
Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production


Abstract
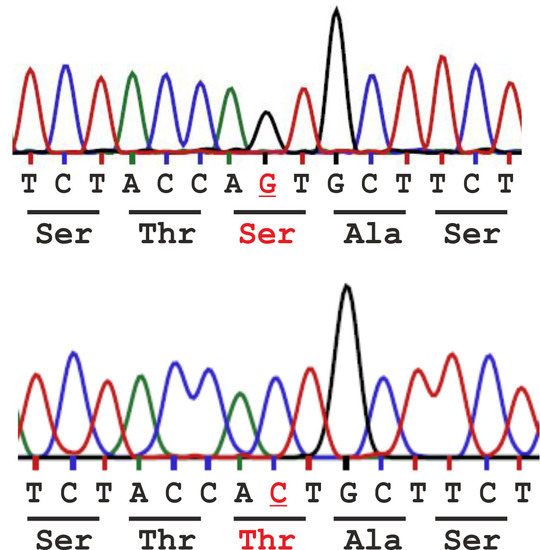
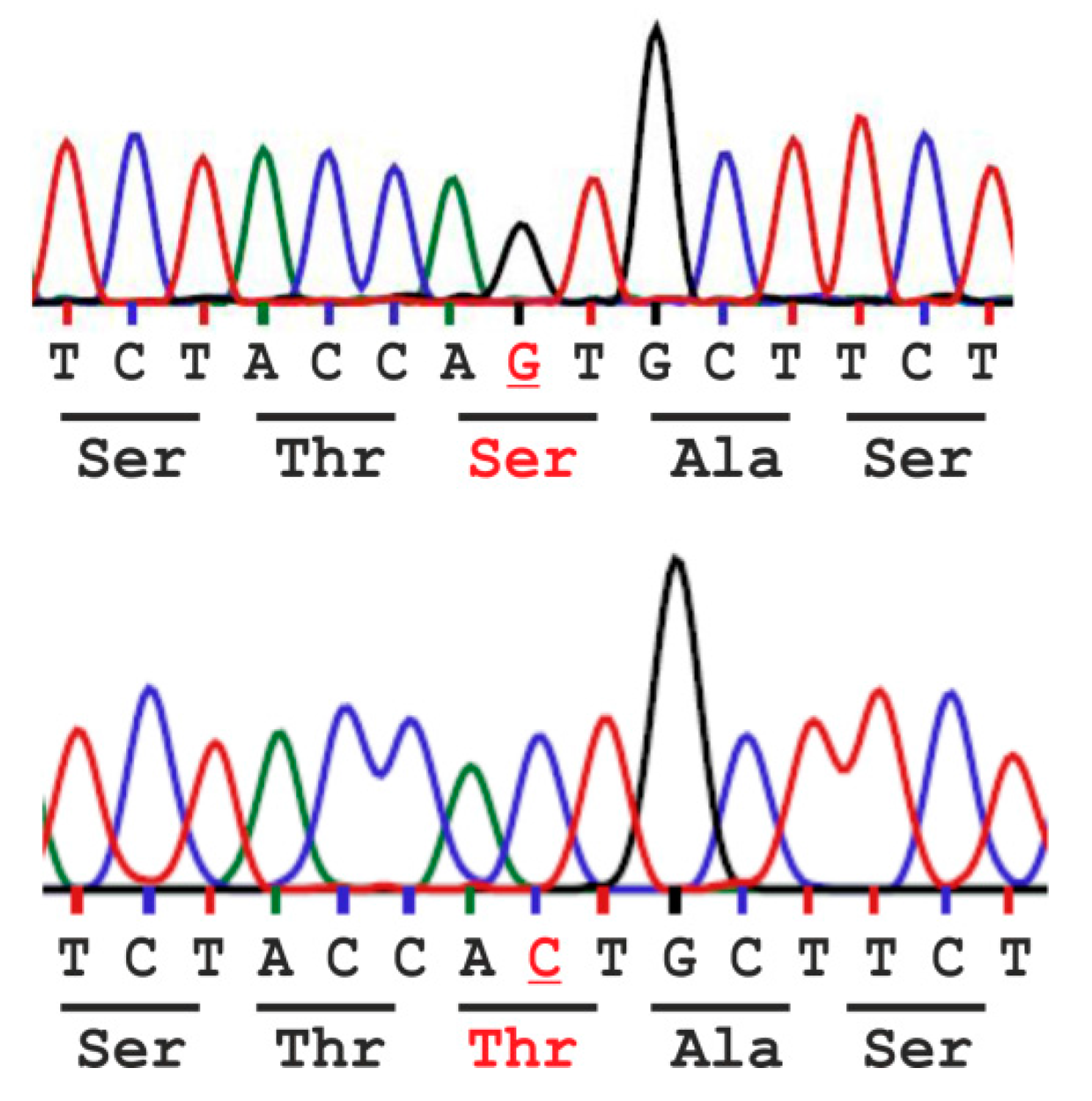
:
1. Introduction
2. Results
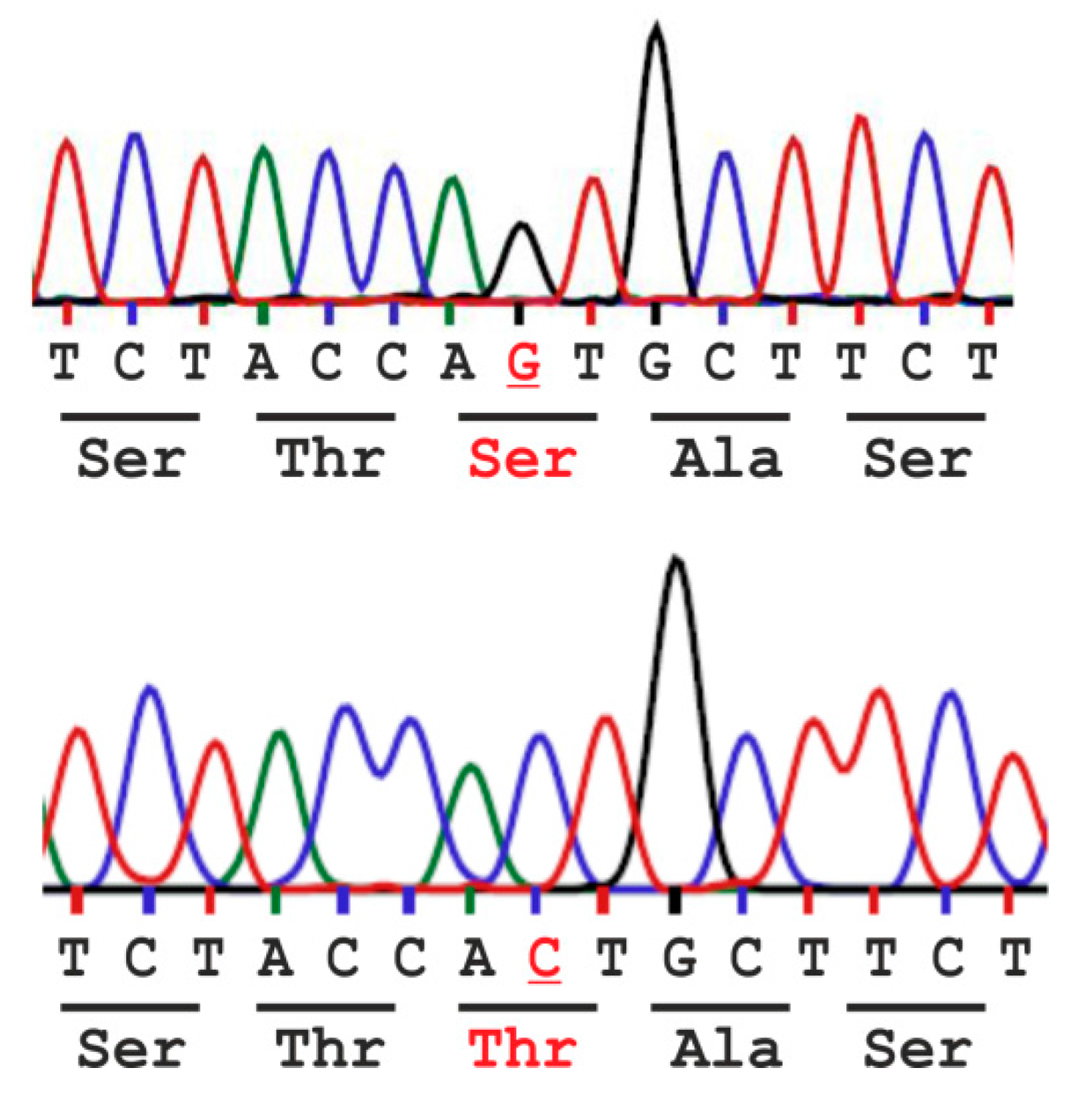
2.1. Analysis of the Population Frequency of a Natural AGA Variant NM_000027.3:c.446C>G - p.Thr149Ser
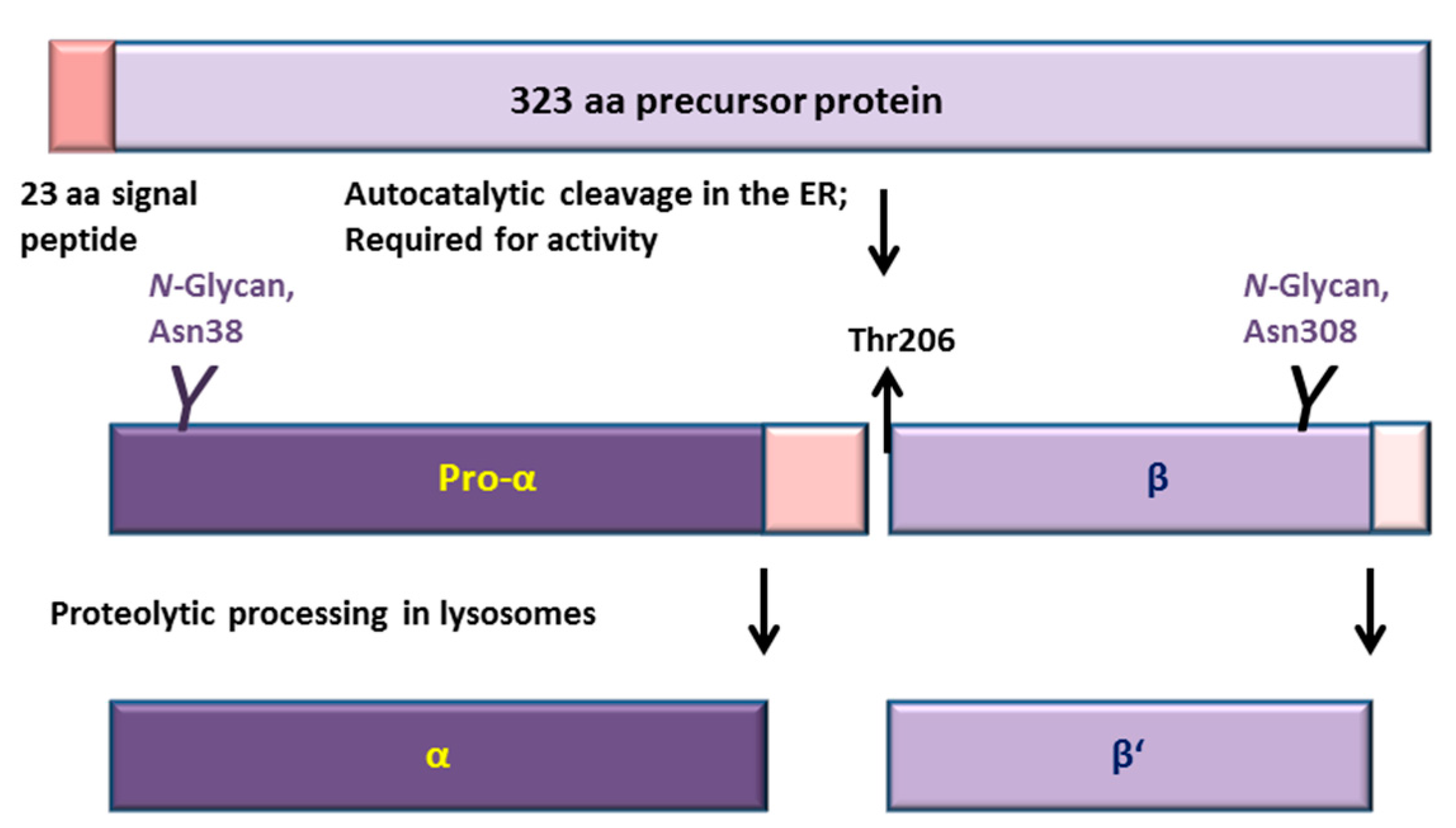
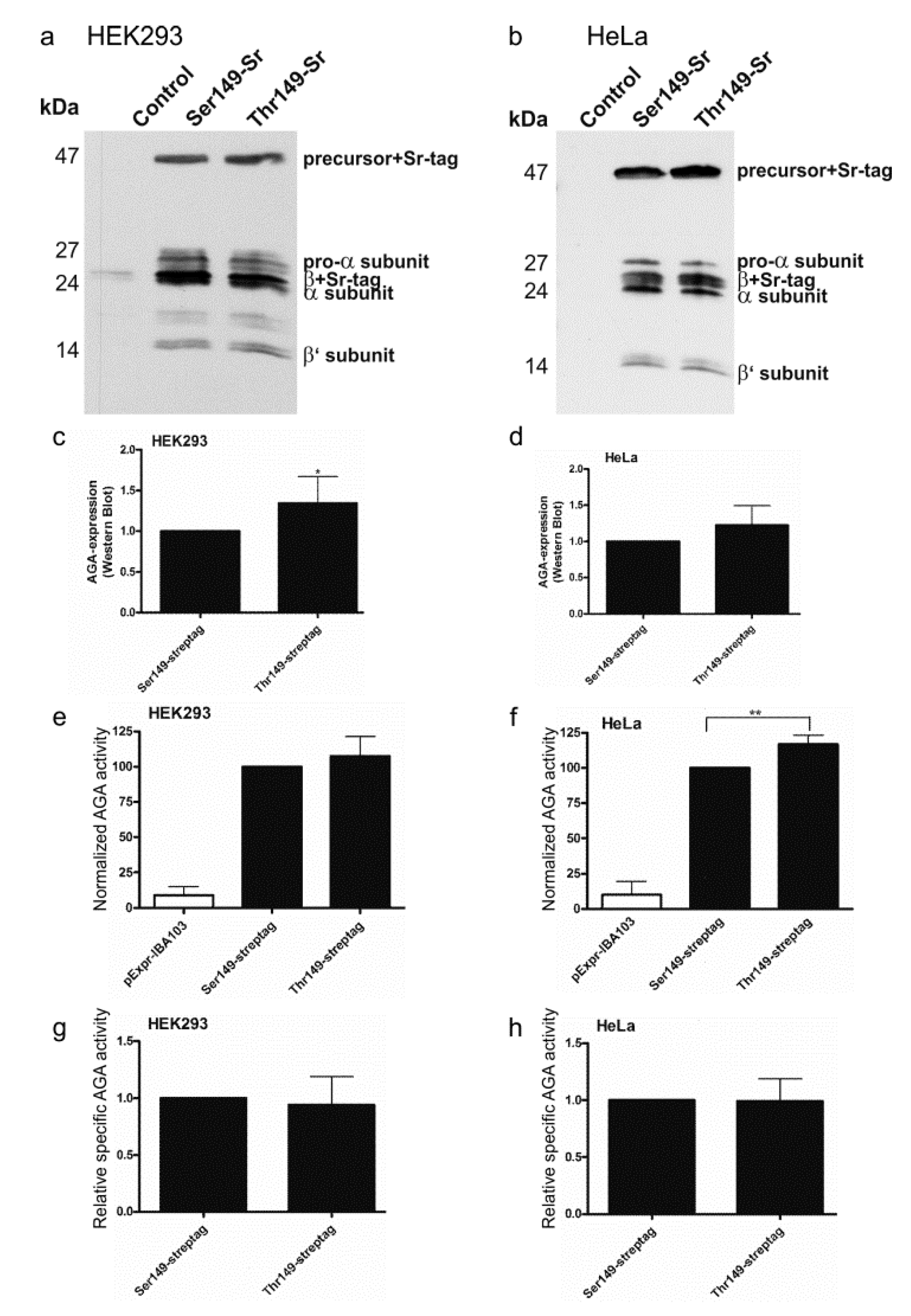
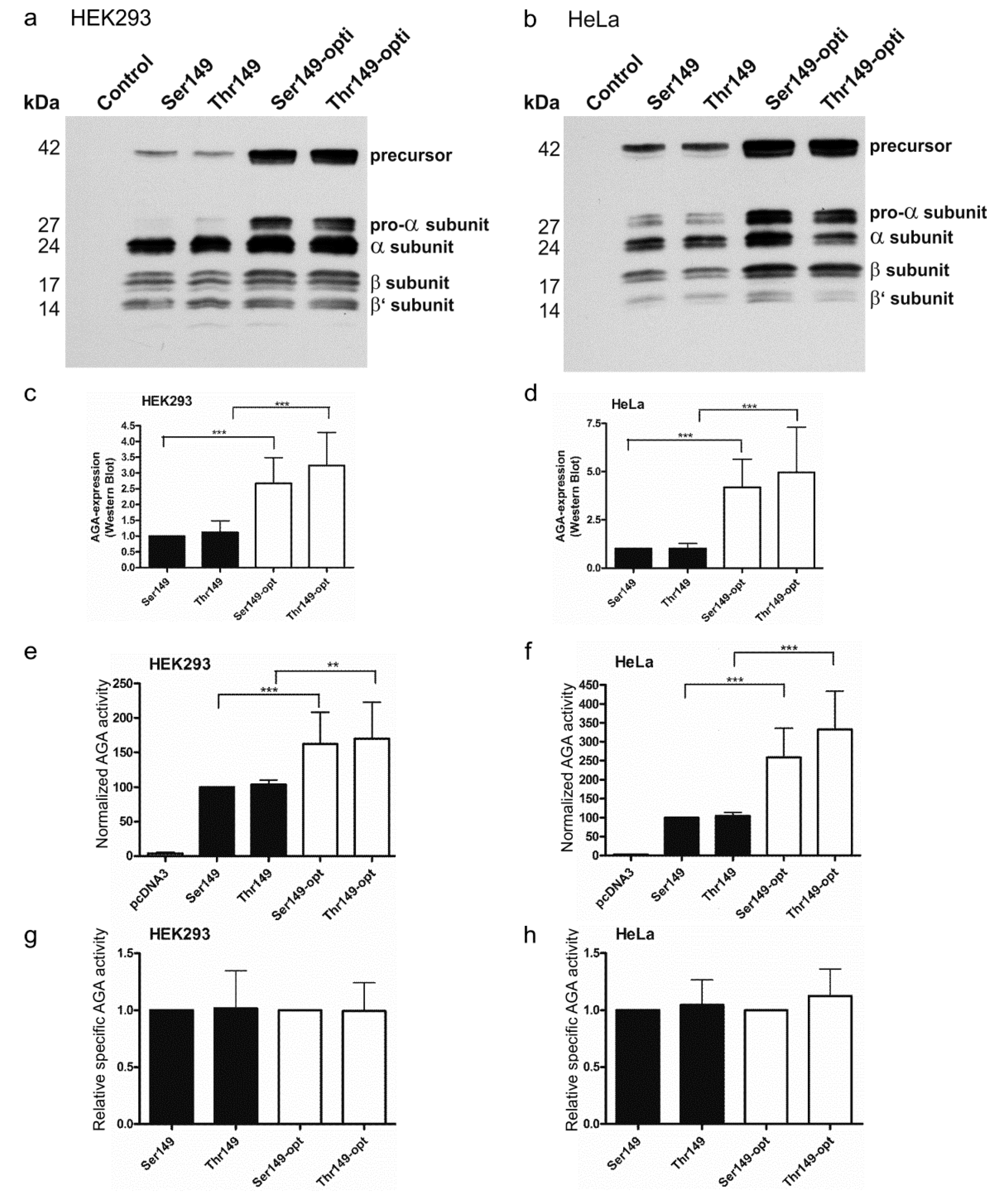
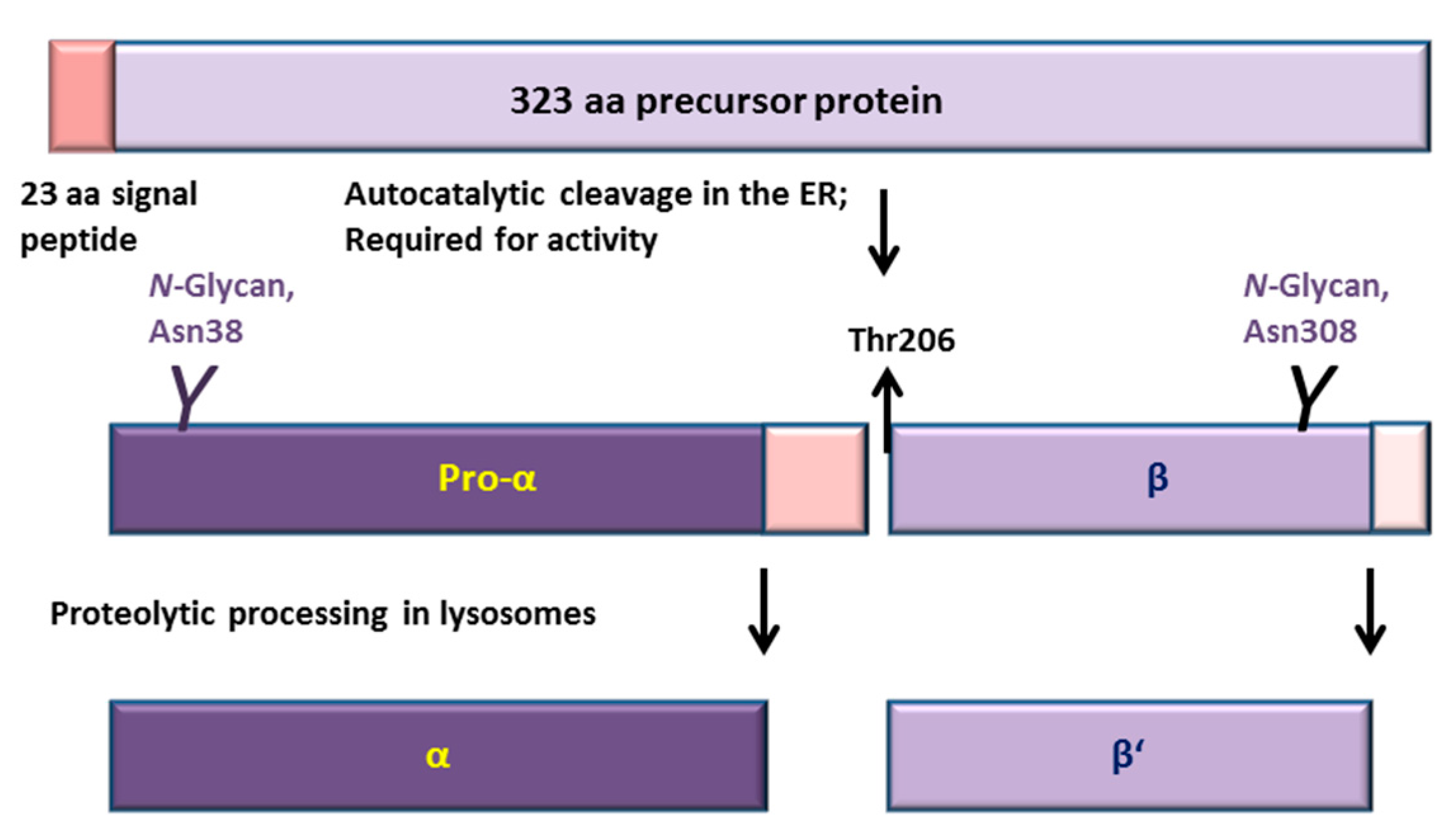
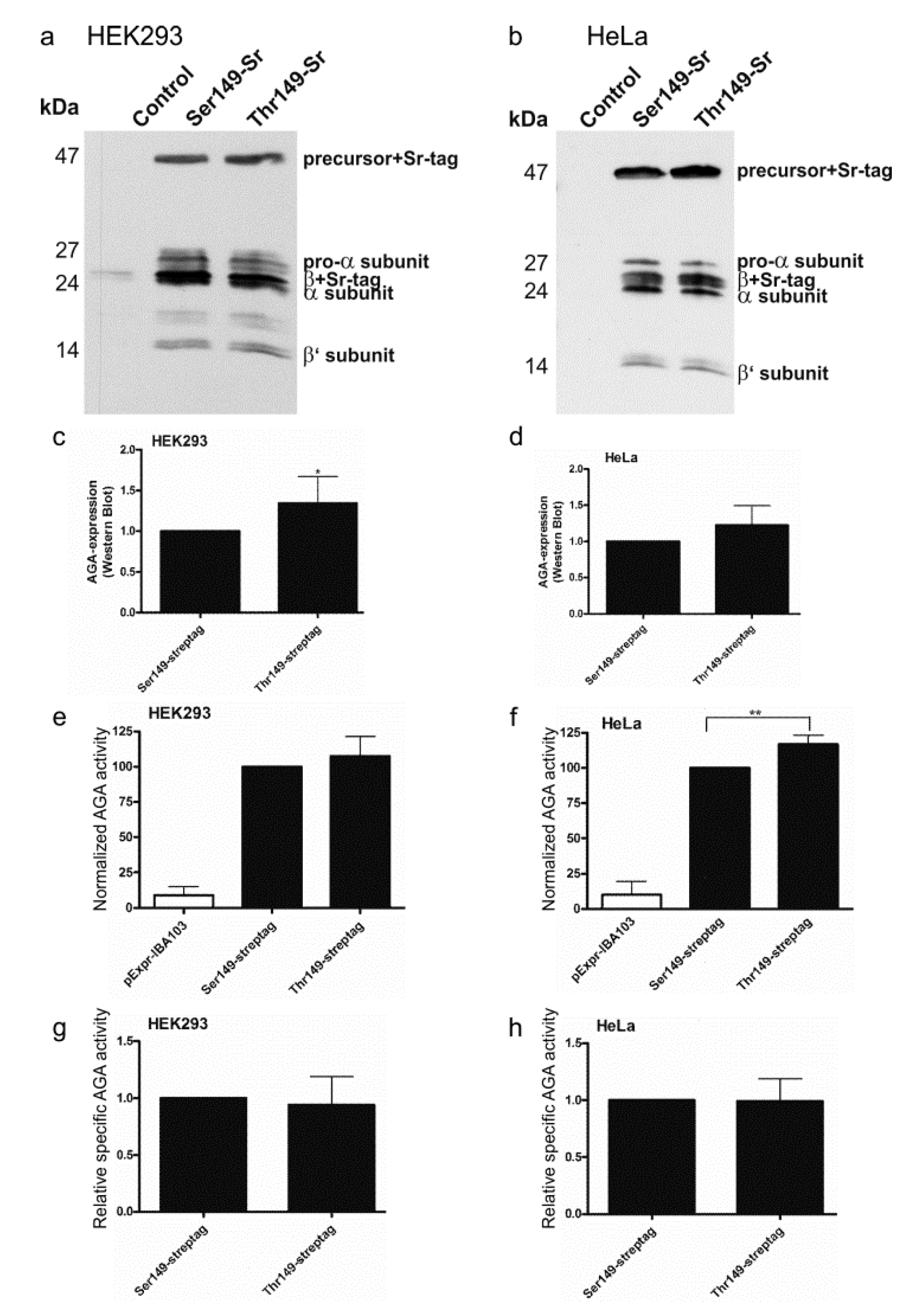
2.2. Consequences of the Ser/Thr149 Variants on the Activity and Processing of the AGA Enzyme
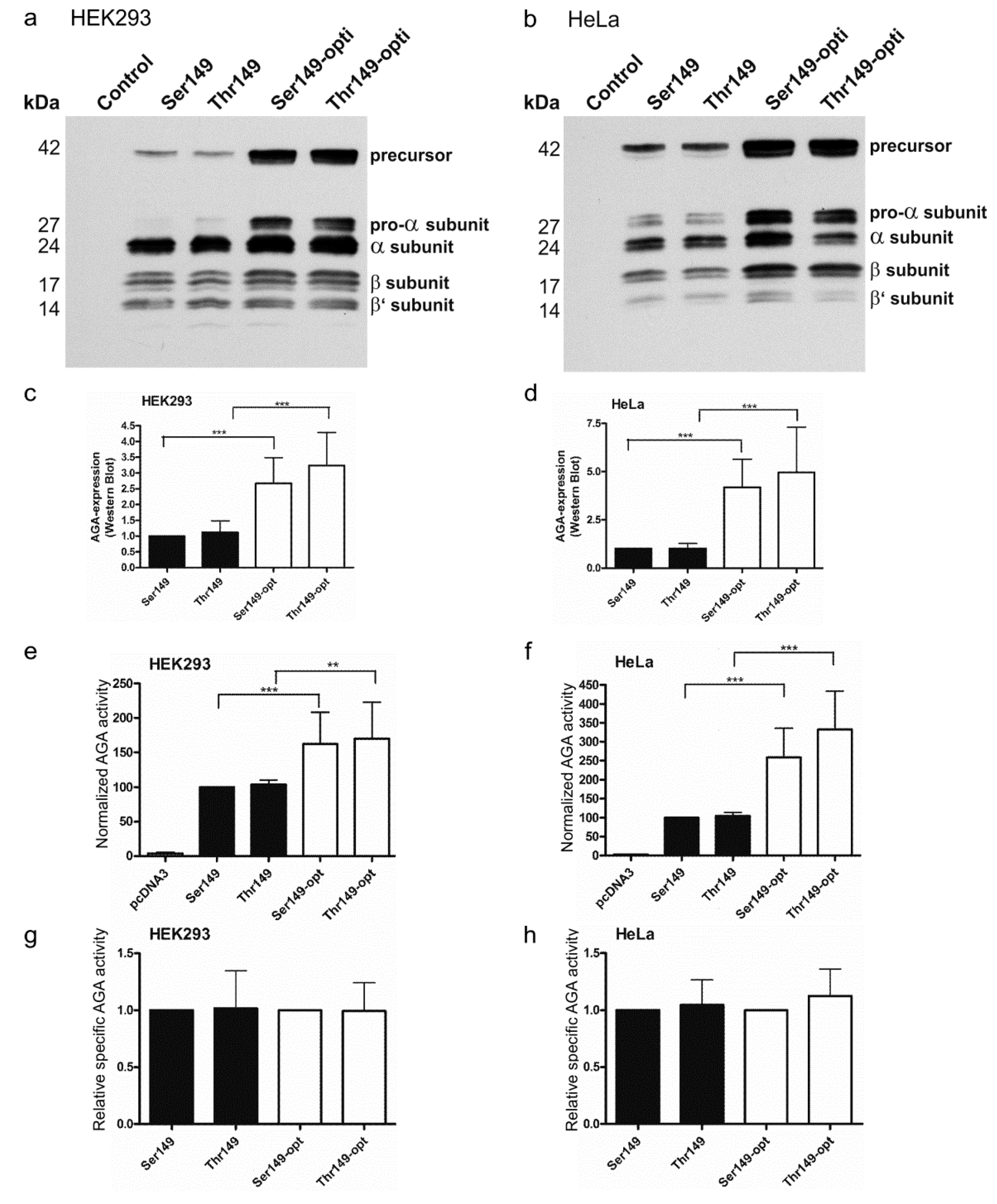
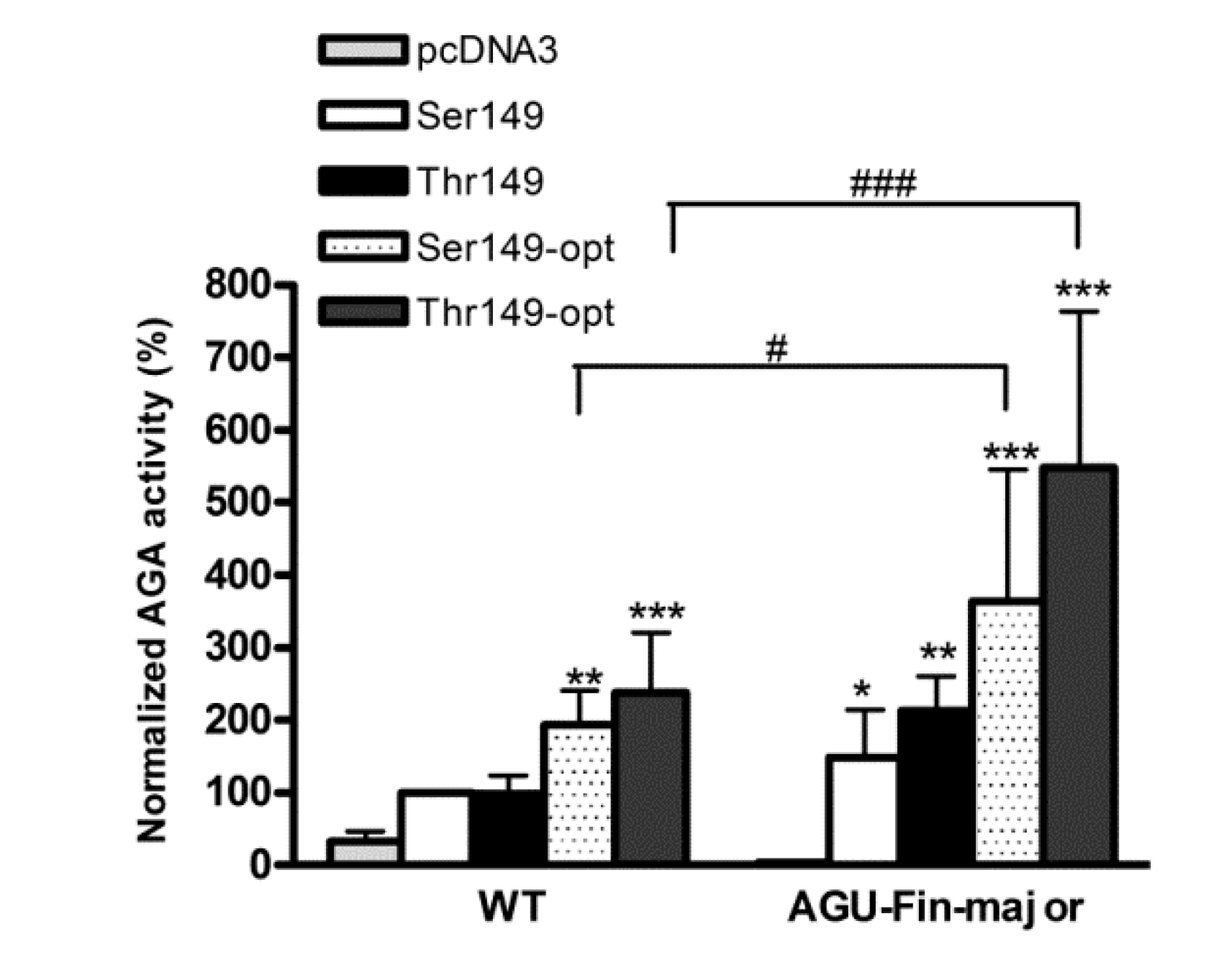
2.3. Expression of Codon-Optimized Variants of Ser149 and Thr149 AGA
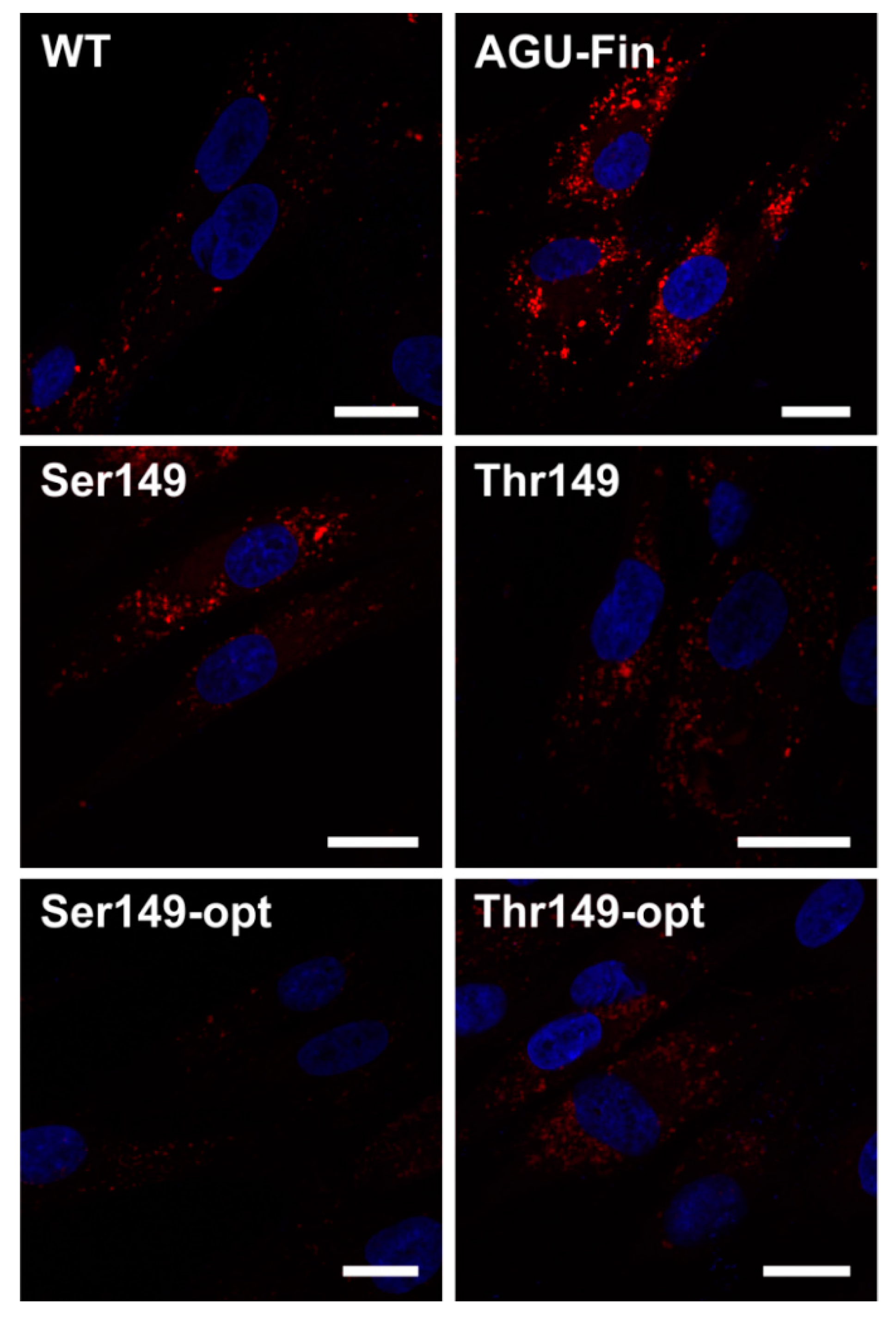
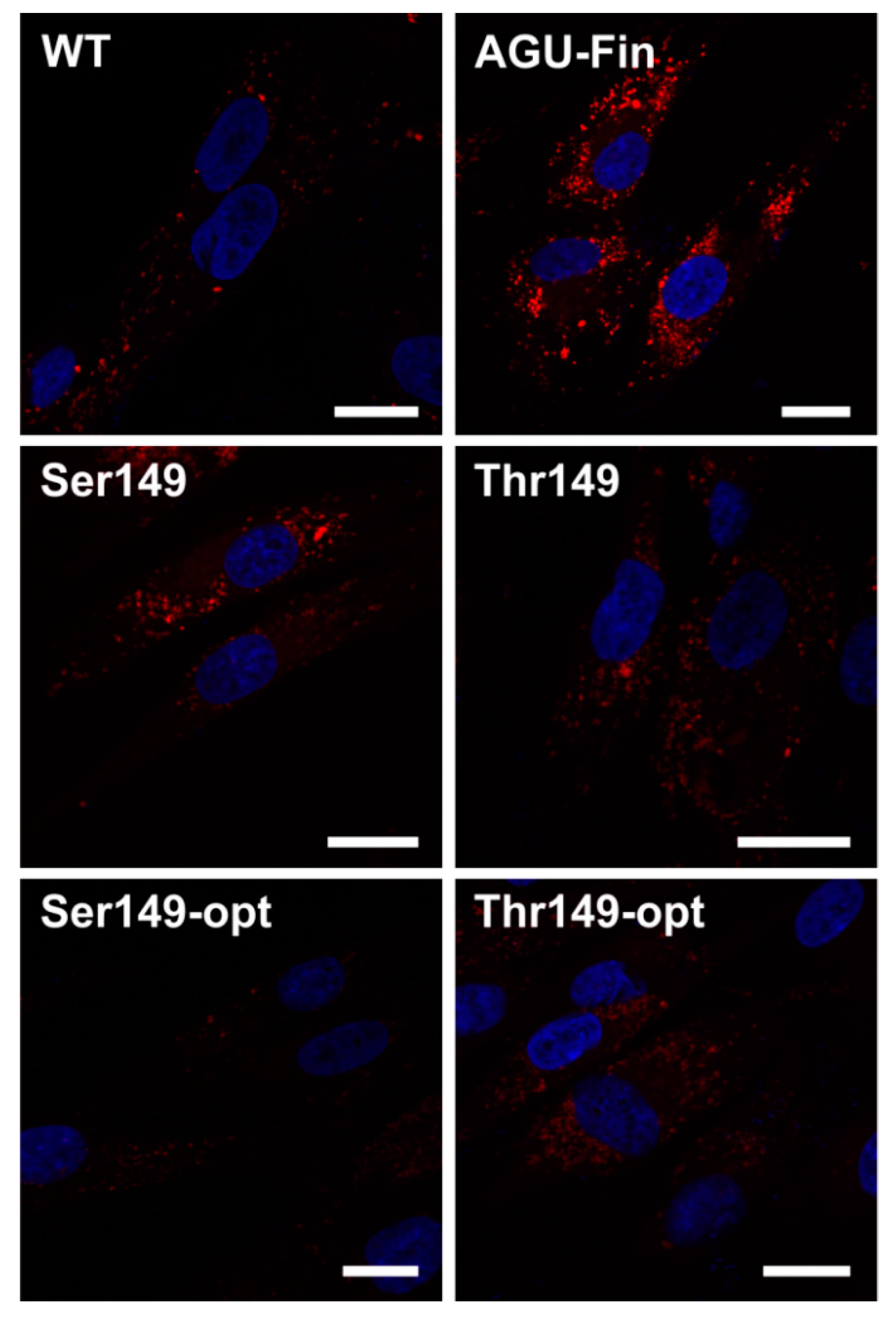
2.4. Expression of AGA Variants in Patient Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Analysis of the Genomic Variant NM_000027.3:c.446C>G - p.Thr149Ser
4.2. Cell Culture
4.3. Plasmids
- AGA-pcDNA3-fwd: 5′-CTATAGGATCCATGGCGCGGAAGTCGAACTTG-3′;
- AGA-pcDNA3-rev: 5′-CTATACTCGAGTTAGATGCAGTCCACTTTTTCC-3′;
- AGA-IBA-fwd: 5′-CTATATCTAGAATGGCGCGGAAGTCGAACTTG-3′;
- AGA-IBA-rev: 5′-CTATAGGATCCGATGCAGTCCACTTTTTCCTC-3′;
