
Dysregulated IER3 Expression is Associated with Enhanced Apoptosis in Titin-Based Dilated Cardiomyopathy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
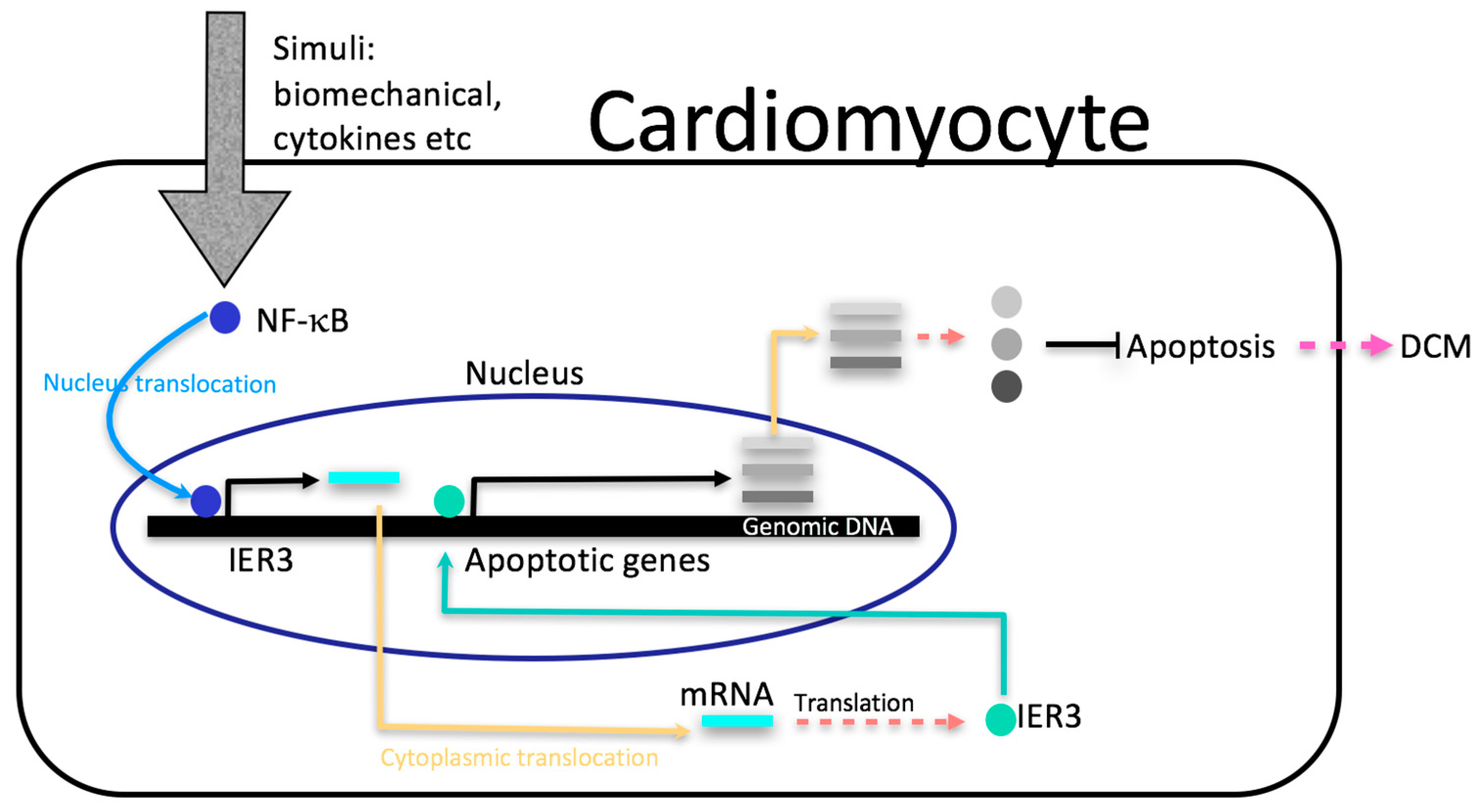
:1. Introduction
2. Results
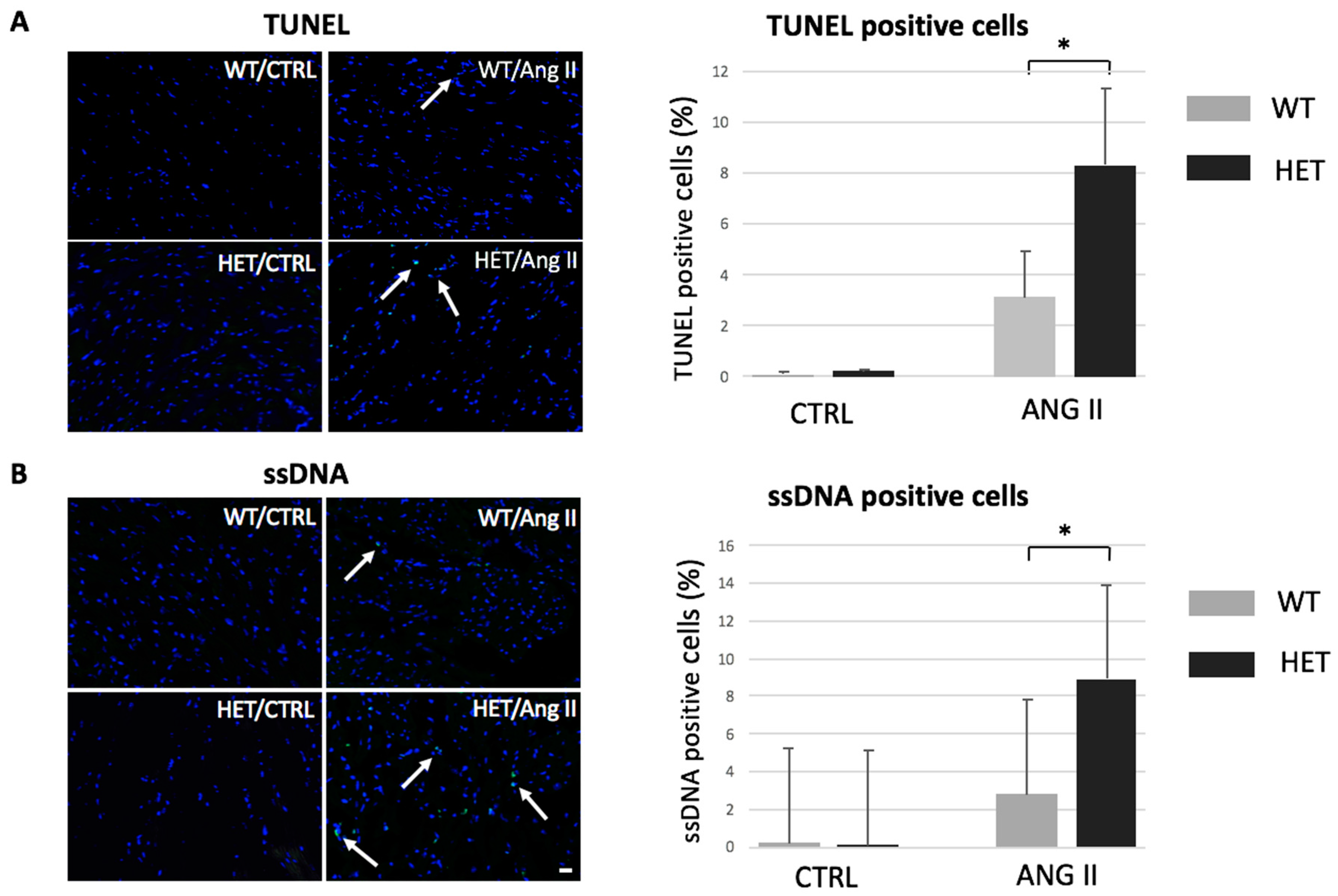
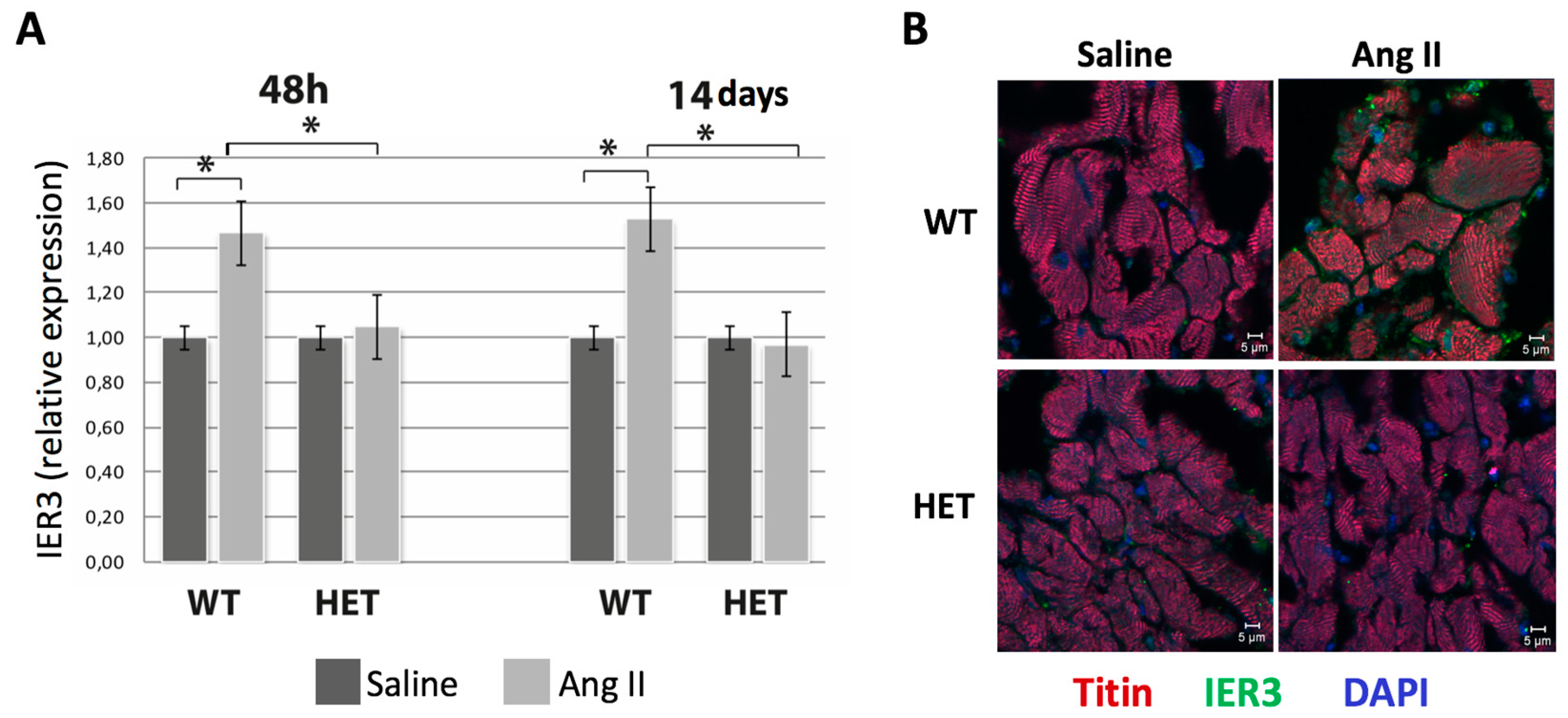
2.1. Heterozygous Ttn Knock-In Mice Exhibit Enhanced Apoptosis during Dilated Cardiomyopathy Development
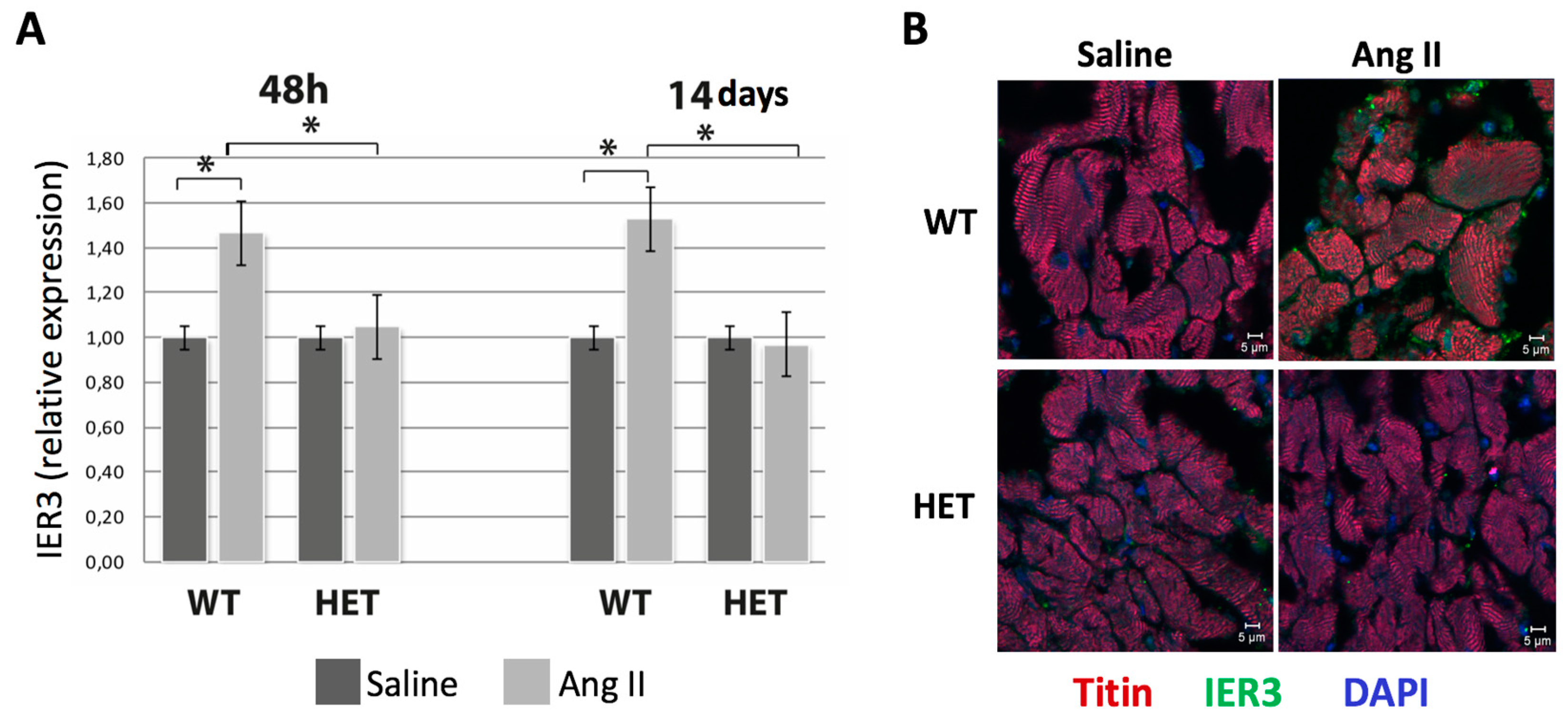
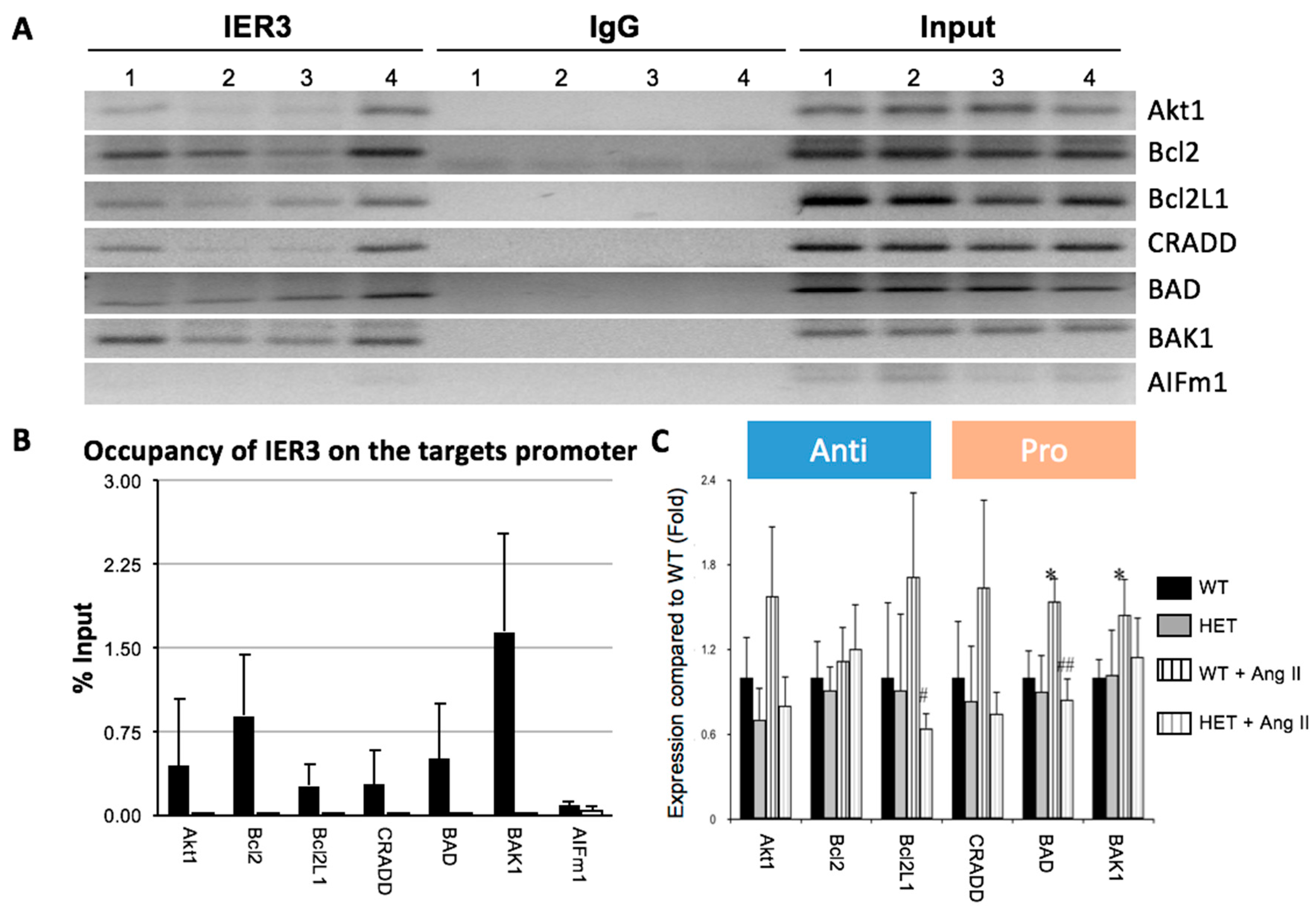
2.2. Heterozygous Ttn-Deficient Mice Show Impaired IER3 Signaling
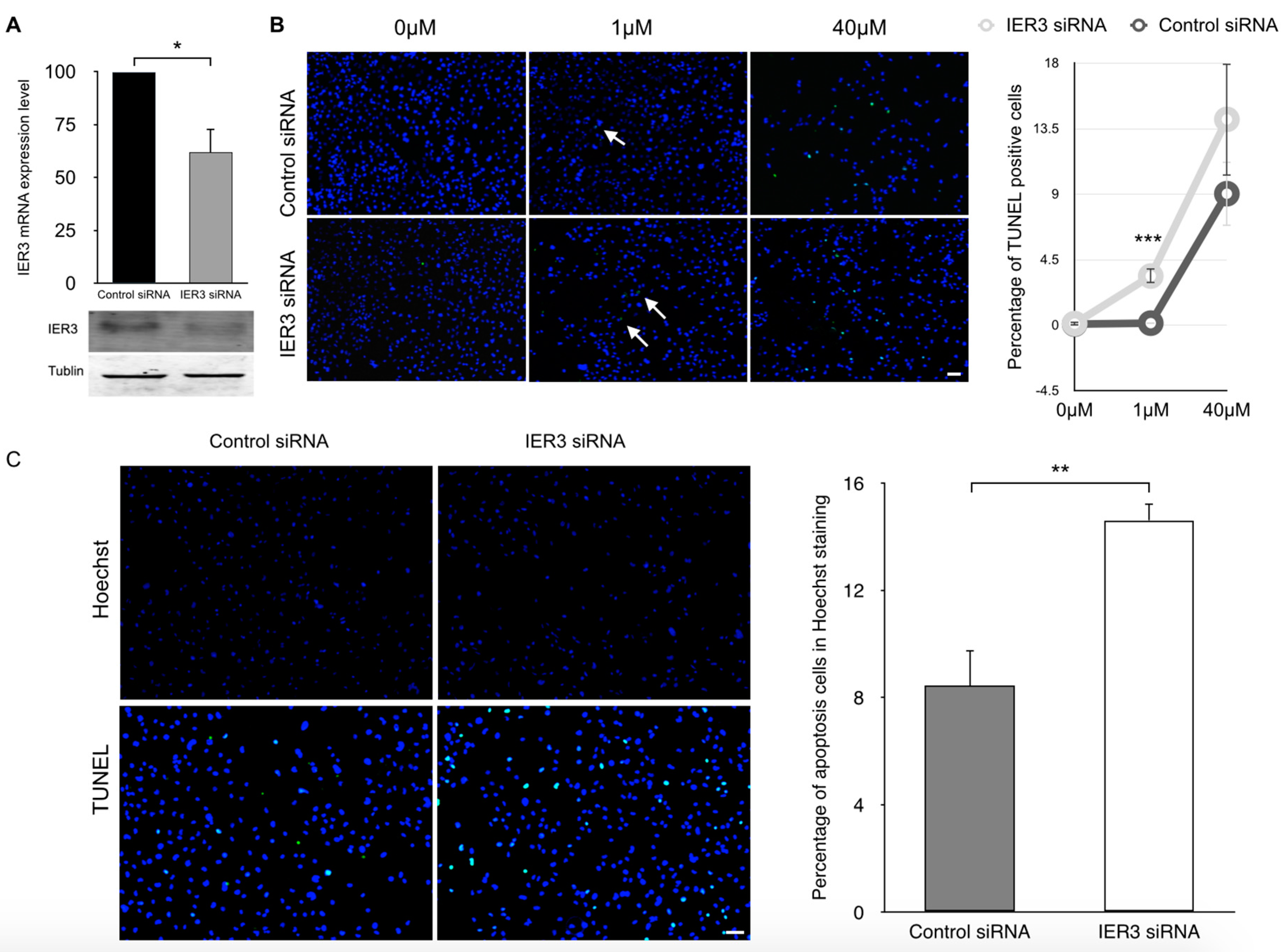
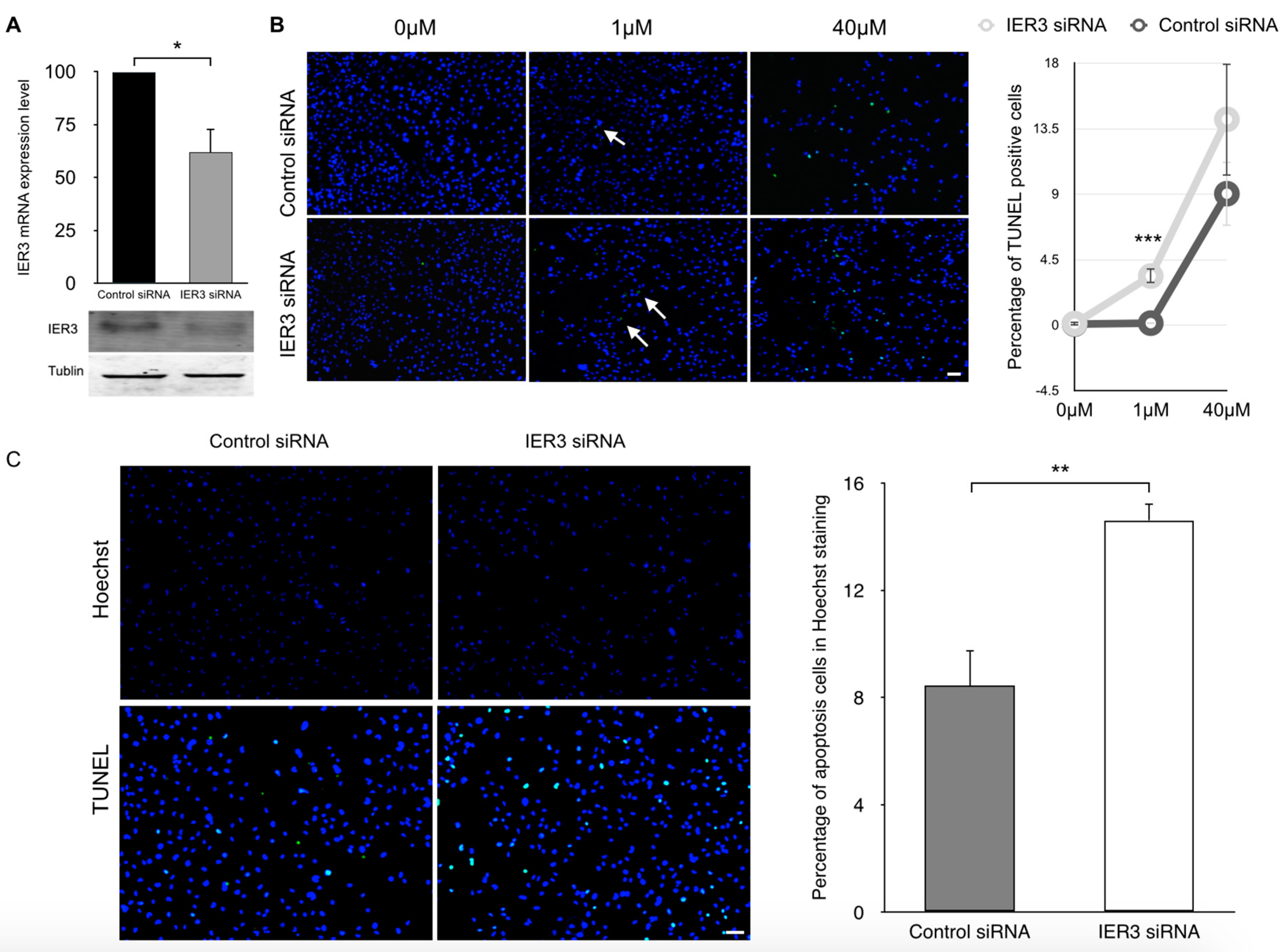
2.3. IER3 Is Involved in the Regulation of Apoptosis in Cardiomyocytes
2.4. IER3 Is Anti-Apoptotic in HL-1 Cardiomyocytes
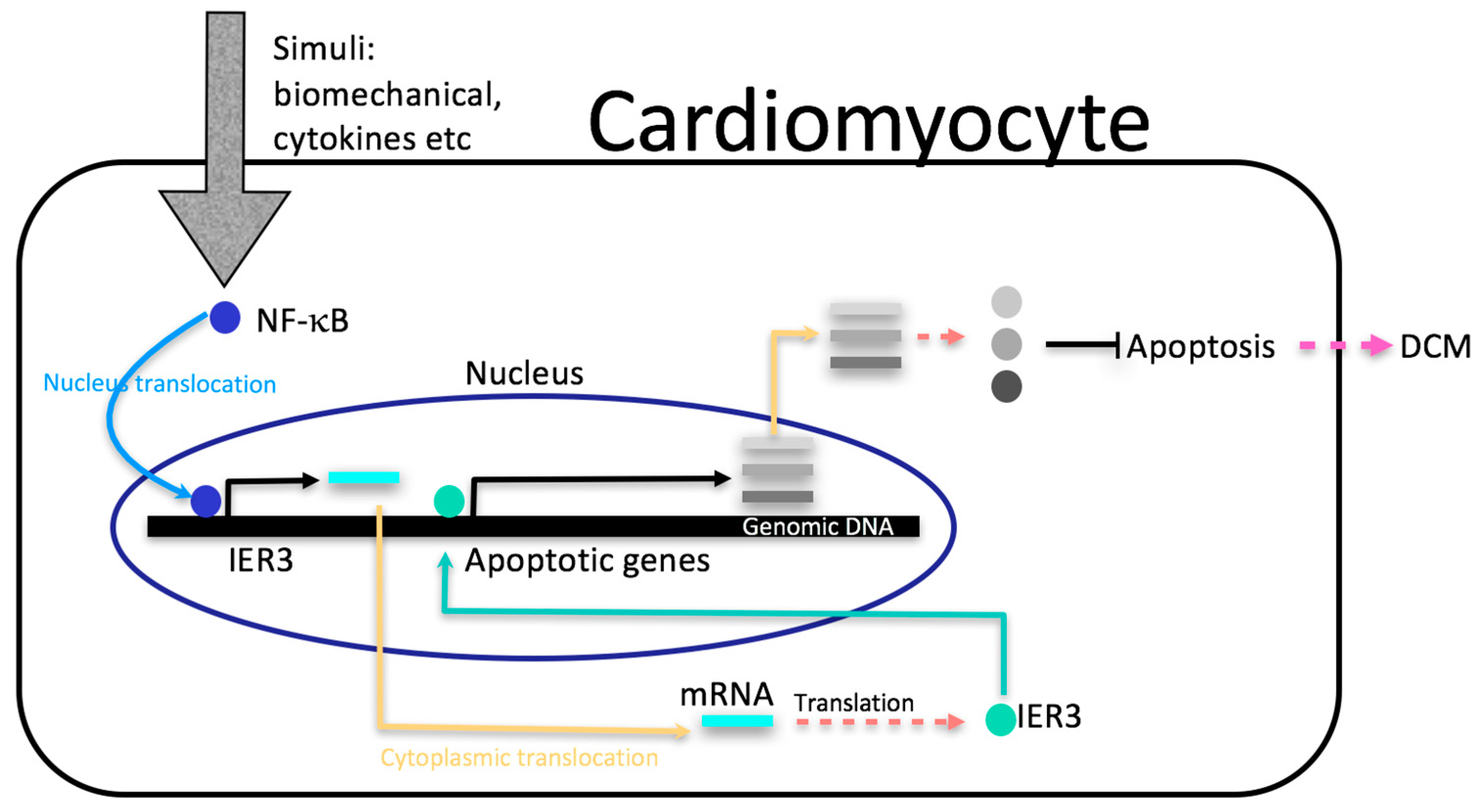
3. Discussion
4. Materials and Methods
4.1. SiRNA Transfections in HL-1 Cells
4.2. Real-Time PCR
4.3. Animal Experiments
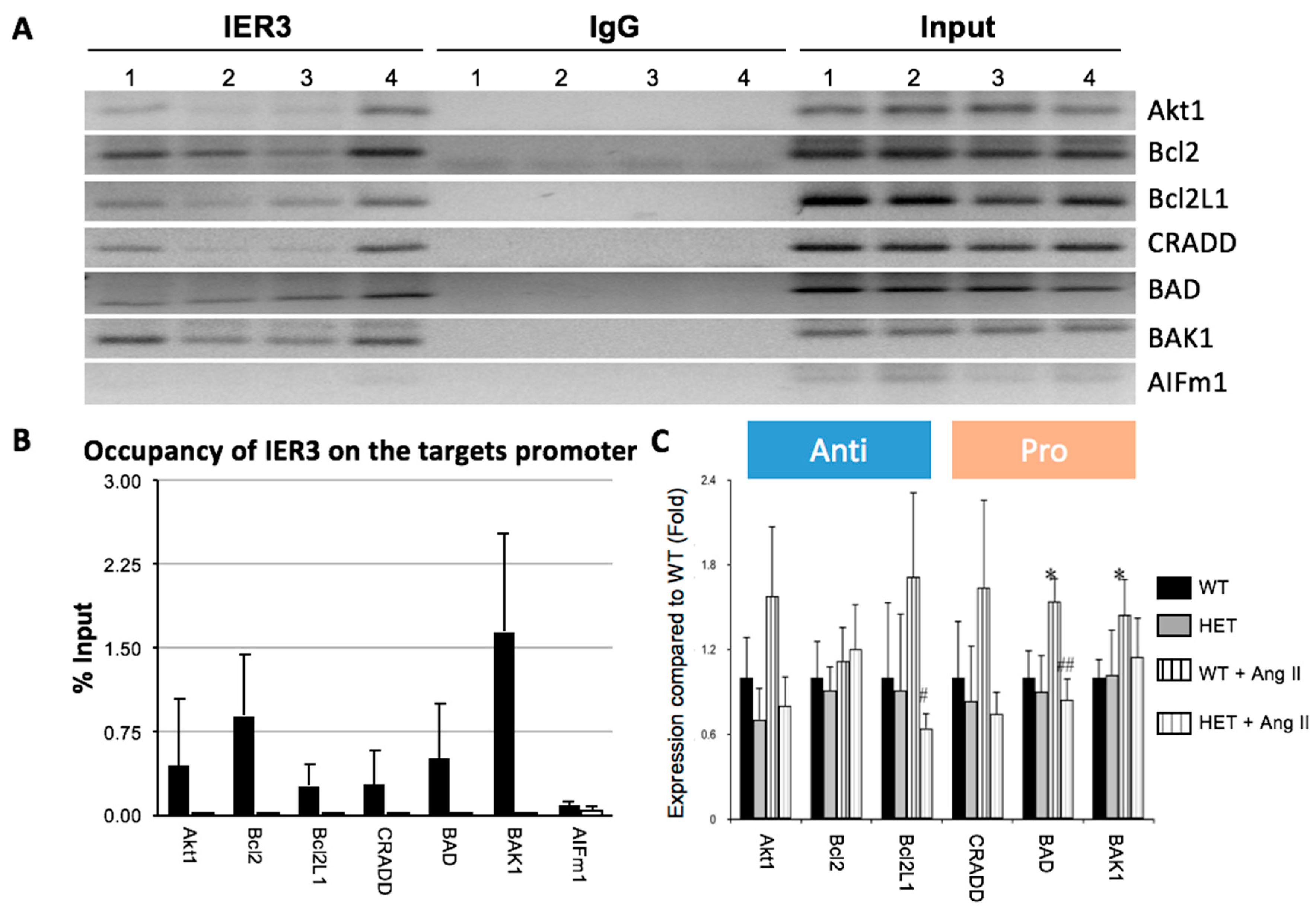
4.4. Chromatin Immunoprecipitation
4.5. ChIP Sequencing and Analysis
4.6. TUNEL Stainings
4.7. Single-Stranded DNA Stainings
