
Adiponectin Is Involved in Connective Tissue Growth Factor-Induced Proliferation, Migration and Overproduction of the Extracellular Matrix in Keloid Fibroblasts

Abstract
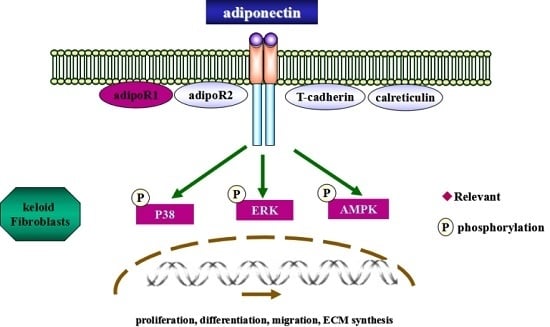
:
1. Introduction
2. Results
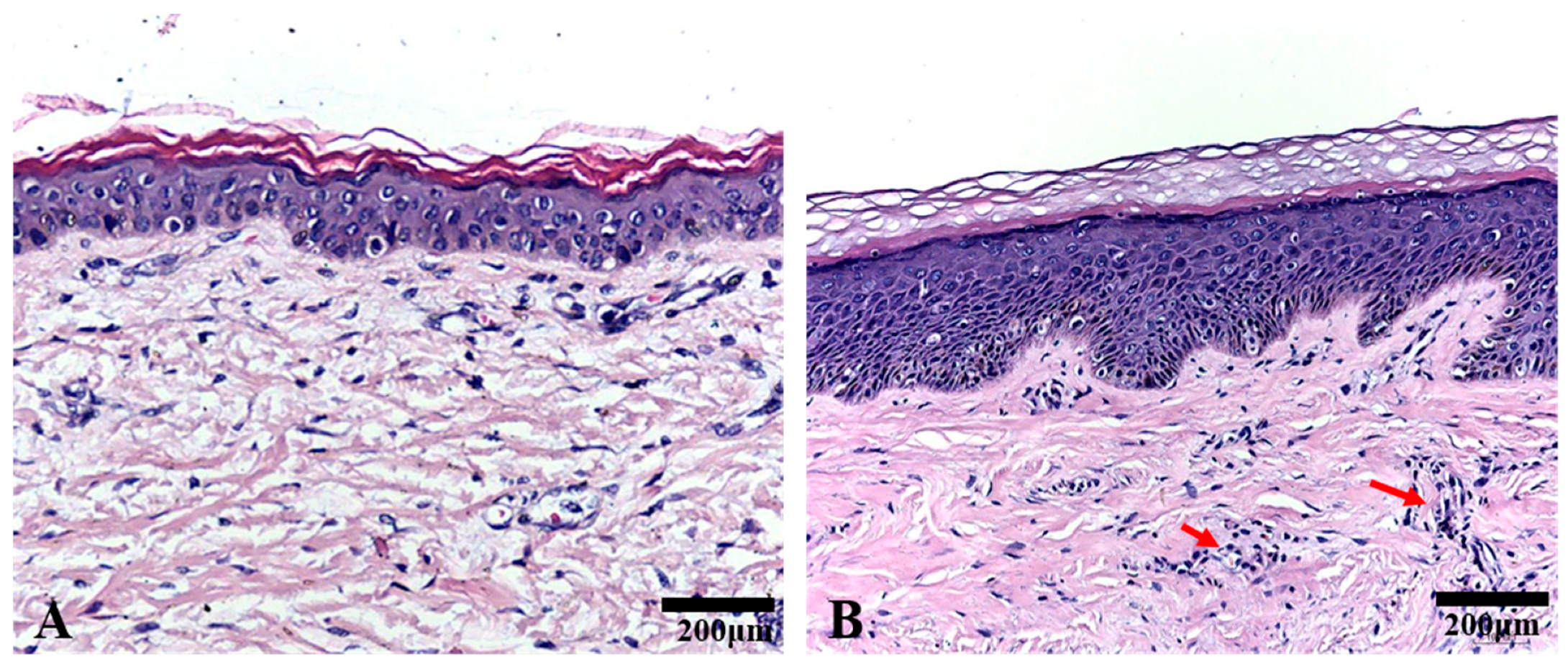
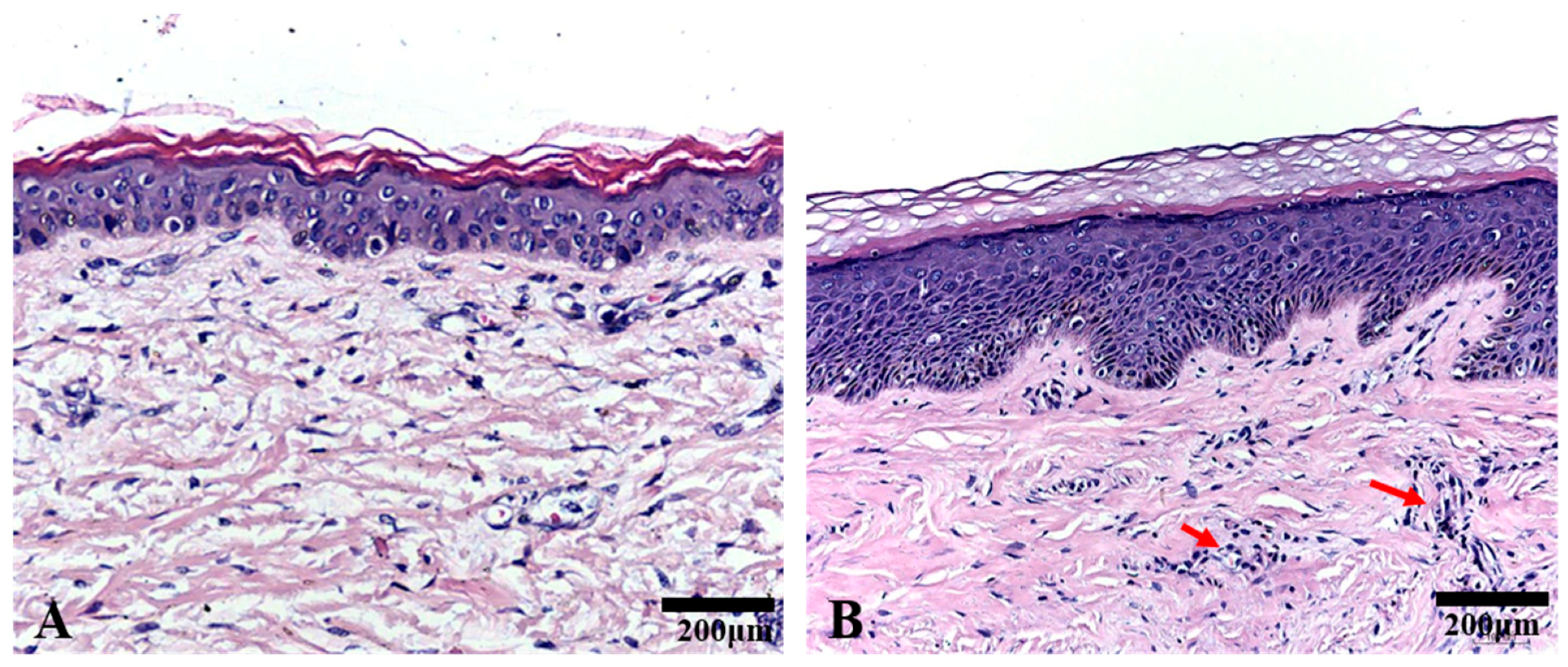
2.1. Histological Analysis
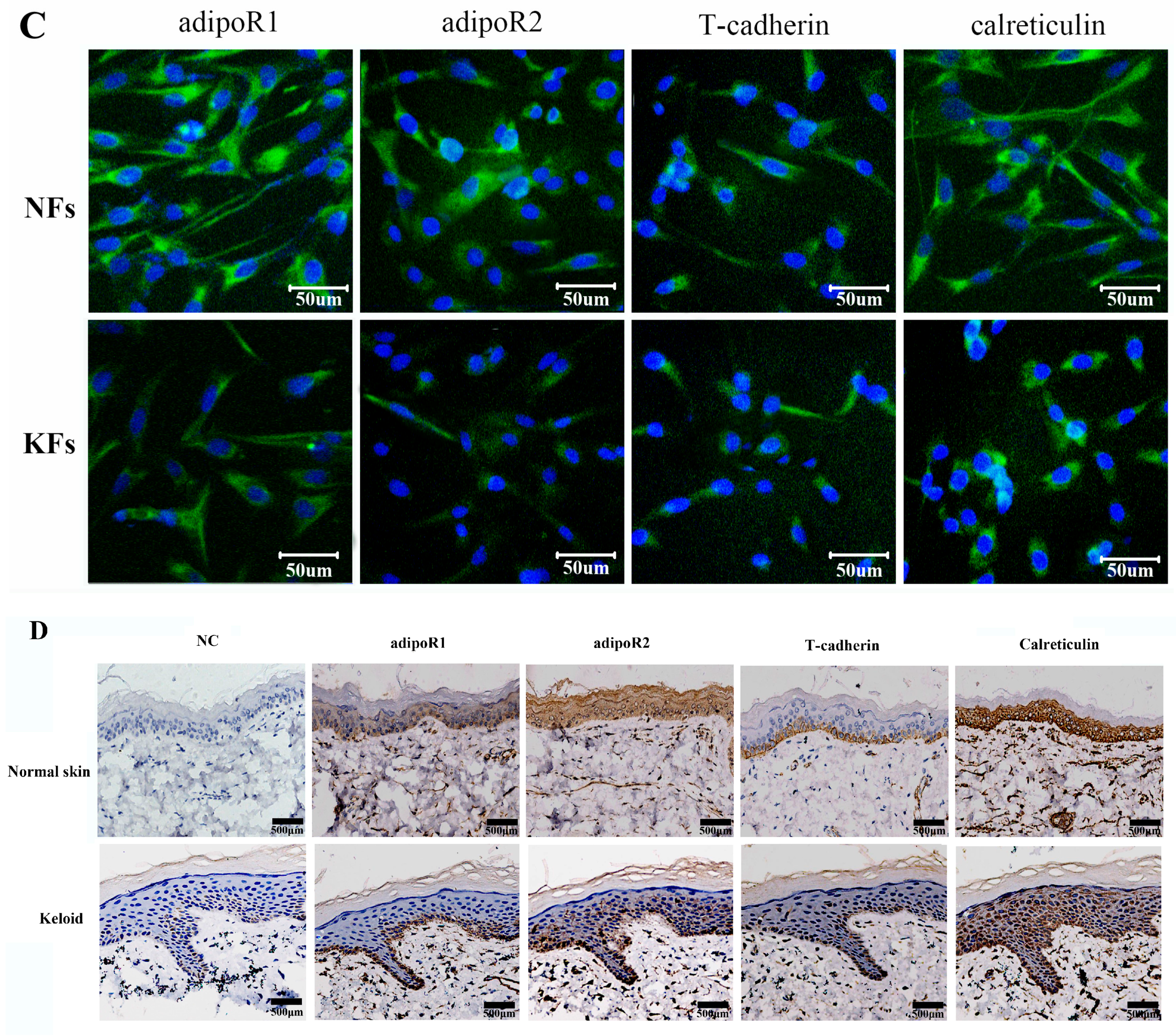
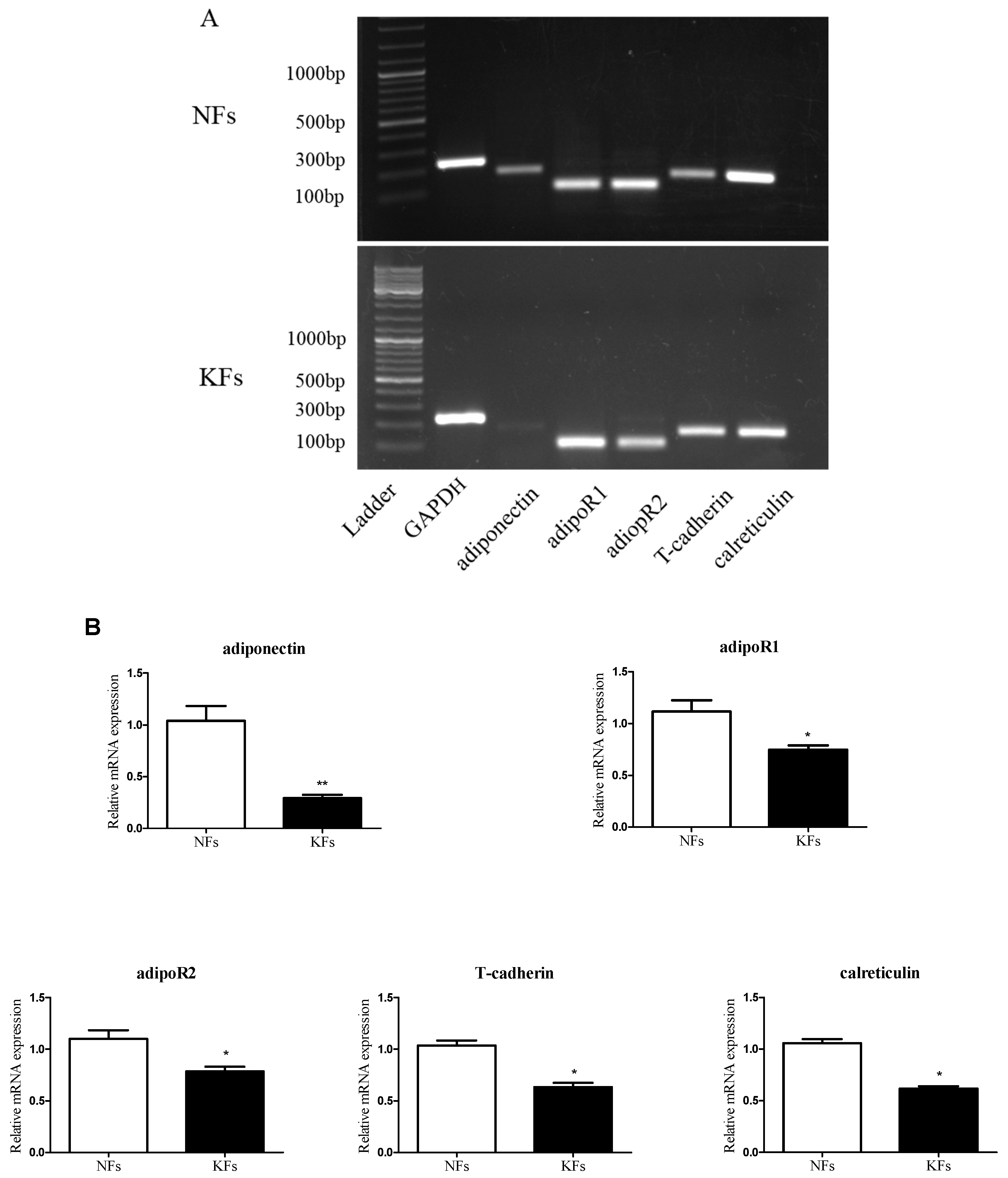
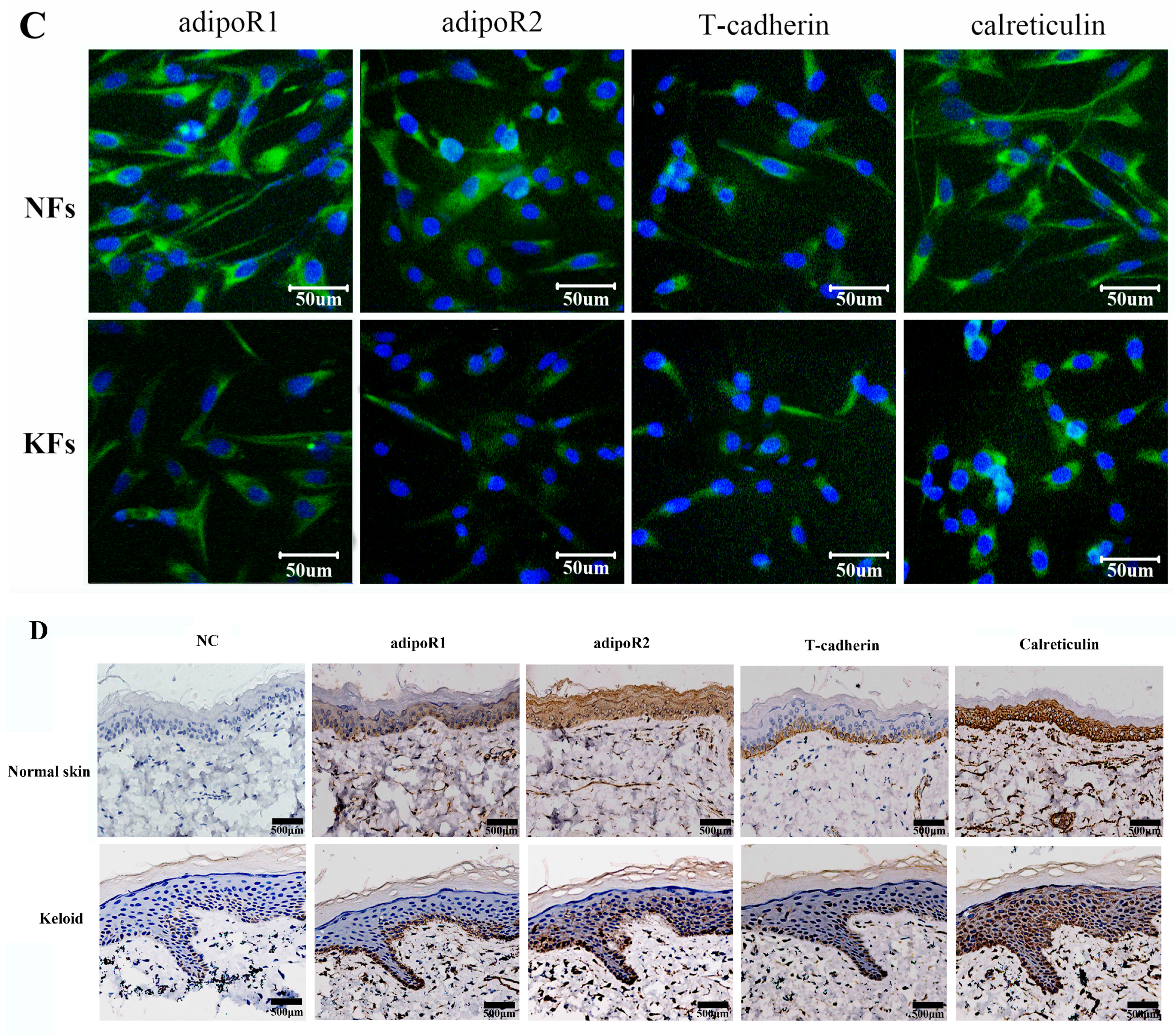
2.2. Expression of Adiponectin and AdipoRs in Keloids
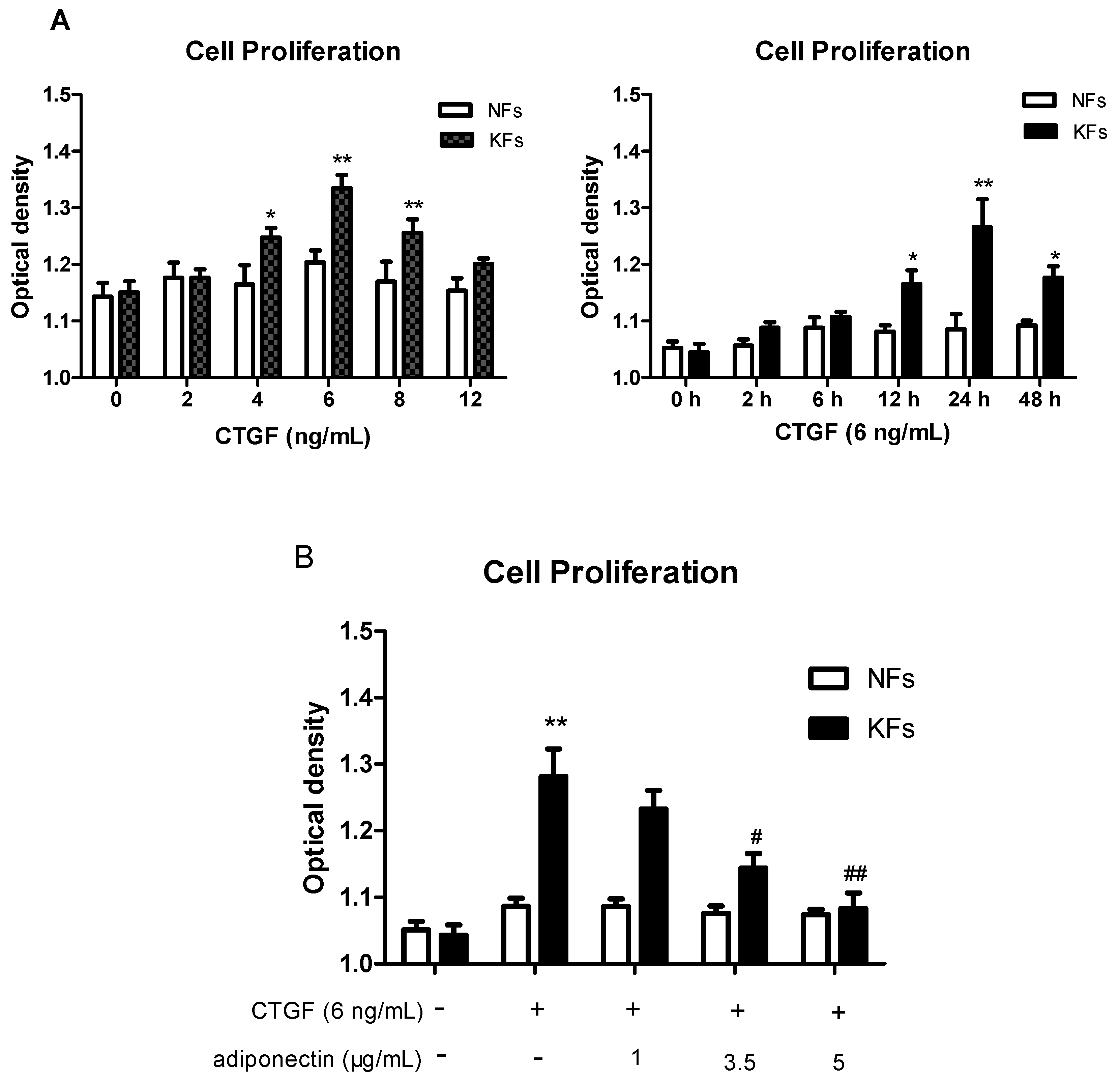
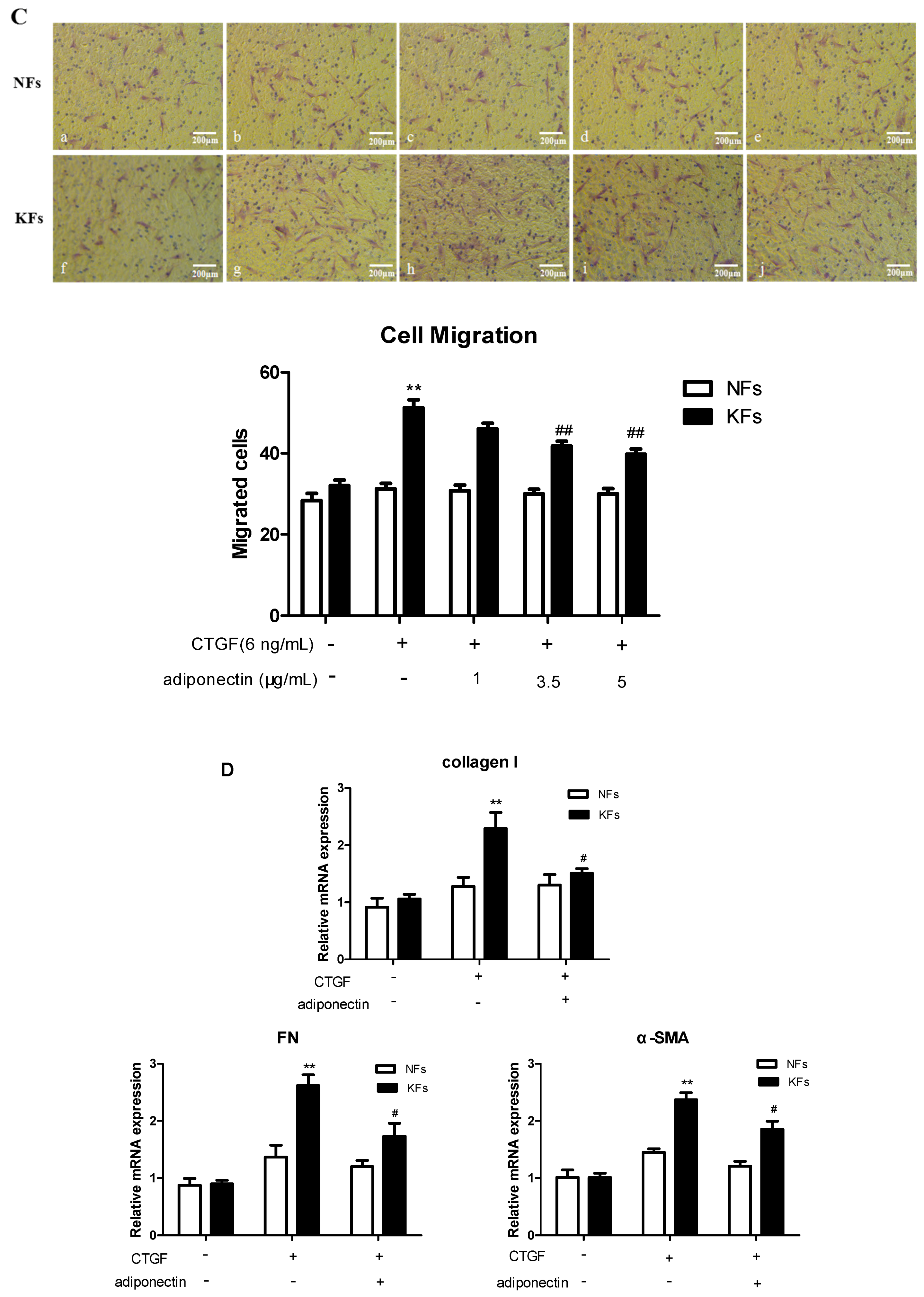
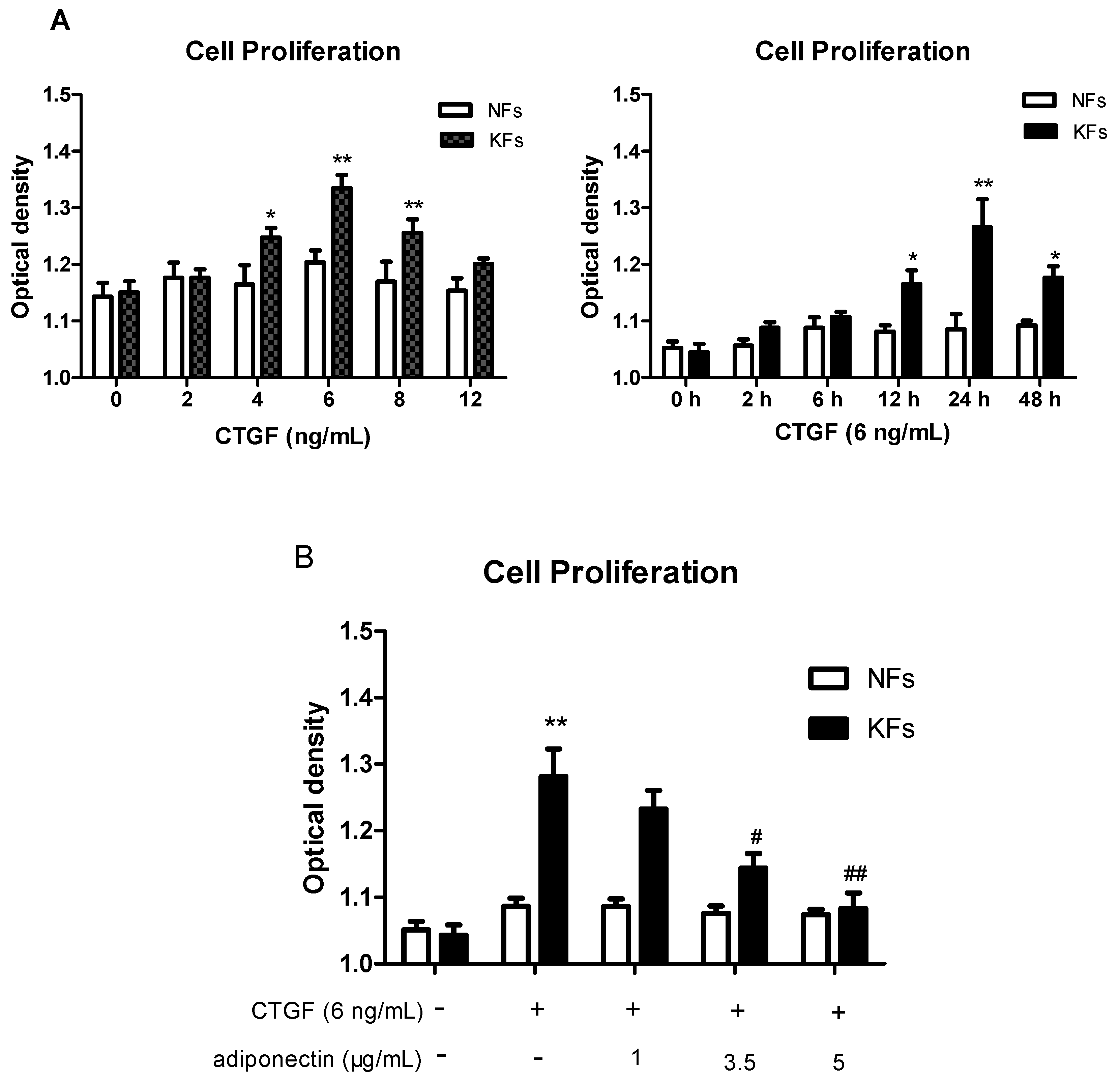
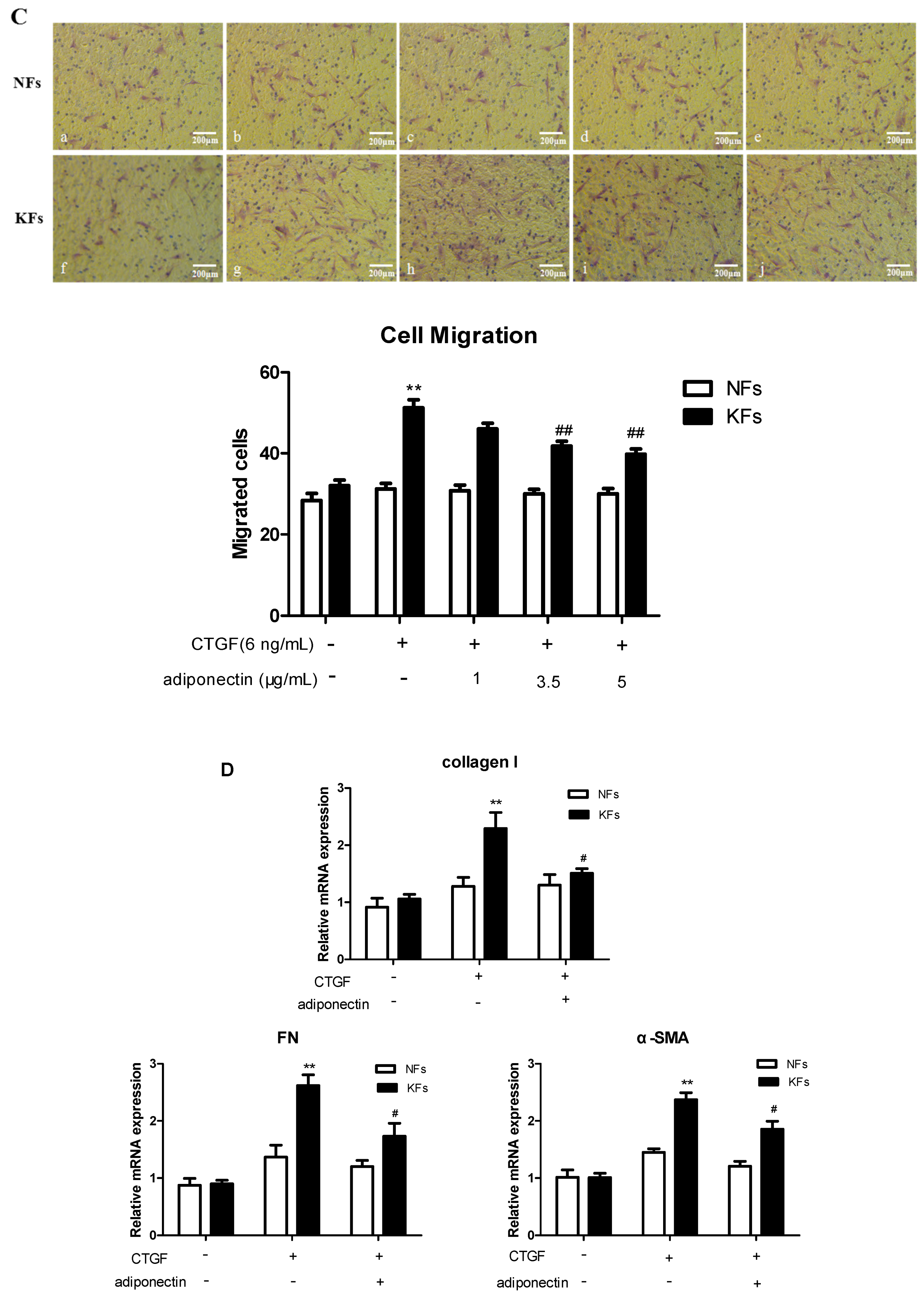