- optiAGA-pcDNA3-fwd: 5′-CTATAGGATCCATGGCACGCAAGTCAAACCTC-3′;
- optiAGA-pcDNA3-rev: 5′-CTATACTCGAGTTAGATGCAATCGACCTTCTCC-3′.
4.4. Transfection
4.5. Enzyme Activity Measurements
4.6. Western Blotting Analysis
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| AGA | Aspartylglucosaminidase |
| AGU | Aspartylglucosaminuria |
| DAPI | 4′,6-Diamidin-2-phenylindol |
| DMEM | Dulbecco’s modified Eagle’s medium |
| ER | Endoplasmic reticulum |
| ERT | Enzyme replacement therapy |
| FCS: | Fetal calf serum |
| HEK293T | Human embryonic kidney cells |
| HeLa | Human cervix carcinoma cells |
| NTN | N-terminal nucleophile |
| SNP | Single nucleotide polymorphism |
References
- Makino, M.; Kojima, T.; Yamashina, I. Enzymatic cleavage of glycopeptides. Biochem. Biophys. Res. Commun. 1966, 24, 961–966. [Google Scholar] [CrossRef]
- Ikonen, E.; Julkunen, I.; Tollersrud, O.K.; Kalkkinen, N.; Peltonen, L. Lysosomal aspartylglucosaminidase is processed to the active subunit complex in the endoplasmic reticulum. EMBO J. 1993, 12, 295–302. [Google Scholar] [PubMed]
- Oinonen, C.; Tikkanen, R.; Rouvinen, J.; Peltonen, L. Three-dimensional structure of human lysosomal aspartylglucosaminidase. Nat. Struct. Biol. 1995, 2, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, H.C. Two-step dimerization for autoproteolysis to activate glycosylasparaginase. J. Biol. Chem. 2003, 278, 3210–3219. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, R.; Enomaa, N.; Riikonen, A.; Ikonen, E.; Peltonen, L. Intracellular sorting of aspartylglucosaminidase: The role of N-linked oligosaccharides and evidence of Man-6-P-independent lysosomal targeting. DNA Cell Biol. 1995, 14, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, R.; Riikonen, A.; Oinonen, C.; Rouvinen, R.; Peltonen, L. Functional analyses of active site residues of human lysosomal aspartylglucosaminidase: Implications for catalytic mechanism and autocatalytic activation. EMBO J. 1996, 15, 2954–2960. [Google Scholar] [PubMed]
- Palotie, L.; Ikonen, E.; Syvanen, A.C.; Halila, R.; Enomaa, N.; Heiskanen, T.; Gron, K.; Aula, P. Molecular genetics of aspartylglucosaminuria. Duodecim 1991, 107, 1916–1925. [Google Scholar] [PubMed]
- Pollitt, R.J.; Jenner, F.A.; Merskey, H. Aspartylglycosaminuria. An inborn error of metabolism associated with mental defect. Lancet 1968, 2, 253–255. [Google Scholar] [CrossRef]
- Autio, S. Aspartylglycosaminuria. Analysis of thirty-four patients. J. Ment. Defic. Res. 1972, 1, 1–93. [Google Scholar] [PubMed]
