.R.; Zhu, T.; Taylor, J.; Barber, T.D.; Vojtek, A.B. Negative regulation of the serine/threonine kinase B-Raf by AKT. J. Biol. Chem. 2000, 275, 27354–27359. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia—Reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Park, D.B. Metformin promotes apoptosis but suppresses autophagy in glucose-deprived H4IIE hepatocellular carcinoma cells. Diabetes Metab. J. 2015, 39, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Patockova, J.; Marhol, P.; Tumova, E.; Krsiak, M.; Rokyta, R.; Stipek, S.; Crkovska, J.; Andel, M. Oxidative stress in the brain tissue of laboratory mice with acute post insulin hypoglycemia. Physiol. Res. 2003, 52, 131–135. [Google Scholar] [PubMed]
- Hino, K.; Nishikawa, M.; Sato, E.; Inoue, M. l-carnitine inhibits hypoglycemia-induced brain damage in the rat. Brain Res. 2005, 1053, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Del’guidice, T.; Sotnikova, T.D.; Lemasson, M.; Gainetdinov, R.R. Beyond camp: The regulation of AKT and GSK3β by dopamine receptors. Front. Mol. Neurosci. 2011, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Kyosseva, S.V. Mitogen-activated protein kinase signaling. Int. Rev. Neurobiol. 2004, 59, 201–220. [Google Scholar] [PubMed]
- Roux, P.P.; Blenis, J. Erk and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Biol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Santos, C.; Ferrer, I.; Reiriz, J.; Vinals, F.; Barrachina, M.; Ambrosio, S. MPP+ increases α-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res. 2002, 935, 32–39. [Google Scholar] [CrossRef]
- Medina, M.G.; Ledesma, M.D.; Dominguez, J.E.; Medina, M.; Zafra, D.; Alameda, F.; Dotti, C.G.; Navarro, P. Tissue plasminogen activator mediates amyloid-induced neurotoxicity via ERK1/2 activation. EMBO J. 2005, 24, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.R.; Zhang, P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: Role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J. Neurochem. 1999, 72, 112–119. [Google Scholar] [CrossRef] [PubMed]
- King, M.K.; Pardo, M.; Cheng, Y.; Downey, K.; Jope, R.S.; Beurel, E. Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol. Ther. 2014, 141, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nito, C.; Kamada, H.; Nishi, T.; Chan, P.H. Activation of the AKT/GSK3β signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2006, 26, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly (ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [PubMed]
- Sosnicki, S.; Kapral, M.; Weglarz, L. Molecular targets of metformin antitumor action. Pharmacol. Rep. 2016, 68, 918–925. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamichhane, S.; Bastola, T.; Pariyar, R.; Lee, E.-S.; Lee, H.-S.; Lee, D.H.; Seo, J. ROS Production and ERK Activity Are Involved in the Effects of d-β-Hydroxybutyrate and Metformin in a Glucose Deficient Condition. Int. J. Mol. Sci. 2017, 18, 674. https://doi.org/10.3390/ijms18030674
Lamichhane S, Bastola T, Pariyar R, Lee E-S, Lee H-S, Lee DH, Seo J. ROS Production and ERK Activity Are Involved in the Effects of d-β-Hydroxybutyrate and Metformin in a Glucose Deficient Condition. International Journal of Molecular Sciences. 2017; 18(3):674. https://doi.org/10.3390/ijms18030674
Chicago/Turabian StyleLamichhane, Santosh, Tonking Bastola, Ramesh Pariyar, Eun-Sol Lee, Ho-Sub Lee, Dae Ho Lee, and Jungwon Seo. 2017. "ROS Production and ERK Activity Are Involved in the Effects of d-β-Hydroxybutyrate and Metformin in a Glucose Deficient Condition" International Journal of Molecular Sciences 18, no. 3: 674. https://doi.org/10.3390/ijms18030674