
Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage

Abstract
:
1. Introduction
2. Results
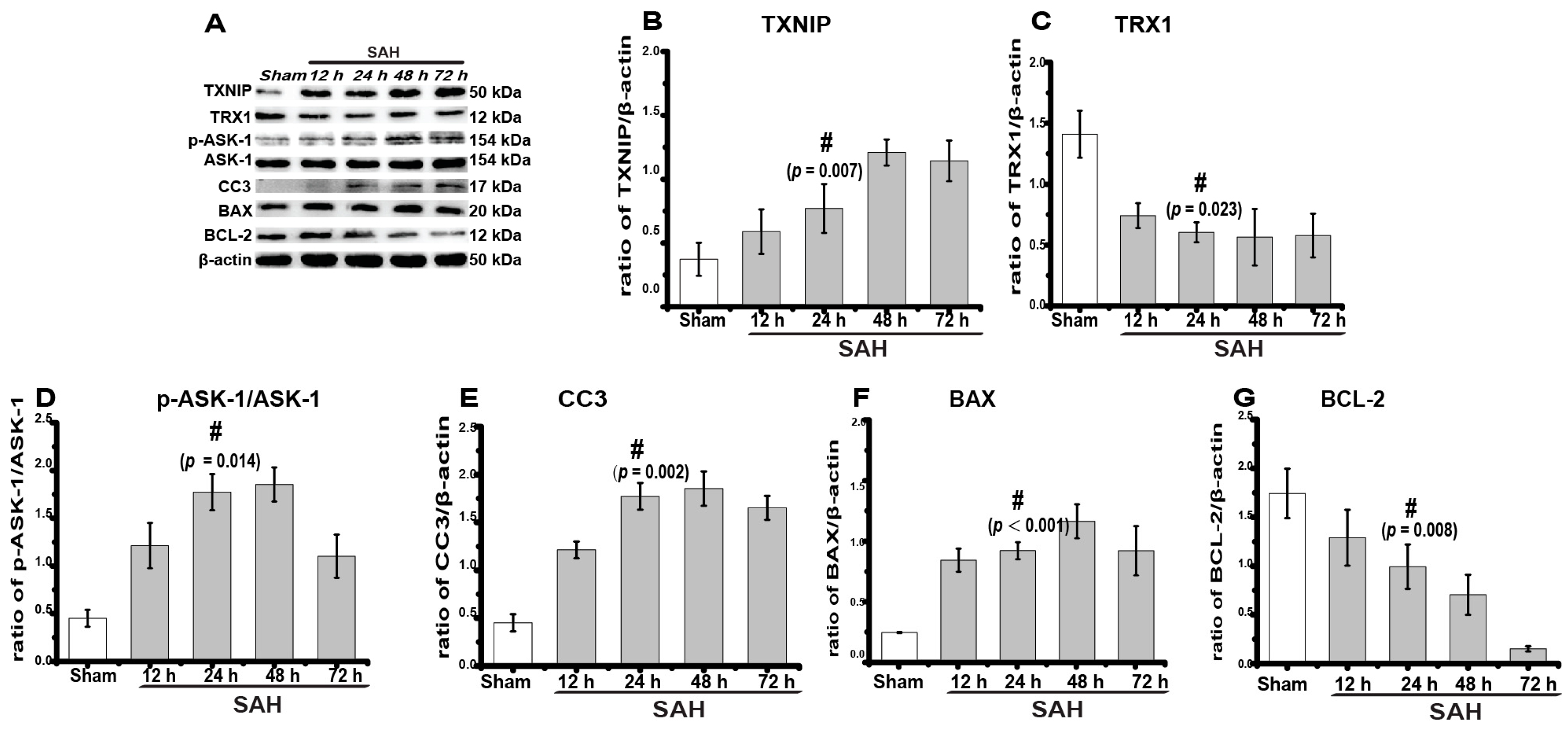
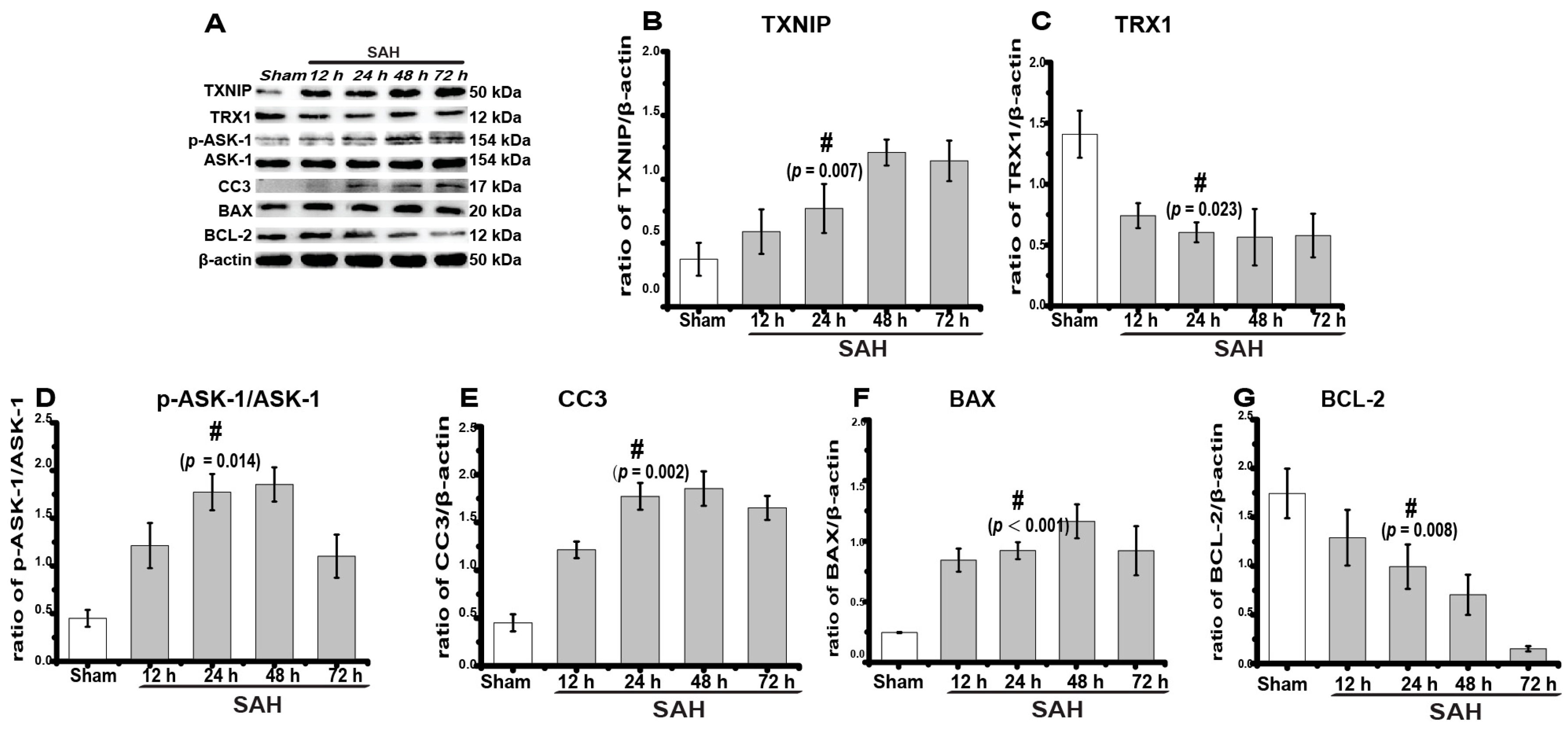
2.1. TXNIP Expression Increased Obviously after SAH
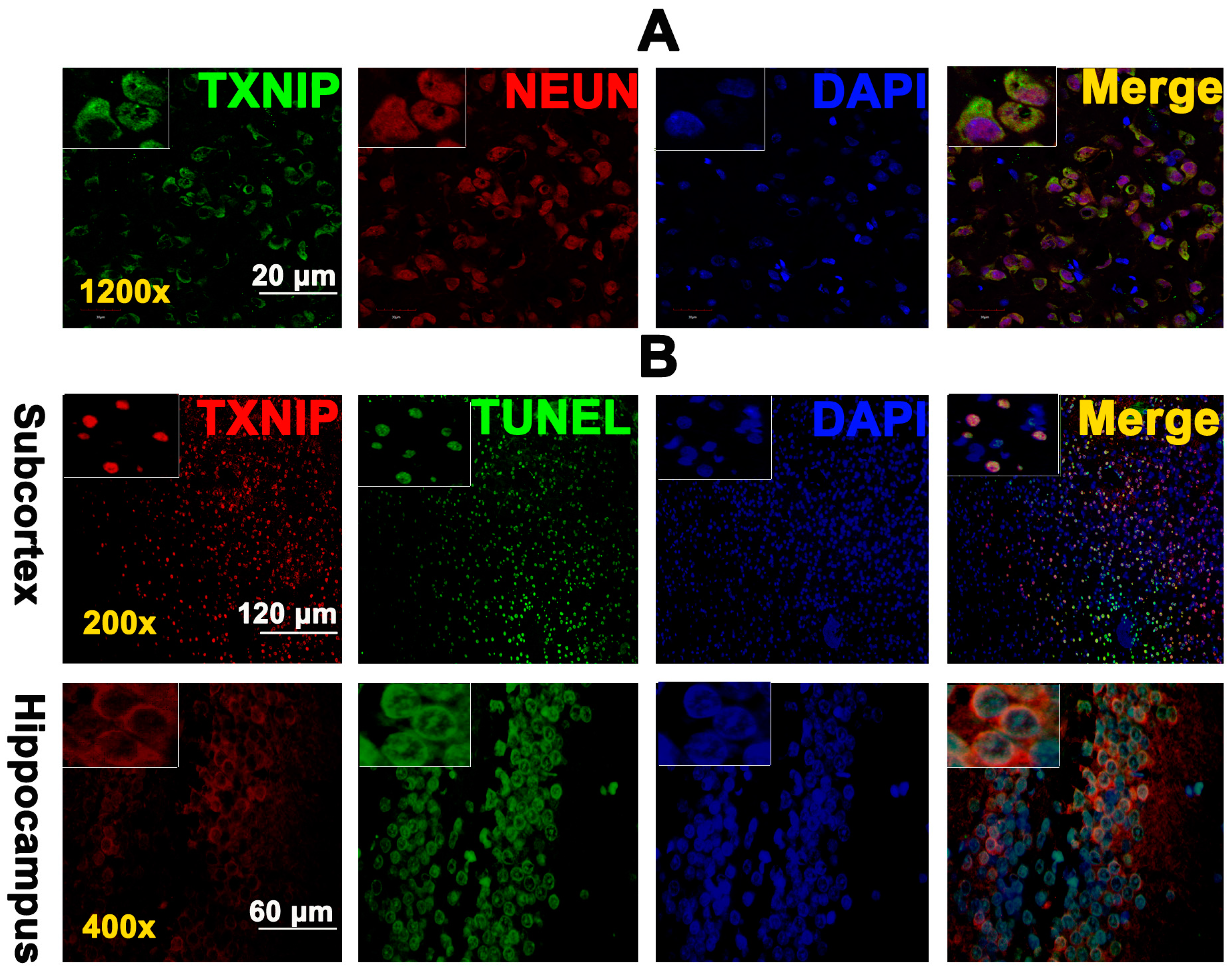
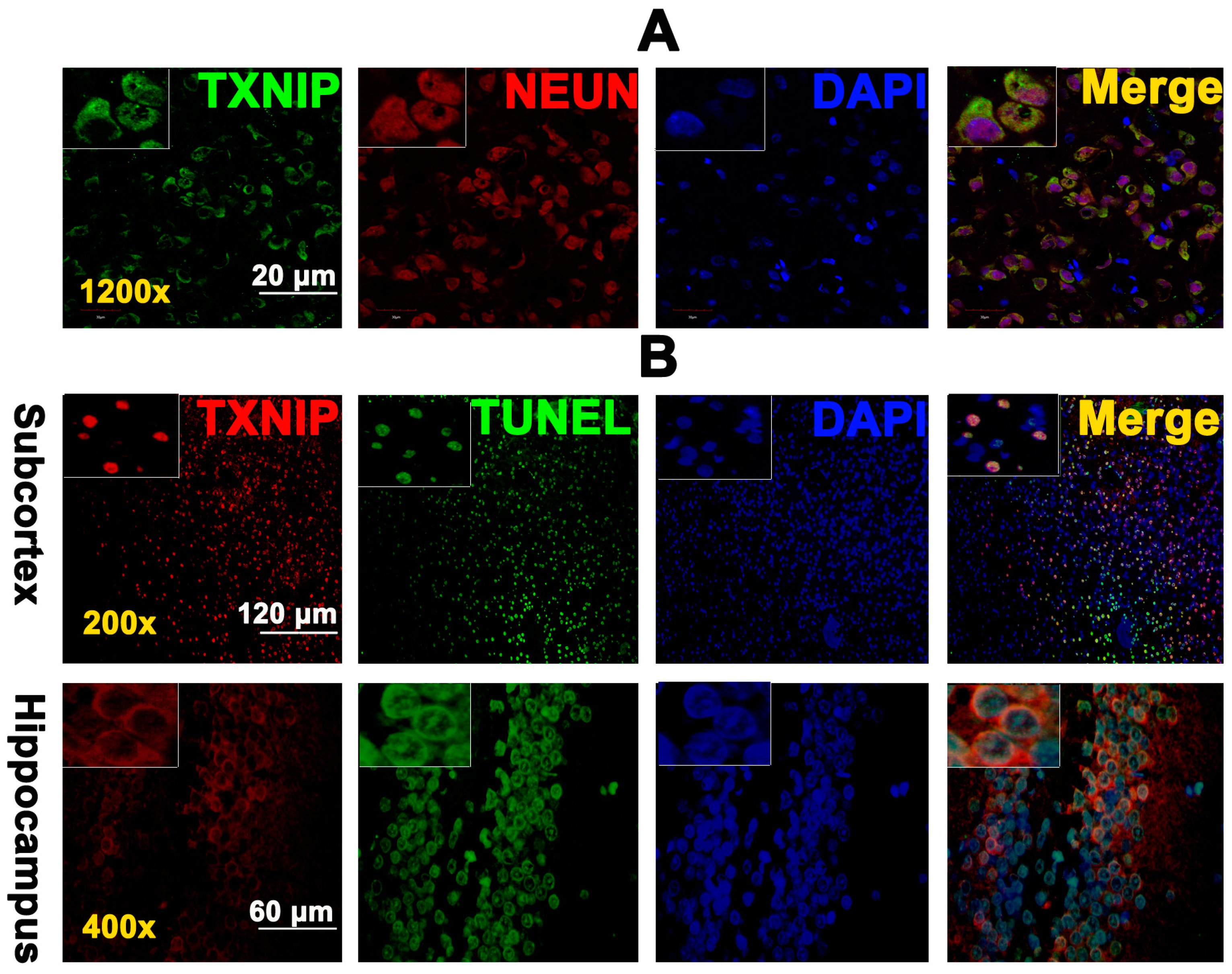
2.2. TXNIP Was Expressed in Neurons and Is Colocalized with TUNEL Positive Cells
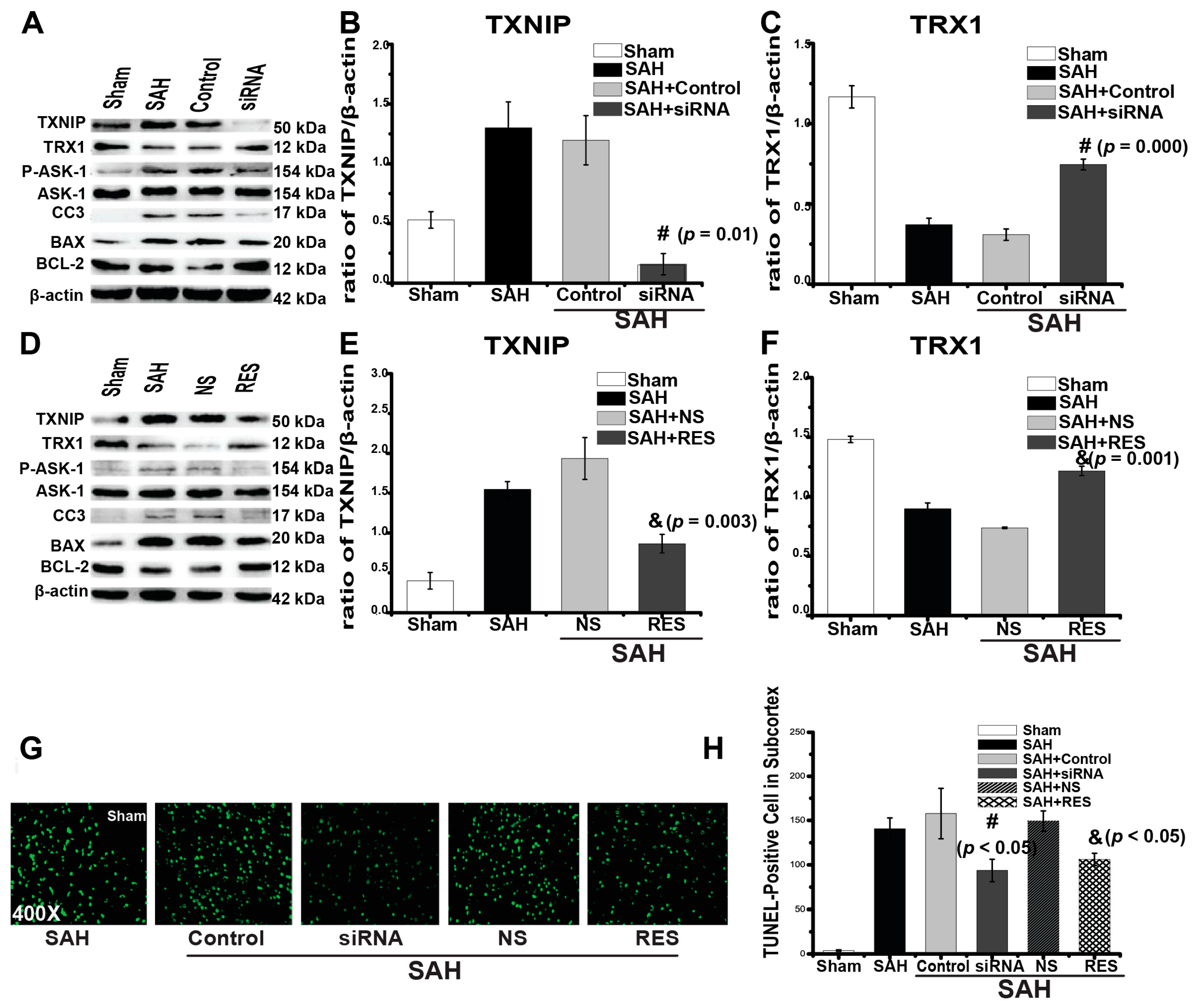
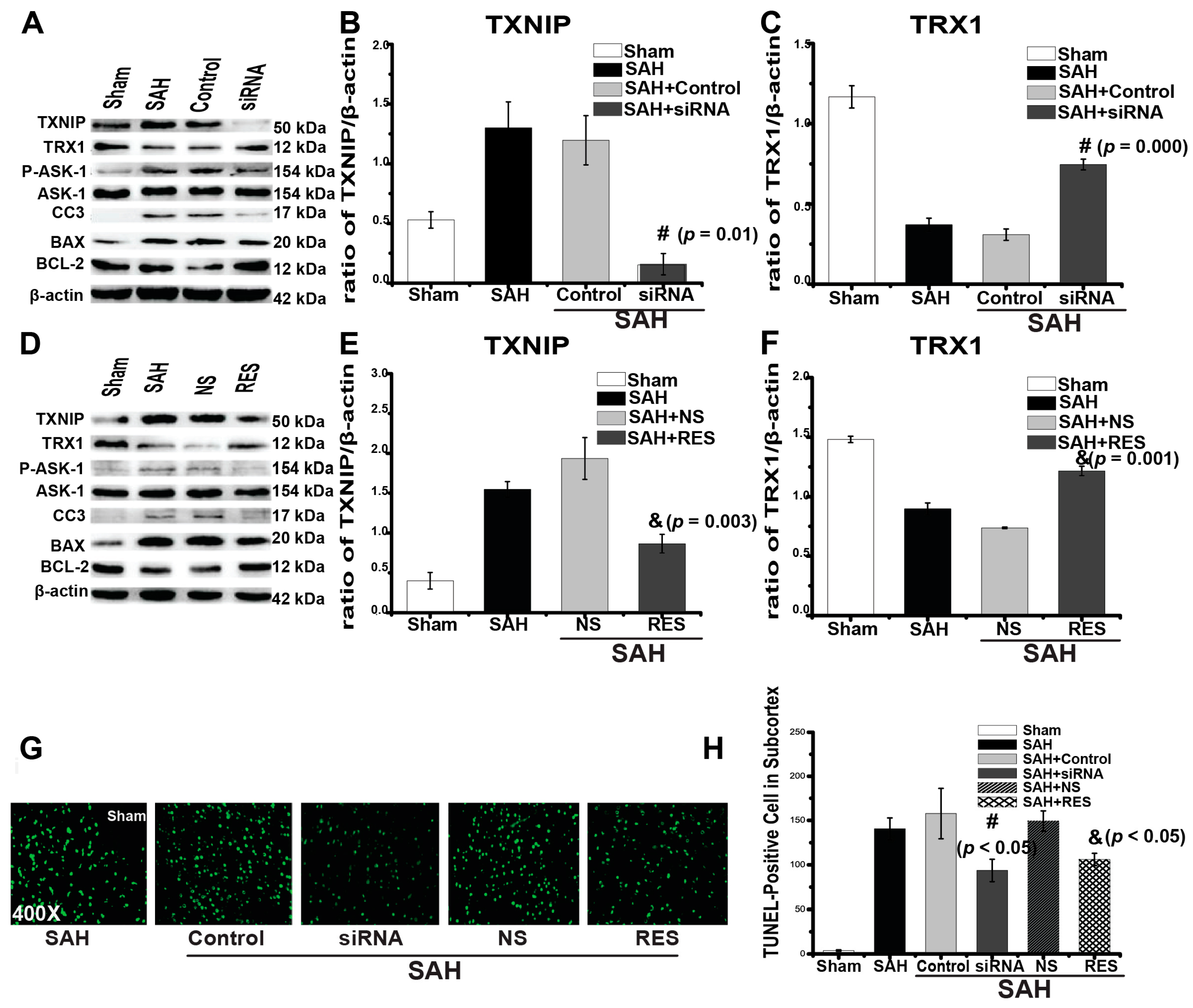
2.3. Downregulation of TXNIP by Resveratrol (RES) and siRNA
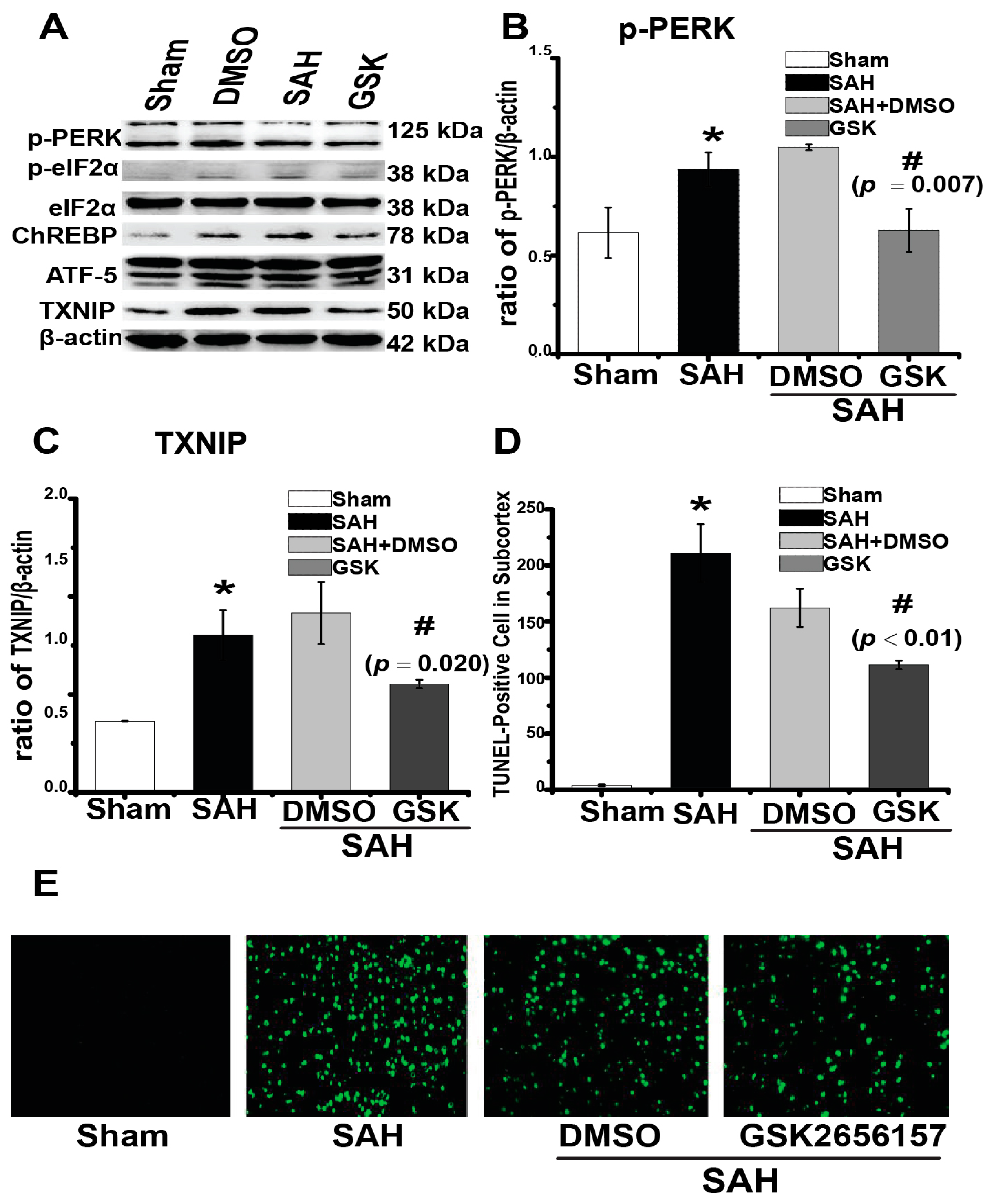
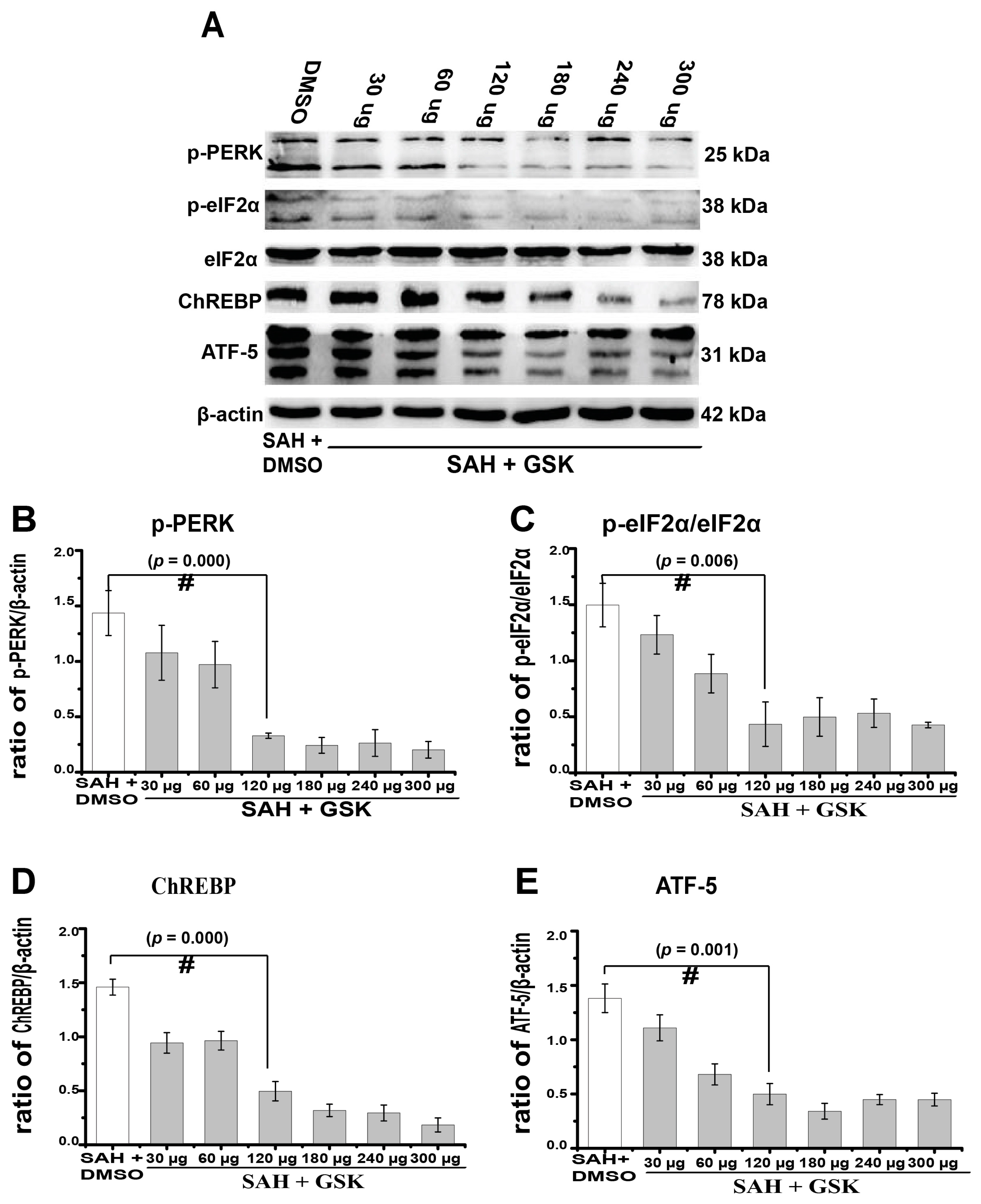
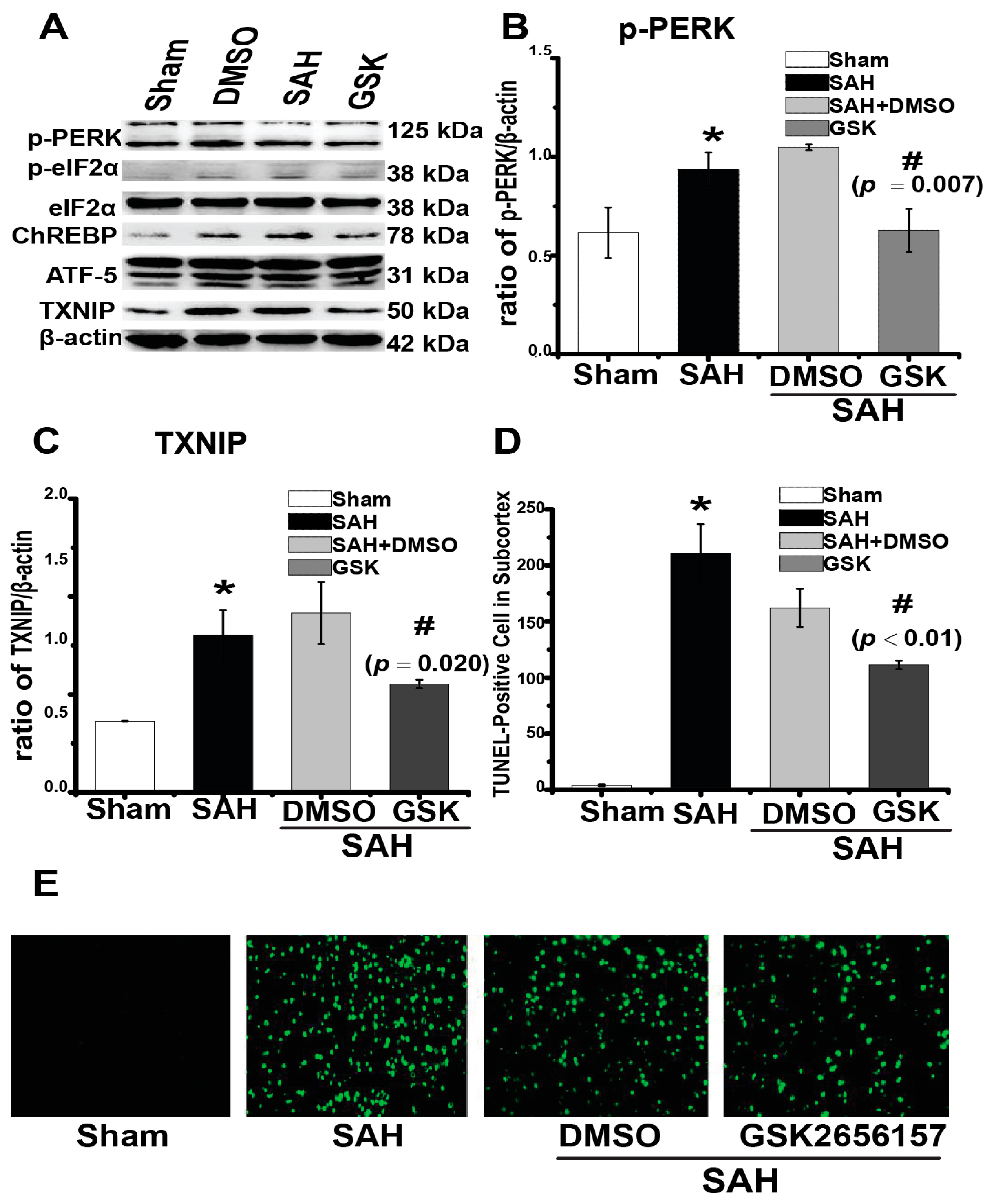
2.4. Downregulation of TXNIP by PERK Inhibition
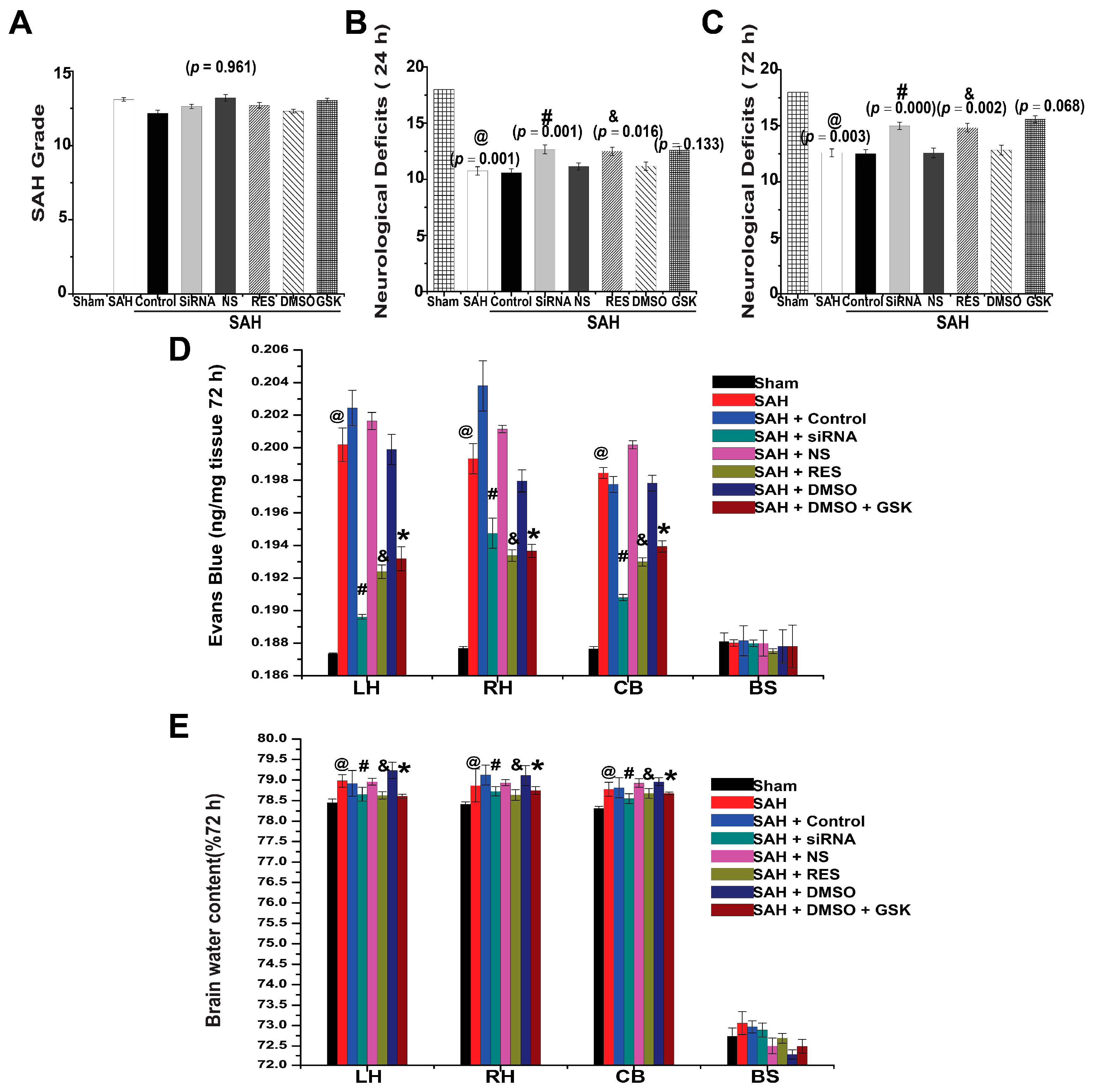
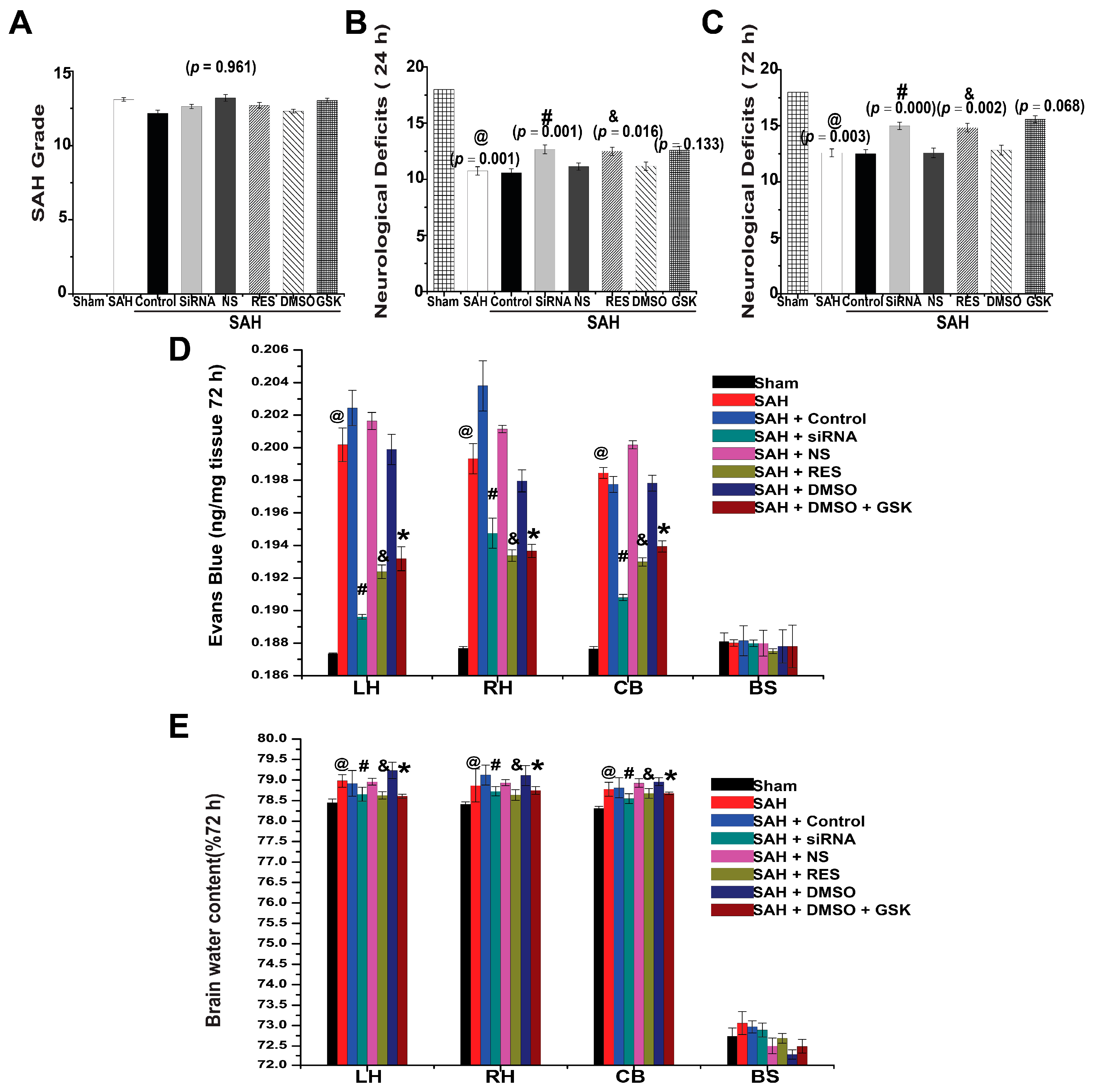
2.5. SAH Grade and Mortality
2.6. Neurological Deficits
2.7. BBB Permeability
2.8. Brain Water Content
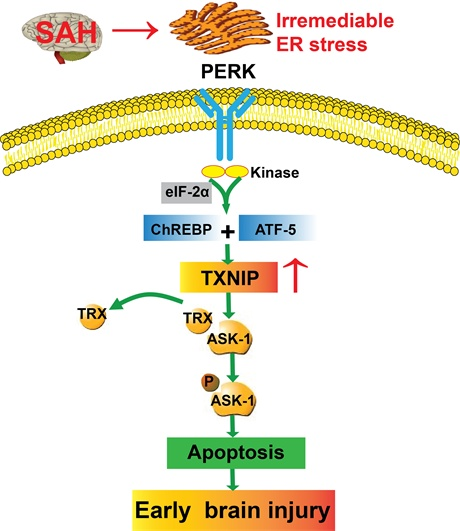
3. Discussion
4. Materials and Methods
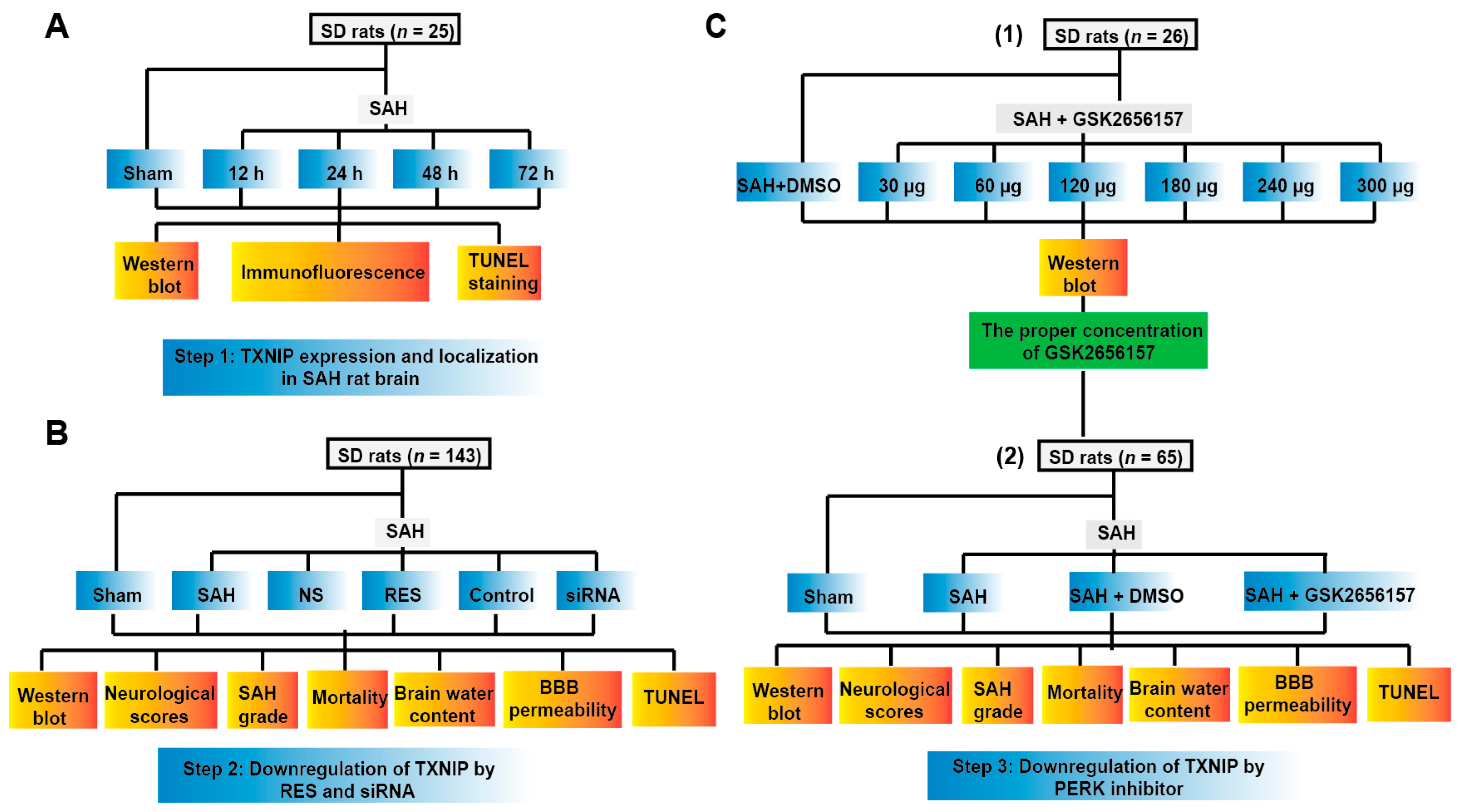
4.1. Animals
4.2. Endovascular Perforation Model of SAH
4.3. Drug Administration
4.3.1. Resveratrol and TXNIP siRNA Injection