- Autio, S.; Visakorpi, J.K.; Jarvinen, H. Aspartylglycosaminuria (AGU). Further aspects on its clinical picture, mode of inheritance and epidemiology based on a series of 57 patients. Ann. Clin. Res. 1973, 5, 149–155. [Google Scholar] [PubMed]
- Mononen, T.; Mononen, I.; Matilainen, R.; Airaksinen, E. High prevalence of aspartylglycosaminuria among school-age children in eastern Finland. Hum. Genet. 1991, 87, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E.; Baumann, M.; Gron, K.; Syvanen, A.C.; Enomaa, N.; Halila, R.; Aula, P.; Peltonen, L. Aspartylglucosaminuria: cDNA encoding human aspartylglucosaminidase and the missense mutation causing the disease. EMBO J. 1991, 10, 51–58. [Google Scholar] [PubMed]
- Mononen, I.; Heisterkamp, N.; Kaartinen, V.; Williams, J.C.; Yates, J.R., 3rd; Griffin, P.R.; Hood, L.E.; Groffen, J. Aspartylglycosaminuria in the Finnish population: Identification of two point mutations in the heavy chain of glycoasparaginase. Proc. Natl. Acad. Sci. USA 1991, 88, 2941–2945. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E.; Enomaa, N.; Ulmanen, I.; Peltonen, L. In vitro mutagenesis helps to unravel the biological consequences of aspartylglucosaminuria mutation. Genomics 1991, 11, 206–211. [Google Scholar] [CrossRef]