4.8. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart disease and stroke statistics-2016 update: A report from the american heart association. Circulation 2016, 133, 38–360. [Google Scholar]
- Parvari, R.; Levitas, A. The mutations associated with dilated cardiomyopathy. Biochem. Res. Int. 2012, 2012, 639250. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, M.; Michely, B.; Krohne, C.; Heuser, A.; Erdmann, B.; Klaassen, S.; Hudson, B.; Magarin, M.; Kirchner, F.; Todiras, M.; et al. Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J. Mol. Cell. Cardiol. 2009, 47, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Haider, N.; Virmani, R.; DiSalvo, T.G.; Kolodgie, F.D.; Hajjar, R.J.; Schmidt, U.; Semigran, M.J.; Dec, G.W.; Khaw, B.A. Apoptosis in myocytes in end-stage heart failure. N. Engl. J. Med. 1996, 335, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.M.; Izumo, S. Apoptosis and heart failure: A critical review of the literature. Circ. Res. 2000, 86, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Oka, T.; Hunter, B.; Robinson, A.; Papp, S.; Nakamura, K.; Srisakuldee, W.; Nickel, B.E.; Light, P.E.; Dyck, J.R.; et al. Calreticulin induces dilated cardiomyopathy. PLoS ONE 2013, 8, e56387. [Google Scholar] [CrossRef] [PubMed]