4.3.2. GSK2656157 Injection
4.4. Neurological Scores
4.5. SAH Grade and Mortality
4.6. Blood-Brain Barrier (BBB) Permeability
4.7. Brain Water Content
4.8. Immunofluorescence
4.9. TUNEL Staining
4.10. Western Blot
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Suzuki, H.; Sozen, T.; Altay, O.; Zhang, J.H. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir. Suppl. 2011, 110, 43–48. [Google Scholar] [PubMed]
- He, Z.; Ostrowski, R.P.; Sun, X.; Ma, Q.; Huang, B.; Zhan, Y.; Zhang, J.H. Chop silencing reduces acute brain injury in the rat model of subarachnoid hemorrhage. Stroke J. Cereb. Circ. 2012, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.E. On the origins of arrestin and rhodopsin. BMC Evol. Biol. 2008, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Ji, L.; Xing, W.; Zhang, W.; Zhou, H.; Qian, X.; Wang, X.; Gao, F.; Sun, X.; Zhang, H. Acute hyperglycaemia enhances oxidative stress and aggravates myocardial ischaemia/reperfusion injury: Role of thioredoxin-interacting protein. J. Cell. Mol. Med. 2013, 17, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, T.; Mohamed, I.N.; Pillai, B.; Soliman, S.; Fouda, A.Y.; Ergul, A.; El-Remessy, A.B.; Fagan, S.C. Thioredoxin-interacting protein: A novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol. Neurobiol. 