
Exome Sequencing Identified a Novel FBN2 Mutation in a Chinese Family with Congenital Contractural Arachnodactyly


Abstract
:1. Introduction
2. Results
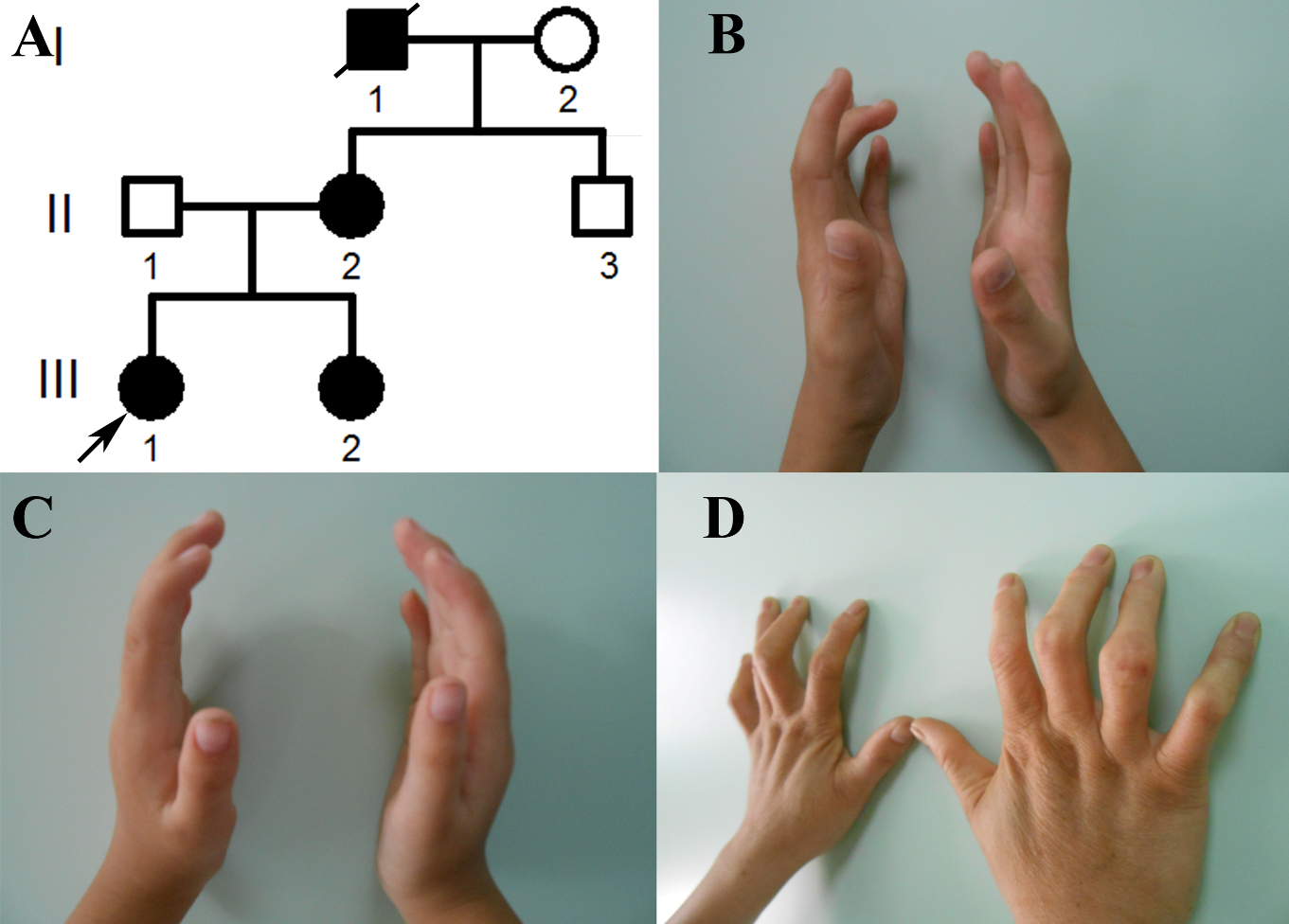
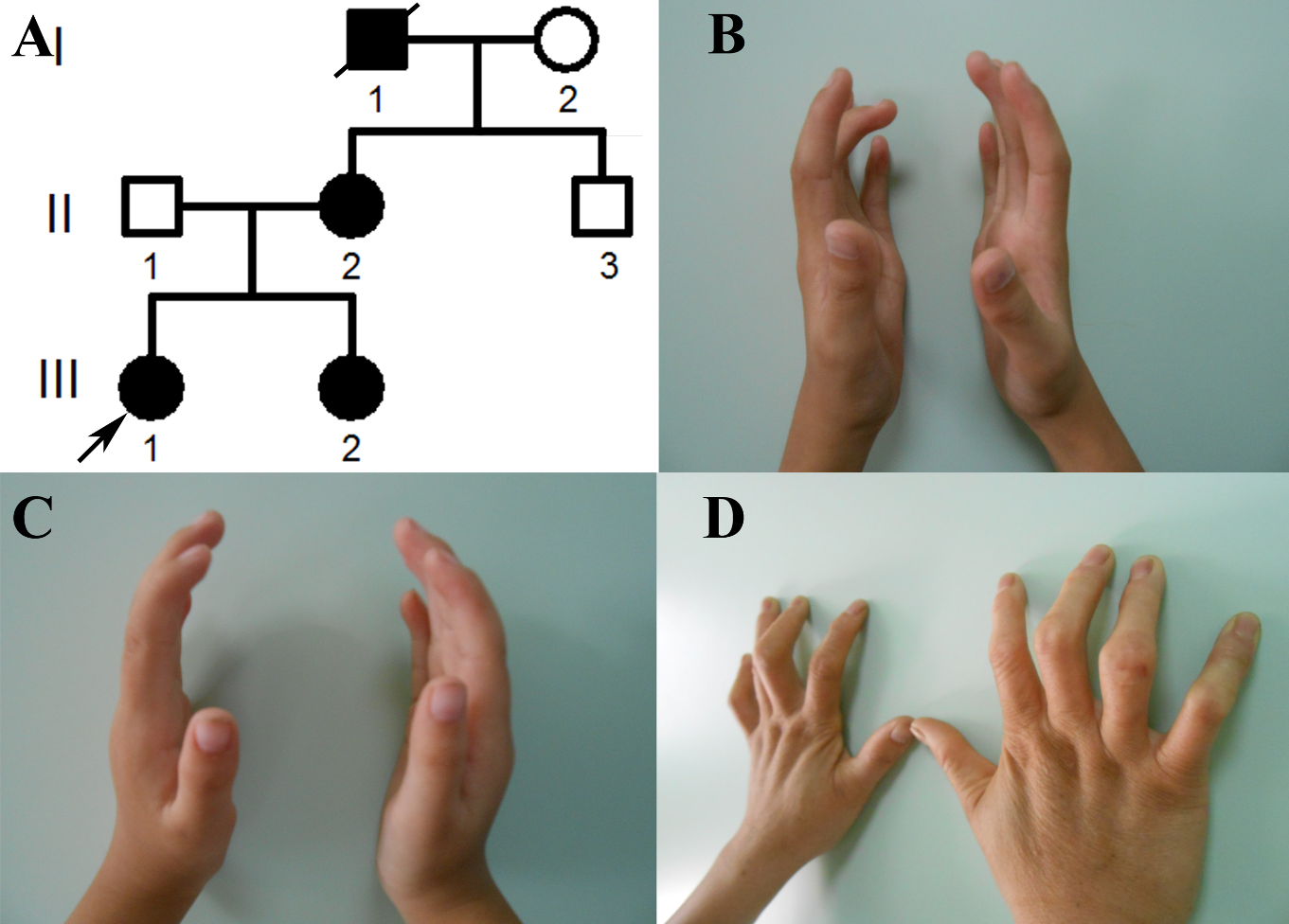
2.1. The Clinical Phenotypes of the Congenital Contractural Arachnodactyly (CCA) Pedigree
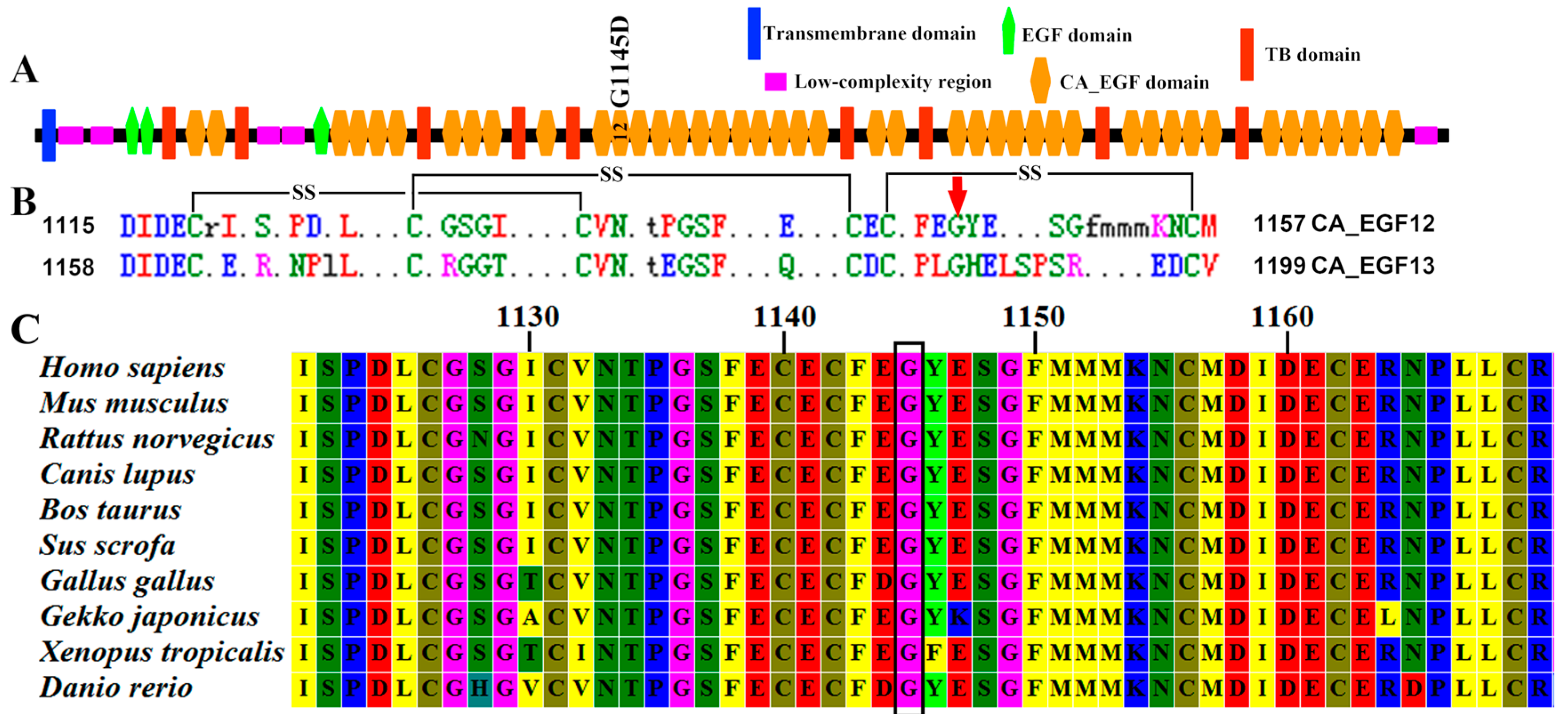
2.2. Mutation Screening for the CCA Pedigree
2.3. Co-Segregation Analysis and Mutation Validation
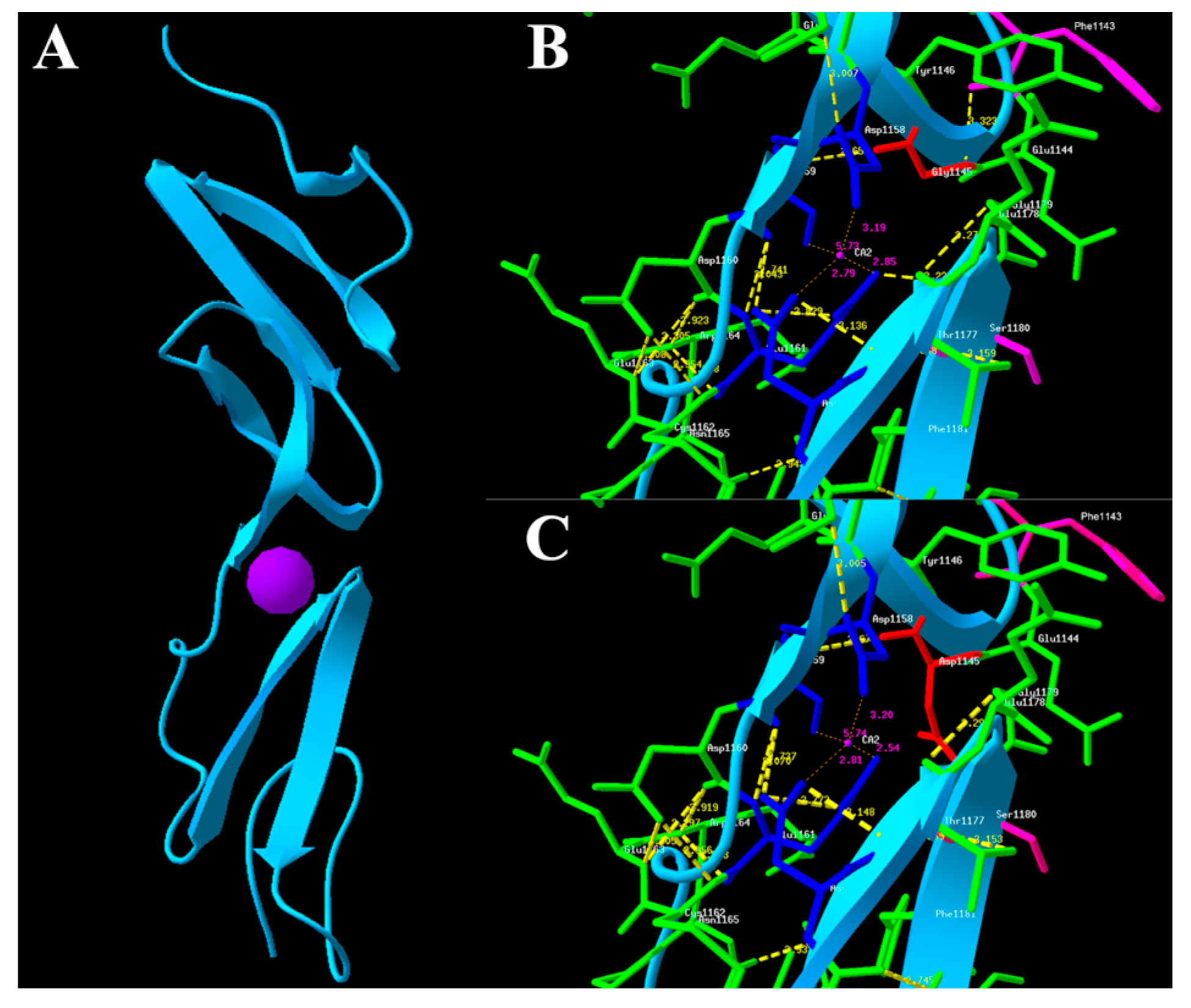
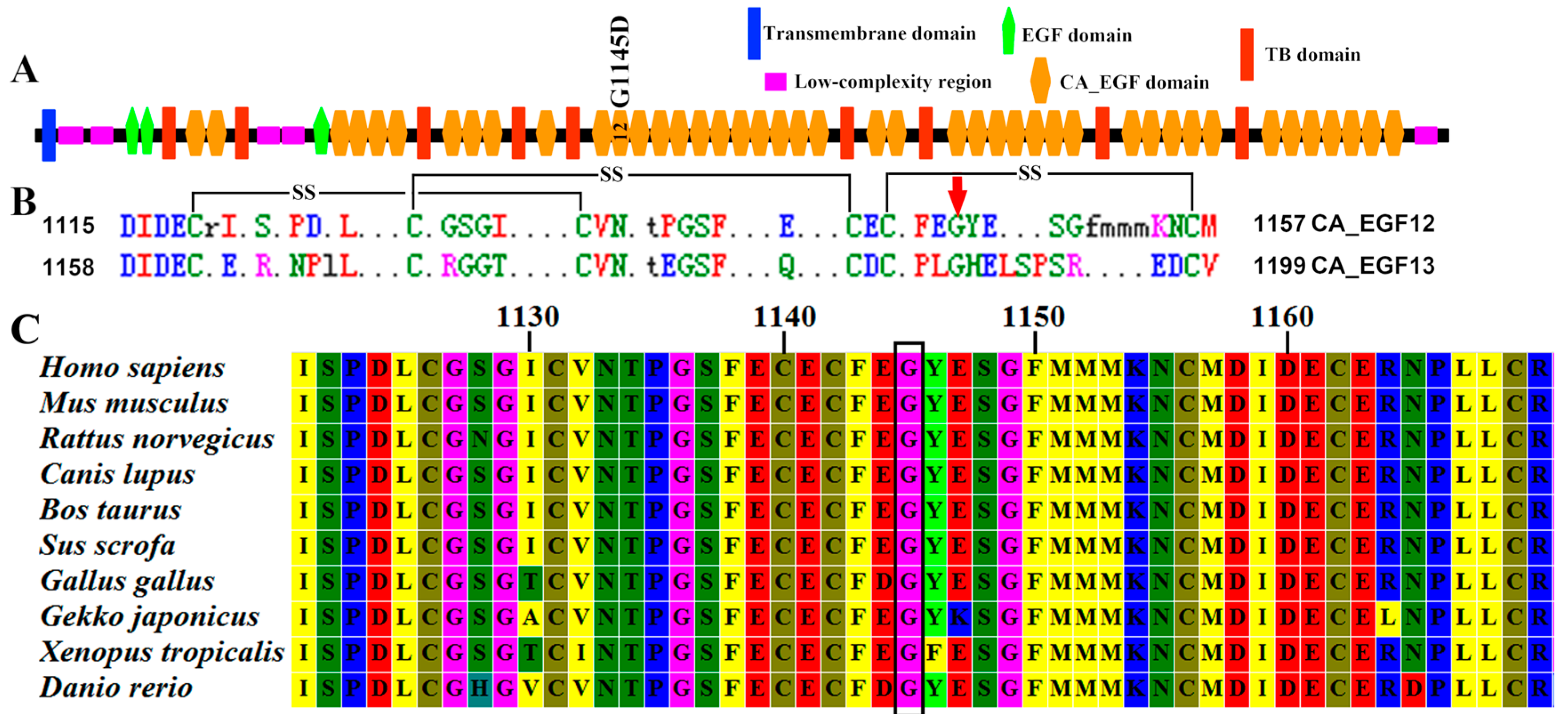
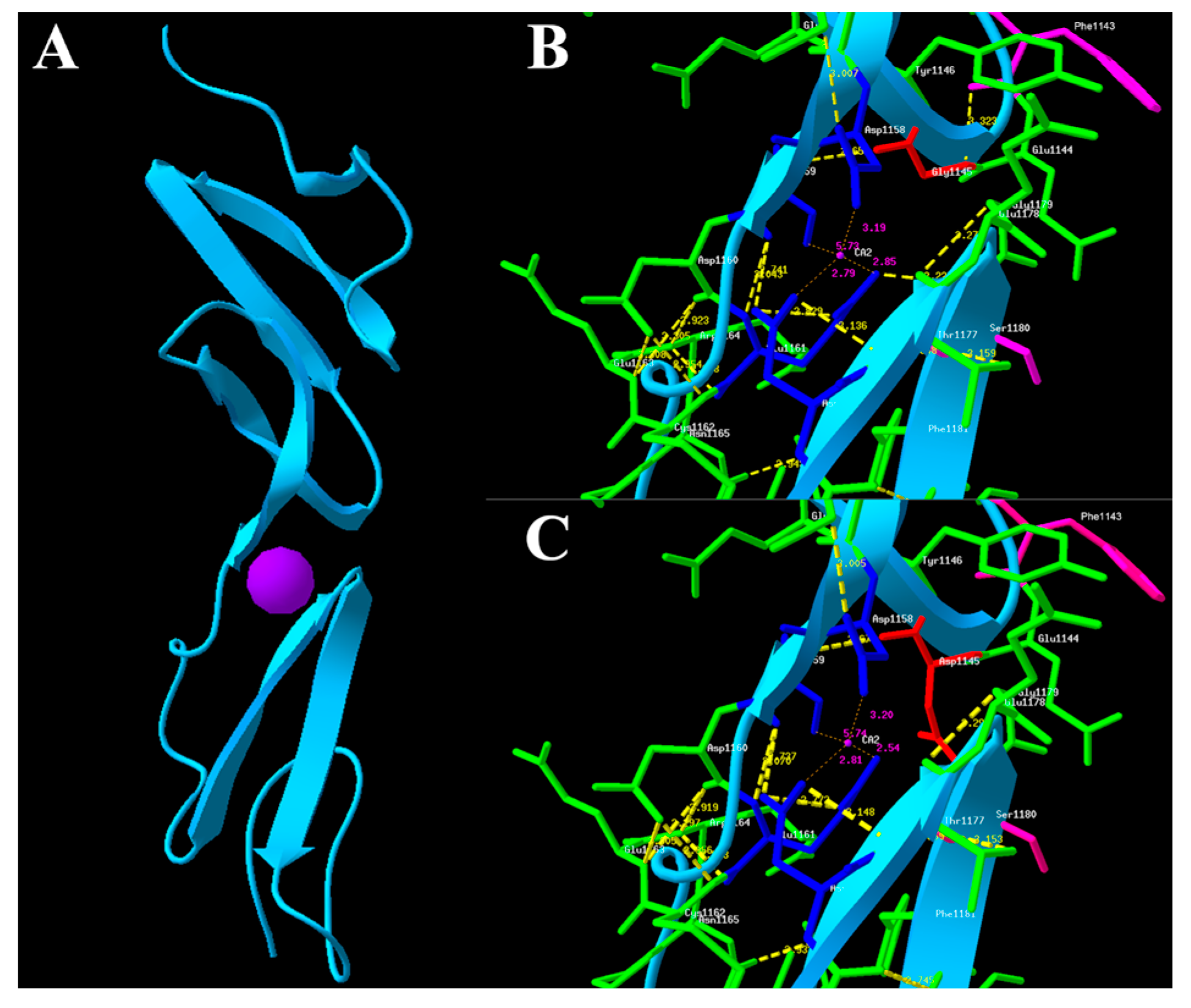
2.4. In Silico Functional Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Genomic DNA Preparation
4.3. Whole Exome Capture and Sequencing
4.4. Analysis of Sequencing Data
4.5. Mutation Validation by Sanger Sequencing