- Kondratyev, A.D.; Chung, K.N.; Jung, M.O. Identification and characterization of a radiation-inducible glycosylated human early-response gene. Cancer Res. 1996, 56, 1498–1502. [Google Scholar] [PubMed]
- Schilling, D.; Pittelkow, M.R.; Kumar, R. IEX-1, an immediate early gene, increases the rate of apoptosis in keratinocytes. Oncogene 2001, 20, 7992–7997. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Grobe, O.; Sieke, A.; Kruse, M.L.; Folsch, U.R.; Schmidt, W.E.; Schafer, H. Expression of the NF-κB target gene IEX-1 (p22/PRG1) does not prevent cell death but instead triggers apoptosis in hela cells. Oncogene 2001, 20, 69–76. [Google Scholar] [CrossRef] [PubMed]
- De Keulenaer, G.W.; Wang, Y.; Feng, Y.; Muangman, S.; Yamamoto, K.; Thompson, J.F.; Turi, T.G.; Landschutz, K.; Lee, R.T. Identification of IEX-1 as a biomechanically controlled nuclear factor-κB target gene that inhibits cardiomyocyte hypertrophy. Circ. Res. 2002, 90, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Cupesi, M.; Yoshioka, J.; Gannon, J.; Kudinova, A.; Stewart, C.L.; Lammerding, J. Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J. Mol. Cell. Cardiol. 2010, 48, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Rosenstiel, P.; Kruse, M.L.; Grohmann, F.; Minkenberg, J.; Perkins, N.D.; Folsch, U.R.; Schreiber, S.; Schafer, H. IEX-1 directly interferes with rela/p65 dependent transactivation and regulation of apoptosis. Biochim. Biophys. Acta 2008, 1783, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, X.; Geng, L.; Li, M.; Zhang, Y.; Wu, M.X. The expression of immediate early response gene x-1 in preeclampsia placenta and its pro-apoptotic role in preeclampsia. Hypertens. Pregnancy 2013, 32, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Satsu, H.; Totsuka, M.; Shimizu, M. IEX-1 suppresses apoptotic damage in human intestinal epithelial Caco-2 cells induced by co-culturing with macrophage-like THP-1 cells. Biosci. Rep. 2011, 31, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Lutz, W.; Frank, E.; Im, H.J. Immediate early gene X-1 interacts with proteins that modulate apoptosis. Biochem. Biophys. Res. Commun. 2004, 323, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.; Mestril, R.; Samali, A. Losing heart: The role of apoptosis in heart disease—A novel therapeutic target? FASEB J. 2002, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Schafer, H.; Diebel, J.; Arlt, A.; Trauzold, A.; Schmidt, W.E. The promoter of human p22/PACAP response gene 1 (PRG1) contains functional binding sites for the p53 tumor suppressor and for NFκB. FEBS Lett. 1998, 436, 139–143. [Google Scholar] [CrossRef]
- Schafer, H.; Lettau, P.; Trauzold, A.; Banasch, M.; Schmidt, W.E. Human pacap response gene 1 (p22/PRG1): Proliferation-associated expression in pancreatic carcinoma cells. Pancreas 1999, 18, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.L.; Berndt, T.J.; Frank, E.; Patel, J.B.; Redfield, M.M.; Dong, X.; Griffin, M.D.; Grande, J.P.; van Deursen, J.M.; Sieck, G.C.; et al. Elevated blood pressure and cardiac hypertrophy after ablation of the gly96/iex-1 gene. J. Appl. Physiol. 