- Isoniemi, A.; Hietala, M.; Aula, P.; Jalanko, A.; Peltonen, L. Identification of a novel mutation causing aspartylglucosaminuria reveals a mutation hotspot region in the aspartylglucosaminidase gene. Hum. Mutat. 1995, 5, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E.; Peltonen, L. Mutations causing aspartylglucosaminuria (AGU): A lysosomal accumulation disease. Hum. Mutat. 1992, 1, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; Ebinger, F.; Zschocke, J.; Sengupta, D.; Ben-Omran, T.; Shahbeck, N.; Moog, U.; Fischer, C.; Burger, F.; Haas, D.; et al. Aspartylglucosaminuria: Unusual neonatal presentation in Qatari twins with a novel aspartylglucosaminidase gene mutation and 3 new cases in a Turkish family. J. Child. Neurol. 2014, 29, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.; Laine, M.; Oinonen, C.; von Schantz, C.; Jalanko, A.; Rouvinen, J.; Peltonen, L. Molecular pathogenesis of a disease: Structural consequences of aspartylglucosaminuria mutations. Hum. Mol. Genet. 2001, 10, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.; Laine, M.; Tikkanen, R.; Oinonen, C.; Jalanko, A.; Rouvinen, J.; Peltonen, L. Activation and oligomerization of aspartylglucosaminidase. J. Biol. Chem. 1998, 273, 25320–25328. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Gulec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of small molecule compounds for pharmacological chaperone therapy of aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef] [PubMed]