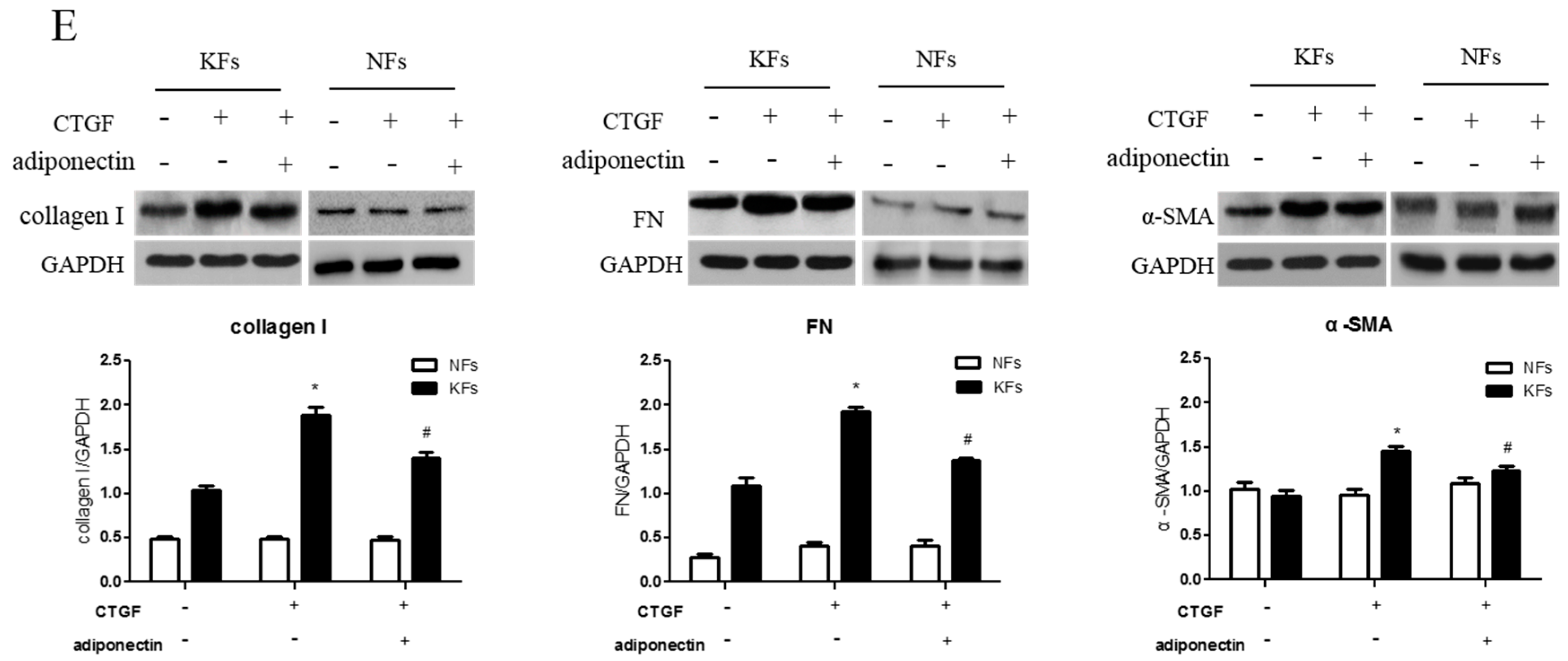
2.3. Adiponectin Suppresses CTGF-Induced Keloid Fibroblast Proliferation, Migration and ECM Production
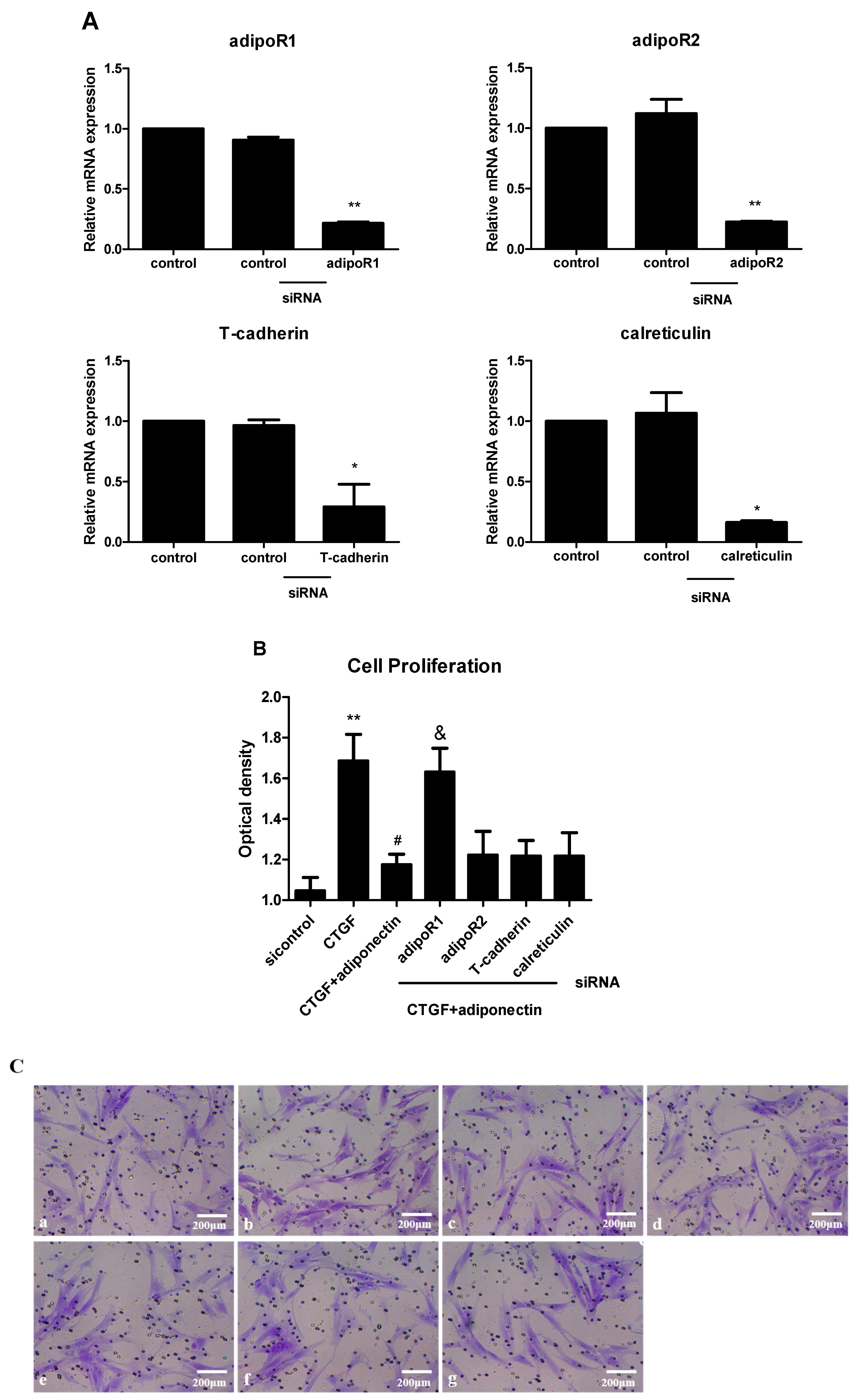
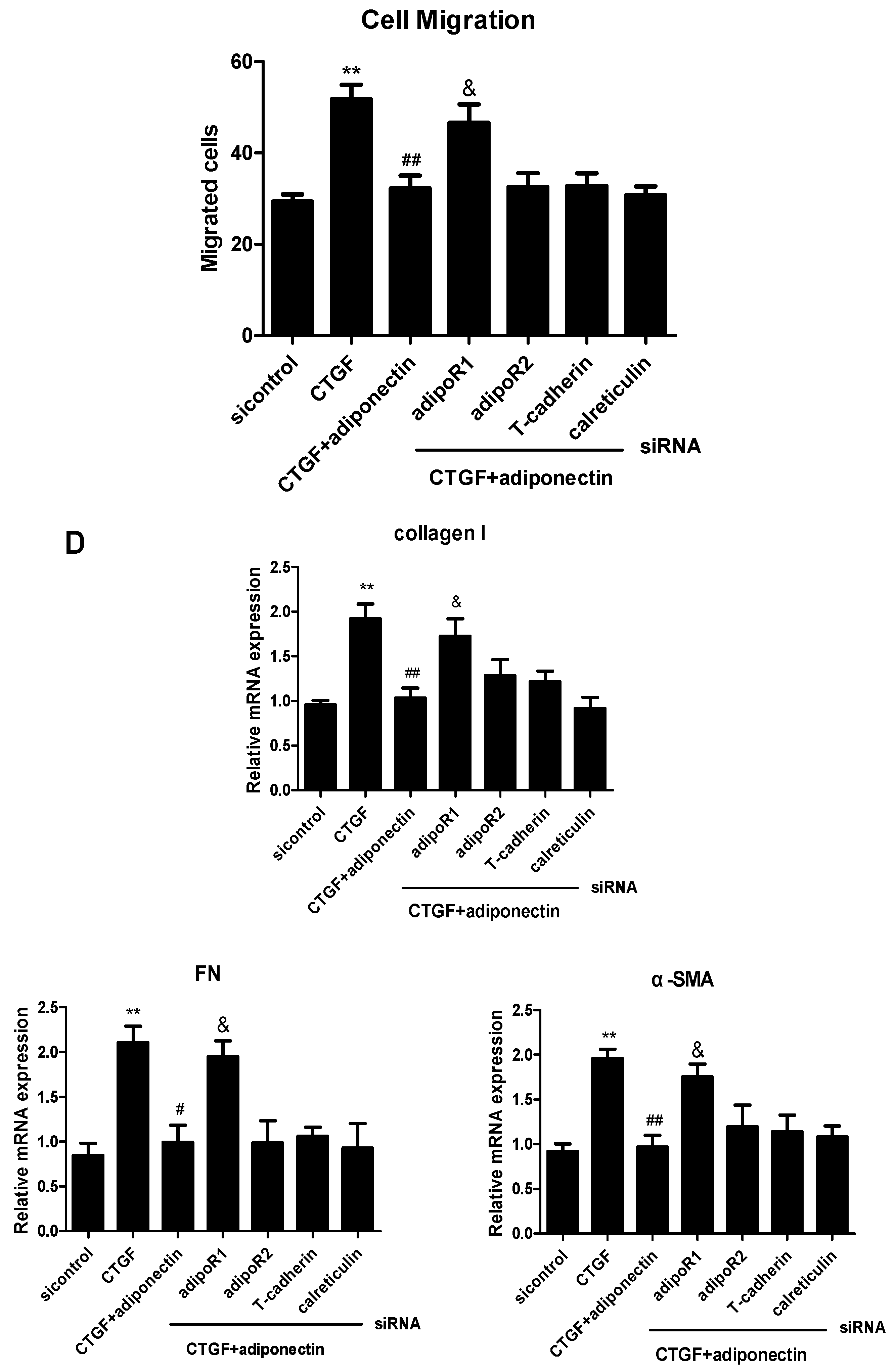
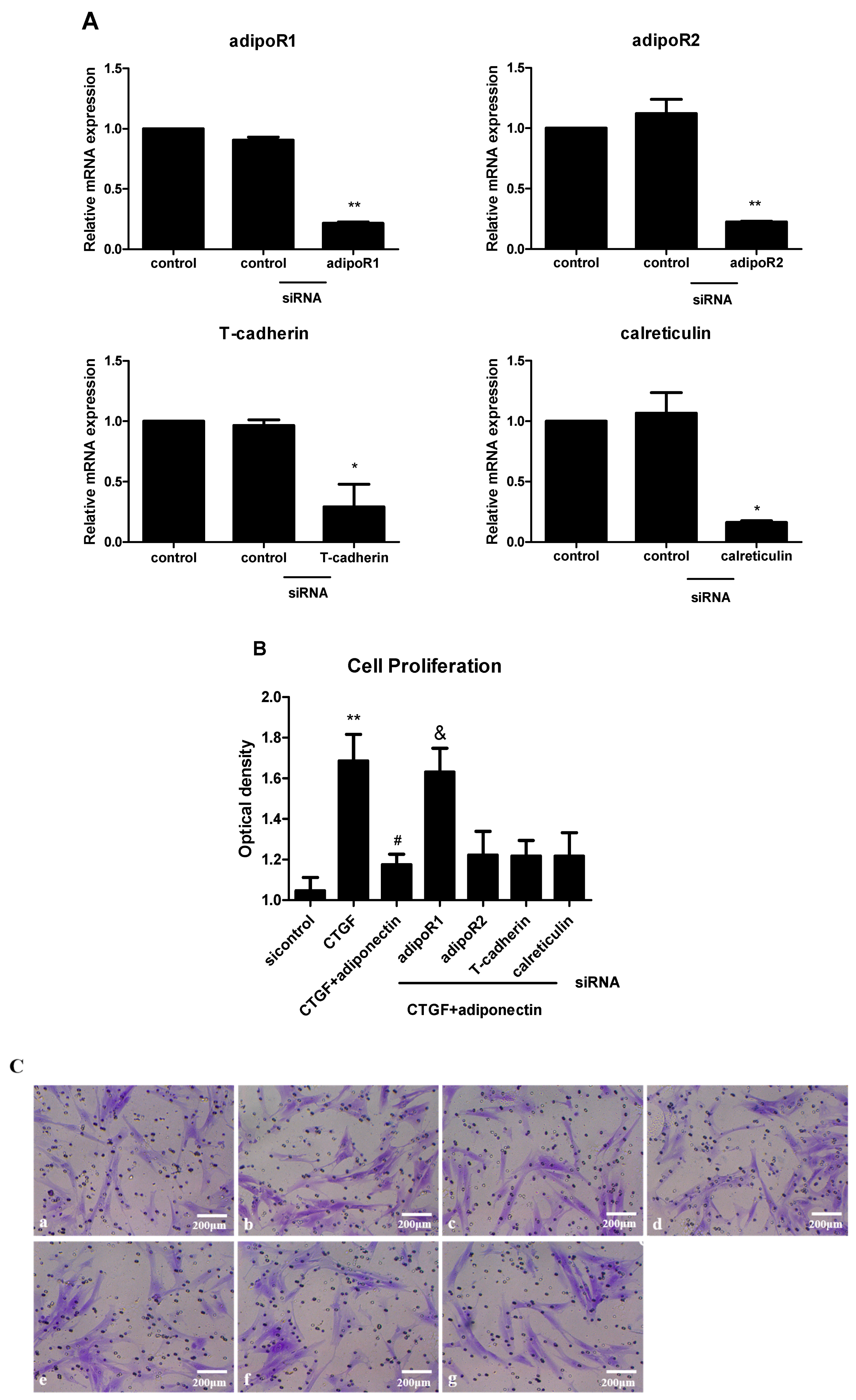
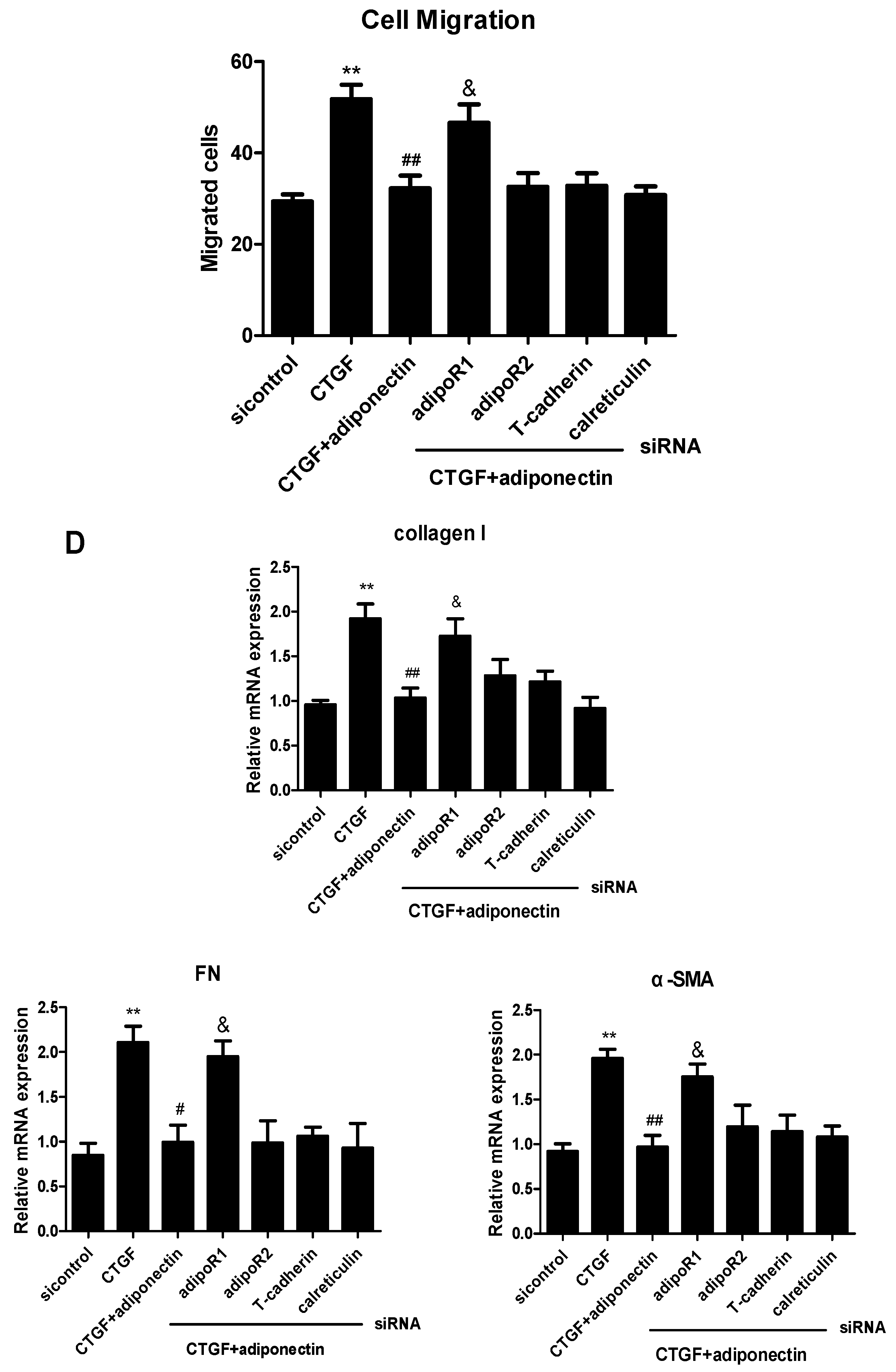
2.4. AdipoR1 Is Involved in Adiponectin-Mediated Signalling Pathways in KFs
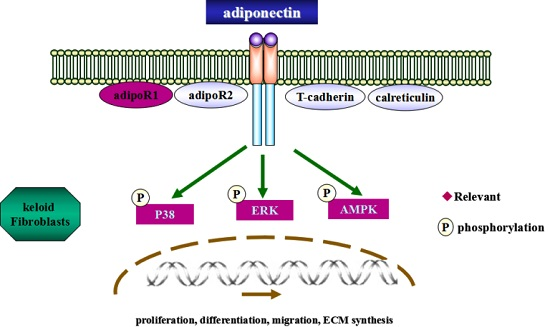
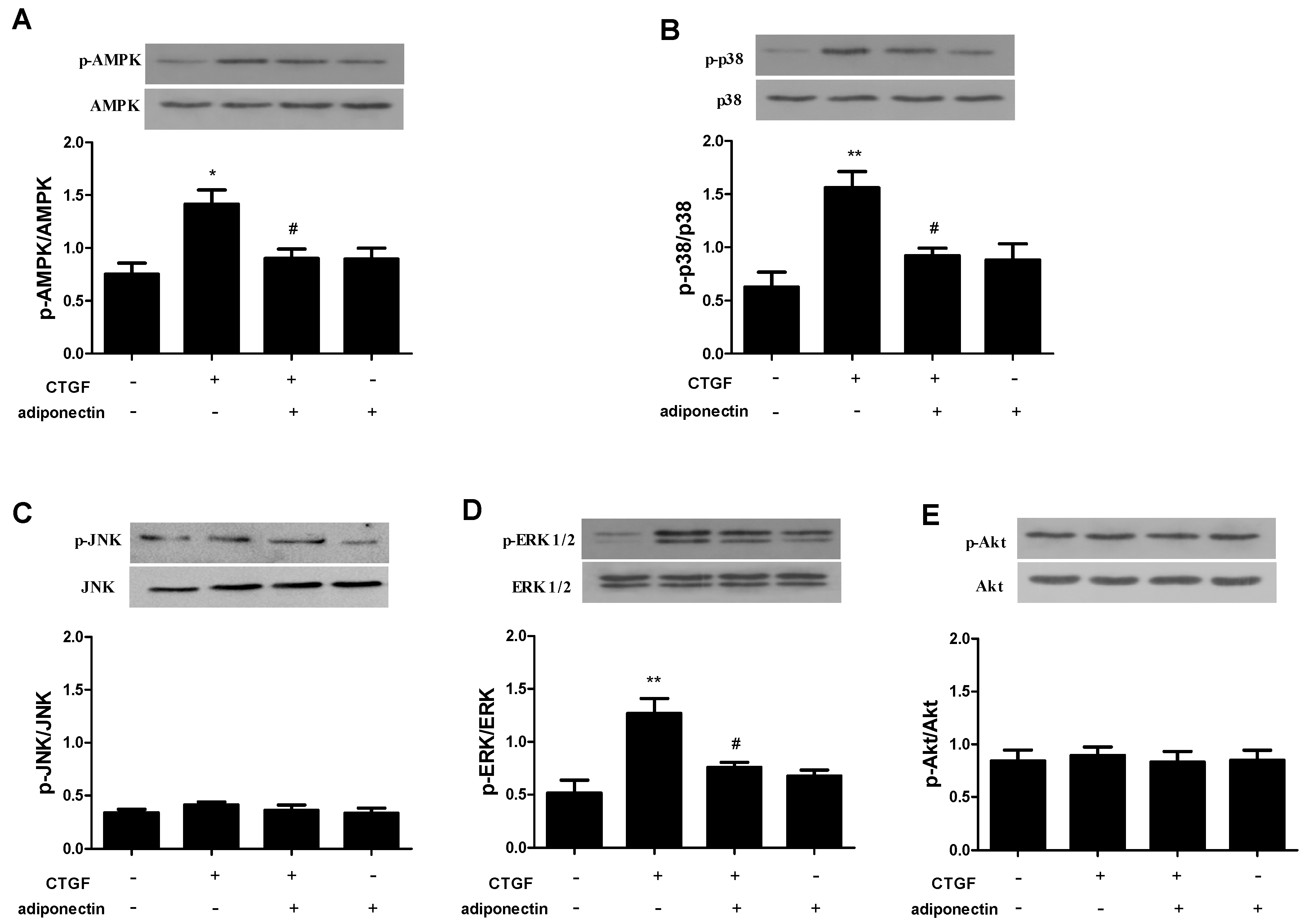
2.5. Adiponectin Attenuates CTGF-Induced Phosphorylation of AMPK, p38 MAPK and ERK in KFs
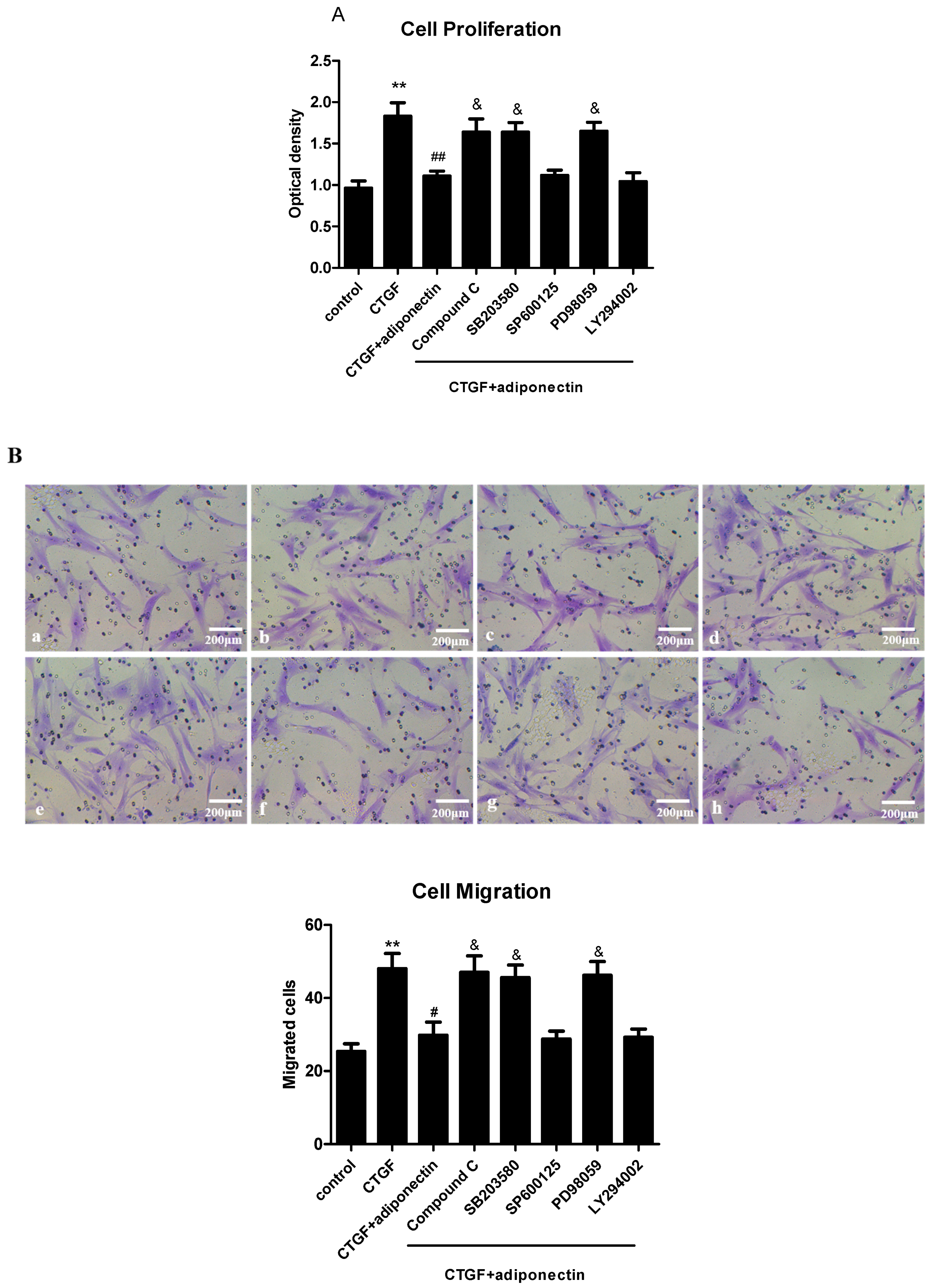
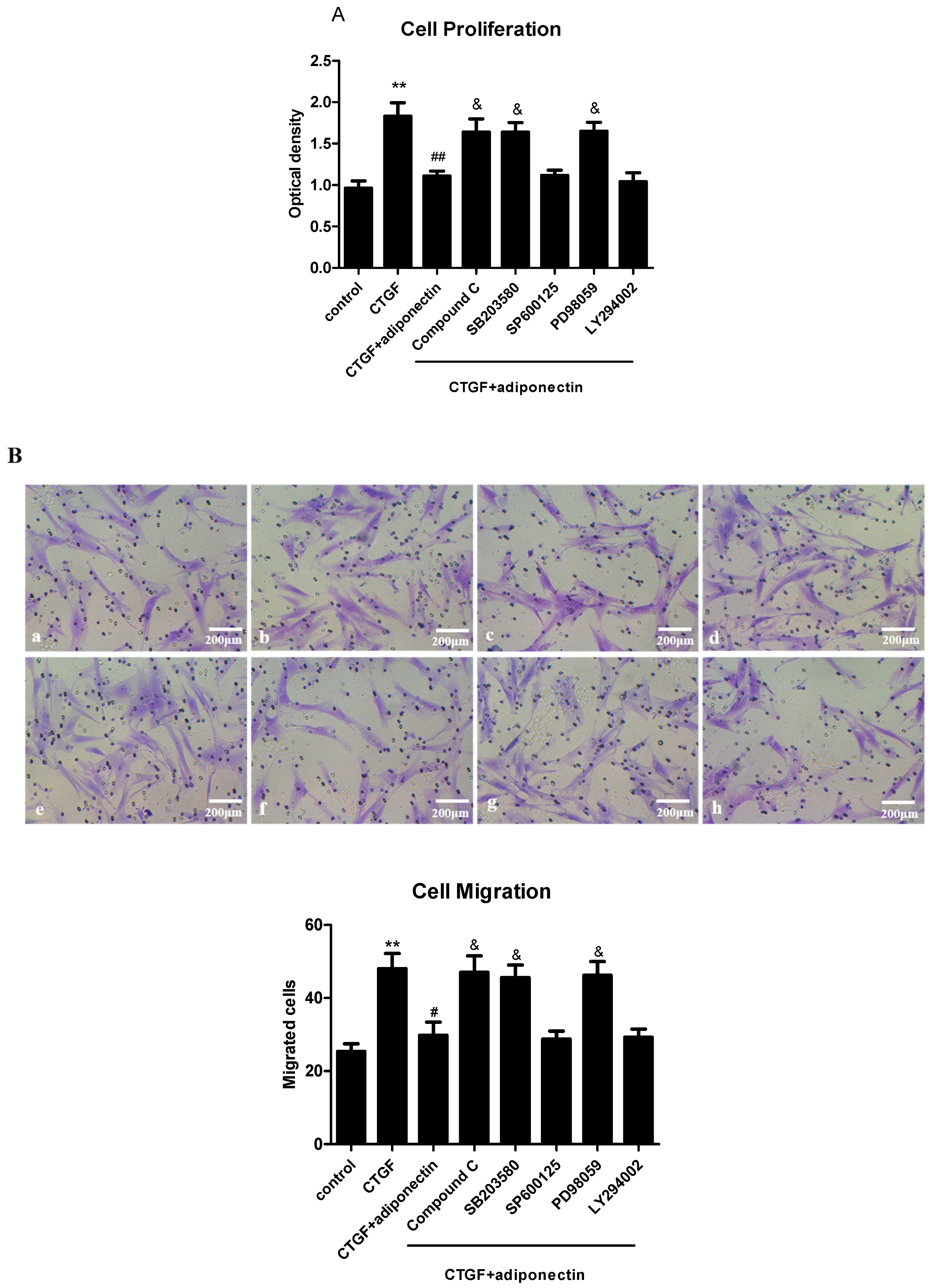
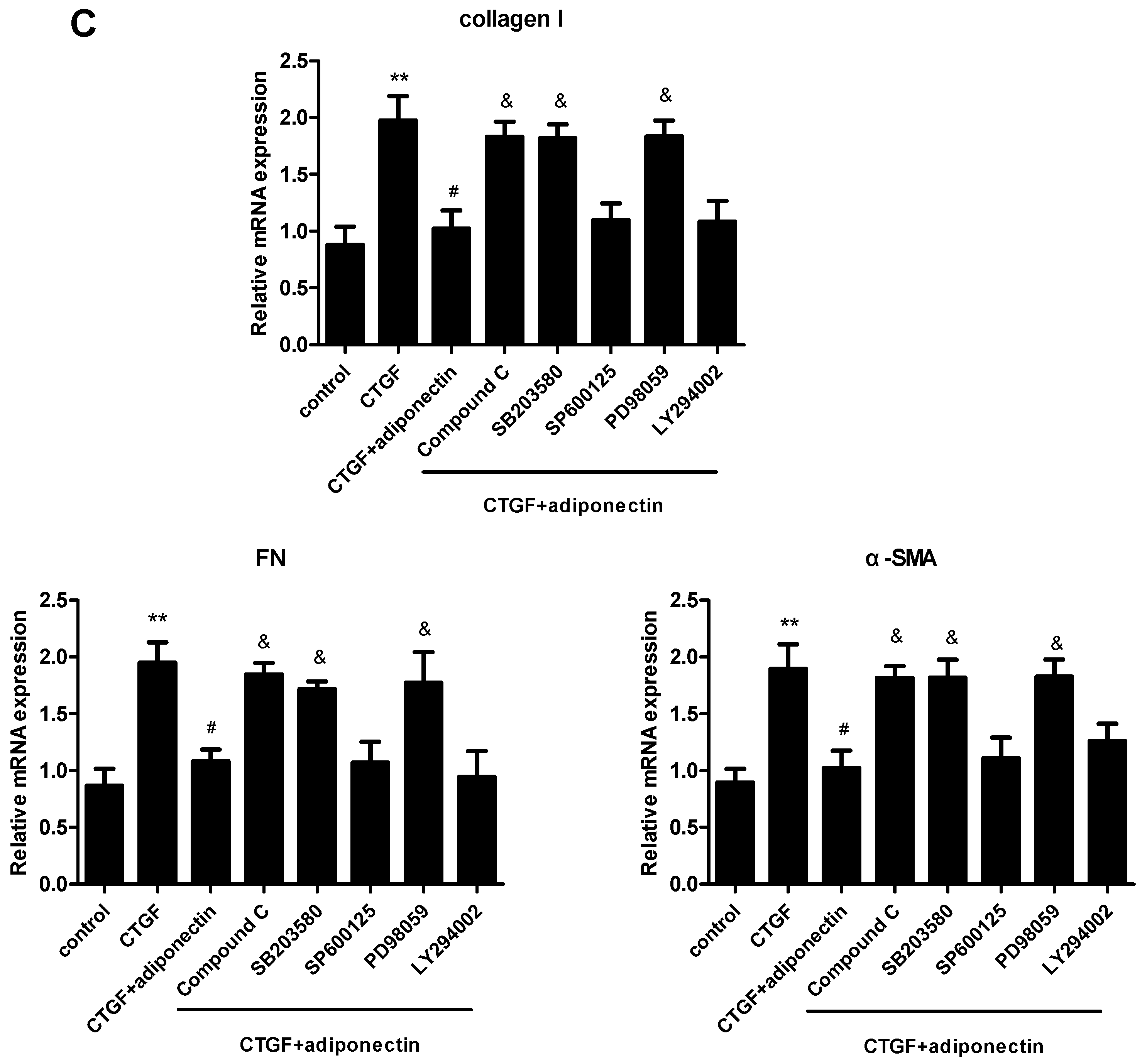