2006, 100, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.M.; Soane, L. Multiple functions of bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nakae, S.; Terry, R.D.; Mokhtari, G.K.; Gunawan, F.; Balsam, L.B.; Kaneda, H.; Kofidis, T.; Tsao, P.S.; Robbins, R.C. Cardiomyocyte-specific Bcl-2 overexpression attenuates ischemia-reperfusion injury, immune response during acute rejection, and graft coronary artery disease. Blood 2004, 104, 3789–3796. [Google Scholar] [CrossRef] [PubMed]
- Hochhauser, E.; Cheporko, Y.; Yasovich, N.; Pinchas, L.; Offen, D.; Barhum, Y.; Pannet, H.; Tobar, A.; Vidne, B.A.; Birk, E. Bax deficiency reduces infarct size and improves long-term function after myocardial infarction. Cell Biochem. Biophys. 2007, 47, 11–20. [Google Scholar] [CrossRef]
- Arlt, A.; Minkenberg, J.; Kruse, M.L.; Grohmann, F.; Folsch, U.R.; Schafer, H. Immediate early gene-X1 interferes with 26 S proteasome activity by attenuating expression of the 19 S proteasomal components S5a/Rpn10 and S1/Rpn2. Biochem. J. 2007, 402, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Sina, C.; Arlt, A.; Gavrilova, O.; Midtling, E.; Kruse, M.L.; Muerkoster, S.S.; Kumar, R.; Folsch, U.R.; Schreiber, S.; Rosenstiel, P.; et