- Peltola, M.; Tikkanen, R.; Peltonen, L.; Jalanko, A. Ser72Pro active-site disease mutation in human lysosomal aspartylglucosaminidase: Abnormal intracellular processing and evidence for extracellular activation. Hum. Mol. Genet. 1996, 5, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Hsi-Yang Fritz, M.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Spiridonov, N.A.; Kashina, A. Sounds of silence: Synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013, 41, 2073–2094. [Google Scholar] [CrossRef] [PubMed]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Mussche, S.; Devreese, B.; Nagabhushan Kalburgi, S.; Bachaboina, L.; Fox, J.C.; Shih, H.J.; van Coster, R.; Samulski, R.J.; Gray, S.J. Restoration of cytoskeleton homeostasis after gigaxonin gene transfer for giant axonal neuropathy. Hum. Gene Ther. 2013, 24, 209–219. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Ser/Ser (%) | (n) | Ser/Thr (%) | (n) | Thr/Thr (%) | (n) |
|---|---|---|---|---|---|---|
| All Populations Studied | 86.3 | (2162) | 11.5 | (288) | 2.2 | (54) |
| Europe | 99.8 | (502) | 0.2 | (1) | - | - |
| Finnish in Finland | 100 | (99) | - | - | - | - |
| British in England and Scotland | 100 | (91) | - | - | - | - |
| Iberian populations in Spain | 99.1 | (106) | 0.9 | (1) | - | - |
| Toscani in Italy | 100 | (107) | - | - | - | - |
| Utah residents with Northern and Western European ancestry | 100 | (99) | - | - | - | - |
| North and South America | 94.8 | (329) | 5.2 | (18) | - | - |
| Colombian in Medellin | 94.7 | (89) | 5.3 | (5) | - | - |
| Mexican Ancestry in Los Angeles | 100 | (64) | - | - | - | - |
| Peruvian in Lima, Peru | 98.8 | (84) | 1.2 | (1) | - | - |
| Puerto Rican in Puerto Rico | 88.5 | (92) | 11.5 | (12) | - | - |
| Africa | 51.1 | (338) | 40.7 | (269) | 8.2 | (54) |
| African Caribean in Barbados | 62.5 | (60) | 32.3 | (31) | 5.2 | (5) |
| African Ancestry in Southwest US | 57.4 | (35) | 39.3 | (24) | 3.3 | (2) |
| Esan in Nigeria | 46.5 | (46) | 44.4 | (44) | 9.1 | (9) |
| Luhya in Webuye, Kenya | 46.5 | (46) | 44.4 | (44) | 9.1 | (9) |
| Mandinka in Gambia | 46.9 | (53) | 39.8 | (45) | 13.3 | (15) |
| Mende in Sierra Leone | 48.2 | (41) | 41.2 | (35) | 10.6 | (9) |
| Yoruba in Ibadan, Nigeria | 52.8 | (57) | 42.6 | (46) | 4.6 | (5) |
| East Asia | 100 | (504) | - | - | - | - |
| Chinese Dai in Xishuangbanna | 100 | (93) | - | - | - | - |