2.6. AMPK, p38 MAPK and ERK Signalling Pathways Are Involved in Adiponectin-Mediated CTGF-Induced KFs Proliferation, Migration and ECM Production
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Source and Culture
4.3. RNA Interference
4.4. RT-PCR and Quantitative Real-Time RT-PCR Analysis
4.5. Western Blot Analysis
4.6. Immunofluorescence Staining
4.7. HE Staining and Immunohistochemistry
4.8. Cell Proliferation Assay
4.9. Cell Migration Assay
4.10. Statistical Aznalysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seo, B.F.; Jung, S.N. The immunomodulatory effects of mesenchymal stem cells in prevention or treatment of excessive scars. Stem Cells Int. 2016, 2016, 6937976. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Wulff, B.C. The importance of mast cells in dermal scarring. Adv. Wound Care 2014, 3, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Mathangi, R.K.; Babu, M.; Lakshmi, M.M.S. Response of keloid fibroblasts to vitamin D3 and quercetin treatment—In vitro study. Ann. Burns Fire Disasters 2015, 28, 187–191. [Google Scholar]
- Mofikoya, B.O.; Adeyemo, W.L.; Ugburo, A.O. An overview of biological basis of pathologic scarring. Niger. Postgrad. Med. J. 2012, 19, 40–45. [Google Scholar] [PubMed]