al. Ablation of Gly96/immediate early gene-x1 (gly96/iex-1) aggravates DSS-induced colitis in mice: Role for gly96/iex-1 in the regulation of NF-κB. Inflamm. Bowel. Dis. 2010, 16, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. Hl-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ortiz, J.A.; Taing, L.; Meyer, C.A.; Lee, B.; Zhang, Y.; Shin, H.; Wong, S.S.; Ma, J.; Lei, Y.; et al. Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 2011, 12, R83. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. Meme: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef] [PubMed]
- Jin, V.X.; O'Geen, H.; Iyengar, S.; Green, R.; Farnham, P.J. Identification of an OCT4 and SRY regulatory module using integrated computational and experimental genomics approaches. Genome Res. 2007, 17, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.; Carlson, J.M.; Khetani, R.S.; Gross, R.H. A novel ensemble learning method for de novo computational identification of DNA binding sites. BMC Bioinform. 2007, 8, 249. [Google Scholar] [CrossRef] [PubMed]
- Leibovich, L.; Paz, I.; Yakhini, Z.; Mandel-Gutfreund, Y. Drimust: A web server for discovering rank imbalanced motifs using suffix trees. Nucleic Acids Res. 2013, 41, W174–W179. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. Fimo: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Hahn, J.K.; Neupane, B.; Aidery, P.; Labeit, S.; Gawaz, M.; Gramlich, M. Dysregulated IER3 Expression is Associated with Enhanced Apoptosis in Titin-Based Dilated Cardiomyopathy. Int. J. Mol. Sci. 2017, 18, 723. https://doi.org/10.3390/ijms18040723
Zhou Q, Hahn JK, Neupane B, Aidery P, Labeit S, Gawaz M, Gramlich M. Dysregulated IER3 Expression is Associated with Enhanced Apoptosis in Titin-Based Dilated Cardiomyopathy. International Journal of Molecular Sciences. 2017; 18(4):723. https://doi.org/10.3390/ijms18040723
Chicago/Turabian StyleZhou, Qifeng, Julia Kelley Hahn, Balram Neupane, Parwez Aidery, Siegfried Labeit, Meinrad Gawaz, and Michael Gramlich. 2017. "Dysregulated IER3 Expression is Associated with Enhanced Apoptosis in Titin-Based Dilated Cardiomyopathy" International Journal of Molecular Sciences 18, no. 4: 723. https://doi.org/10.3390/ijms18040723