2015, 51, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T. Thioredoxin-interacting protein mediates er stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the bcl-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, L.; Zhang, L.; Wu, J.; Zhou, Y.; Zhou, Y.; Zhao, Y.; Zhao, J. Inhibition of thioredoxin-1 with sirna exacerbates apoptosis by activating the ask1-jnk/p38 pathway in brain of a stroke model rats. Brain Res. 2015, 1599, 20–31. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ostrowski, R.P.; Sun, X.; Ma, Q.; Tang, J.; Zhang, J.H. Targeting c/ebp homologous protein with sirna attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. Exp. Neurol. 2012, 238, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.S.; Jung, J.E.; Narasimhan, P.; Sakata, H.; Chan, P.H. Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiol. Dis. 2012, 46, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Negoescu, A.; Lorimier, P.; Labat-Moleur, F.; Drouet, C.; Robert, C.; Guillermet, C.; Brambilla, C.; Brambilla, E. In situ apoptotic cell labeling by the tunel method: Improvement and evaluation on cell preparations. J. Histochem. Cytochem. 1996, 44, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Dis. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Bedarida, T.; Baron, S.; Vibert, F.; Ayer, A.; Henrion, D.; Thioulouse, E.; Marchiol, C.; Beaudeux, J.L.; Cottart, C.H.; Nivet-Antoine, V. Resveratrol decreases txnip mrna and protein nuclear expressions with an arterial function improvement in old mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Zhou, M.L.; Zhang, X.S.; Zhuang, Z.; Li, T.; Shi, J.X.; Zhang, X. Resveratrol prevents neuronal apoptosis in an early brain injury model. J. Surg. Res. 2014, 189, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhu, Y.; Liu, Y.; Tipoe, G.L.; Xing, F.; So, K.-F. Lycium barbarum polysaccharide attenuates alcoholic cellular injury through txnip-nlrp3 inflammasome pathway. Int. J. Biol. Macromol. 2014, 69, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. Nlrp3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. Perk is required at the er-mitochondrial contact sites to convey apoptosis after ros-based er stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yamaguchi, M.; Zhou, C.; Calvert, J.W.; Tang, J.; Zhang, J.H. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke J. Cereb. Circ. 2004, 35, 2412–2417. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke J. Cereb. Circ. 2009, 40, e331–e339. [Google Scholar] [CrossRef] [PubMed]
- Kaya, B.; Erdi, F.; Kilinc, I.; Keskin, F.; Feyzioglu, B.; Esen, H.; Karatas, Y.; Uyar, M.; Kalkan, E. Alterations of the thioredoxin system during subarachnoid hemorrhage-induced cerebral vasospasm. Acta Neurochir. 2015, 157, 793–799; discussion 799–800. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ask) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin d3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Suh, H.W.; Jeon, Y.H.; Hwang, E.; Nguyen, L.T.; Yeom, J.; Lee, S.G.; Lee, C.; Kim, K.J.; Kang, B.S.; et al. The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein. Nat. Commun. 2014, 5, 2958. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M. Ire1α induces thioredoxin-interacting protein to activate the nlrp3 inflammasome and promote programmed cell death under irremediable er stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet. Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.I.; Deshmukh, M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and apaf-1 for execution in primary neurons. Cell Death Differ. 2007, 14, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, S.; Oyadomari, S.; Yano, S.; Morioka, M.; Gotoh, T.; Hamada, J.I.; Ushio, Y.; Mori, M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving chop. Cell Death Differ. 2004, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Cao, S.; Li, J.; Dixon, B.; Yu, X.; Chen, J.; Gu, C.; Lin, W.; Chen, G. Pharmacological inhibition of perk attenuates early brain injury after subarachnoid hemorrhage in rats through the activation of akt. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sagher, O.; Keep, R.; Hua, Y.; Xi, G. Comparison of experimental rat models of early brain injury after subarachnoid hemorrhage. Neurosurgery 2009, 65, 331–343; discussion 343. [Google Scholar] [CrossRef] [PubMed]