4.6. Molecular Modeling of the FBN2 Protein and Conservation Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Godfrey, M. Congenital contractural arachnodactyly. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Putnam, E.A.; Park, E.S.; Aalfs, C.M.; Hennekam, R.C.; Milewicz, D.M. Parental somatic and germ-line mosaicism for a FBN2 mutation and analysis of FBN2 transcript levels in dermal fibroblasts. Am. J. Hum. Genet. 1997, 60, 818–827. [Google Scholar] [PubMed]
- Babcock, D.; Gasner, C.; Francke, U.; Maslen, C. A single mutation that results in an asp to his substitution and partial exon skipping in a family with congenital contractural arachnodactyly. Hum. Genet. 1998, 103, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Jurko, A., Jr.; Krsiakova, J.; Minarik, M.; Tonhajzerova, I. Congenital contractural arachnodactyly (Beals–Hecht syndrome): A rare connective tissue disorder. Wien. Klin. Wochenschr. 2013, 125, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Clericuzio, C.L.; Godfrey, M. Familial occurrence of typical and severe lethal congenital contractural arachnodactyly caused by missplicing of Exon 34 of fibrillin-2. Am. J. Hum. Genet. 1996, 59, 1027–1034. [Google Scholar] [PubMed]
- Frederic, M.Y.; Monino, C.; Marschall, C.; Hamroun, D.; Faivre, L.; Jondeau, G.; Klein, H.G.; Neumann, L.; Gautier, E.; Binquet, C.; et al. The FBN2 gene: New mutations, locus-specific database (universal mutation database FBN2), and genotype-phenotype correlations. Hum. Mutat. 2009, 30, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Godfrey, M.; Vitale, E.; Hori, H.; Mattei, M.G.; Sarfarazi, M.; Tsipouras, P.; Ramirez, F.; Hollister, D.W. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature 1991, 352, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, S.; Ramirez, F. Fibrillin microfibrils in bone physiology. Matrix Biol. 2016, 52, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Bastian, N.A.; Bayne, R.A.; Hummitzsch, K.; Hatzirodos, N.; Bonner, W.M.; Hartanti, M.D.; Irving-Rodgers, H.F.; Anderson, R.A.; Rodgers, R.J. Regulation of fibrillins and modulators of TGFβ in fetal bovine and human ovaries. Reproduction 2016, 152, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Sengle, G.; Carlberg, V.; Tufa, S.F.; Charbonneau, N.L.; Smaldone, S.; Carlson, E.J.; Ramirez, F.; Keene, D.R.; Sakai, L.Y. Abnormal activation of BMP signaling causes myopathy in FBN2 null mice. PLoS Genet. 2015, 11, e1005340. [Google Scholar] [CrossRef] [PubMed]
- Arteaga-Solis, E.; Gayraud, B.; Lee, S.Y.; Shum, L.; Sakai, L.; Ramirez, F. Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 2001, 154, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Collod-Beroud, G.; Loeys, B.L.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of mutation type and location on clinical outcome in 1013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: An international study. Am. J. Hum. Genet. 2007, 81, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Downing, A.K.; Knott, V.; Werner, J.M.; Cardy, C.M.; Campbell, I.D.; Handford, P.A. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: Implications for the marfan syndrome and other genetic disorders. Cell 1996, 85, 597–605. [Google Scholar] [CrossRef]
- Smallridge, R.S.; Whiteman, P.; Werner, J.M.; Campbell, I.D.; Handford, P.A.; Downing, A.K. Solution structure and dynamics of a calcium binding epidermal growth factor-like domain pair from the neonatal region of human fibrillin-1. J. Biol. Chem. 2003, 278, 12199–12206. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using polyphen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. Revel: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Heger, A.; Holm, L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins 2000, 41, 224–237. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | III-1 | III-2 | II-2 |
|---|---|---|---|
| Gender | Female | Female | Female |
| Age (years) | 15 | 12 | 43 |
| Mutation | Gly1145Asp | Gly1145Asp | Gly1145Asp |
| Genotype | Heterozygote | Heterozygote | Heterozygote |
| Tall stature | − | − | − |
| External ear anomalies | − | − | − |
| Arachnodactyly | + | + | + |
| Camptodactyly | + | + | + |
| Contractures of knuckles | + | + | + |
| Contractures of large joints | − | − | − |
| Scoliosis | − | − | − |
| Pectus deformity | − | − | − |
| Ocular complication | − | − | − |
| Cardiovascular complication | − | − | − |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, G.; Zu, B.; Wang, B.; Wang, Z.; Xu, Y.; Fu, Q. Exome Sequencing Identified a Novel FBN2 Mutation in a Chinese Family with Congenital Contractural Arachnodactyly. Int. J. Mol. Sci. 2017, 18, 626. https://doi.org/10.3390/ijms18040626
You G, Zu B, Wang B, Wang Z, Xu Y, Fu Q. Exome Sequencing Identified a Novel FBN2 Mutation in a Chinese Family with Congenital Contractural Arachnodactyly. International Journal of Molecular Sciences. 2017; 18(4):626. https://doi.org/10.3390/ijms18040626
Chicago/Turabian StyleYou, Guoling, Bailing Zu, Bo Wang, Zhigang Wang, Yunlan Xu, and Qihua Fu. 2017. "Exome Sequencing Identified a Novel FBN2 Mutation in a Chinese Family with Congenital Contractural Arachnodactyly" International Journal of Molecular Sciences 18, no. 4: 626. https://doi.org/10.3390/ijms18040626