| Han Chinese in Bejing | 100 | (103) | - | - | - | - |
| Southern Han Chinese | 100 | (105) | - | - | - | - |
| Japanese in Tokyo, Japan | 100 | (104) | - | - | - | - |
| Kinh in Ho Chi Minh City, Vietnam | 100 | (99) | - | - | - | - |
| South Asia | 100 | (489) | - | - | - | - |
| Bengali in Bangladesh | 100 | (86) | - | - | - | - |
| Gujarati Indian in Houston, Tx | 100 | (103) | - | - | - | - |
| Indian Telegu in the UK | 100 | (102) | - | - | - | - |
| Punjabi in Lahore, Pakistan | 100 | (96) | - | - | - | - |
| Sri Lankan Tamil in the UK | 100 | (102) | - | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banning, A.; König, J.F.; Gray, S.J.; Tikkanen, R. Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production. Int. J. Mol. Sci. 2017, 18, 706. https://doi.org/10.3390/ijms18040706
Banning A, König JF, Gray SJ, Tikkanen R. Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production. International Journal of Molecular Sciences. 2017; 18(4):706. https://doi.org/10.3390/ijms18040706
Chicago/Turabian StyleBanning, Antje, Jan F. König, Steven J. Gray, and Ritva Tikkanen. 2017. "Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production" International Journal of Molecular Sciences 18, no. 4: 706. https://doi.org/10.3390/ijms18040706