- Duris, K.; Manaenko, A.; Suzuki, H.; Rolland, W.B.; Krafft, P.R.; Zhang, J.H. A7 nicotinic acetylcholine receptor agonist pnu-282987 attenuates early brain injury in a perforation model of subarachnoid hemorrhage in rats. Stroke J. Cereb. Circ. 2011, 42, 3530–3536. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel perk kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Ayer, R.; Jadhav, V.; Zhang, J.H. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J. Neurosci. Methods 2008, 167, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Tsubokawa, T.; Solaroglu, I.; Yatsushige, H.; Cahill, J.; Yata, K.; Zhang, J.H. Cathepsin and calpain inhibitor e64d attenuates matrix metalloproteinase-9 activity after focal cerebral ischemia in rats. Stroke J. Cereb. Circ. 2006, 37, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Suzuki, H.; Altay, O.; Zhang, J.H. Preservation of tropomyosin-related kinase b (trkb) signaling by sodium orthovanadate attenuates early brain injury after subarachnoid hemorrhage in rats. Stroke J. Cereb. Circ. 2011, 42, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Hasegawa, Y.; Kanamaru, K.; Zhang, J.H. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke J. Cereb. Circ. 2010, 41, 1783–1790. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences |
|---|---|
| siRNA 1 [17] | sense: 5′-GCUGG AUAGACCUAAACAUTT-3′ antisense: 5′-AUGUUUAGGUCUAUCCAGCTT-3′ |
| siRNA 2 [18] | sense: 5′-UGGUCACGUCGAAAUGAAUTT-3′ antisense: 5′-TTACCAGUGCAGCUUUACUUA-3′ |
| Control siRNA | sense: 5′-UUCUCCGAACGUGUCACGUTT-3′ antisense: 5′-ACGUGACACGUUCGGAGAATT-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Che, X.; Zhang, H.; Tan, G.; Liu, L.; Jiang, D.; Zhao, J.; Xiang, X.; Sun, X.; He, Z. Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage. Int. J. Mol. Sci. 2017, 18, 854. https://doi.org/10.3390/ijms18040854
Zhao Q, Che X, Zhang H, Tan G, Liu L, Jiang D, Zhao J, Xiang X, Sun X, He Z. Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage. International Journal of Molecular Sciences. 2017; 18(4):854. https://doi.org/10.3390/ijms18040854
Chicago/Turabian StyleZhao, Qing, Xudong Che, Hongxia Zhang, Guanping Tan, Liu Liu, Dengzhi Jiang, Jun Zhao, Xiang Xiang, Xiaochuan Sun, and Zhaohui He. 2017. "Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage" International Journal of Molecular Sciences 18, no. 4: 854. https://doi.org/10.3390/ijms18040854