- Bradham, D.M.; Igarashi, A.; Potter, R.L. Connective tissue growth factor: A cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1991, 114, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, D.P.; de Farias, G.C.; de Sousa, E.B. New strategy to control cell migration and metastasis regulated by CCN2/CTGF. Cancer Cell Int. 2014, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.F. Cell surface receptors for CCN proteins. J. Cell Commun. Signal. 2016, 10, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, B.R.; Wu, T.S. Input of microenvironmental regulation on colorectal cancer: Role of the CCN family. World J. Gastroenterol. 2014, 20, 6826–6831. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Parapuram, S.K.; Shi-Wen, X. Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal. 2009, 3, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Colwell, A.S.; Phan, T.T.; Kong, W. Hypertrophic scar fibroblasts have increased connective tissue growth factor expression after transforming growth factor-β stimulation. Plast. Reconstr. Surg. 2005, 116, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Grotendorst, G.R. Connective tissue growth factor: A mediator of TGF-β action on fibroblasts. Cytokine Growth Factor Rev. 1997, 8, 171–179. [Google Scholar] [CrossRef]
- Jurzak, M.; Adamczyk, K.; Antonczak, P. Evaluation of genistein ability to modulate CTGF mRNA/protein expression, genes expression of TGFβ isoforms and expression of selected genes regulating cell cycle in keloid fibroblasts in vitro. Acta Pol. Pharm. 2014, 71, 972–986. [Google Scholar] [PubMed]
- Fang, X.; Sweeney, G. Mechanisms regulating energy metabolism by adiponectin in obesity and diabetes. Biochem. Soc. Trans. 2006, 34, 798–801. [Google Scholar]
- Arita, Y.; Kihara, S.; Ouchi, N. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. 1999. Biochem. Biophys. Res. Commun. 2012, 425, 560–564. [Google Scholar]
- Takahashi, M.; Arita, Y.; Yamagata, K. Genomic structure and mutations in adipose-specific gene, adiponectin. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Ito, Y. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Dong, L.Q. Adiponectin signalling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, M. Adiponectin: A versatile player of innate immunity. J. Mol. Cell Biol. 2016, 8, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G. Adiponectin and inflammation: Consensus and controversy. J. Allergy Clin. Immunol. 2008, 121, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Liu, L.; Yang, Y. The adipokine adiponectin has potent anti-fibrotic effects mediated via adenosine monophosphate-activated protein kinase: Novel target for fibrosis therapy. Arthritis Res. Ther. 2012, 14, R229. [Google Scholar] [CrossRef] [PubMed]
- Reinke, L.; Lam, A.P.; Flozak, A.S. Adiponectin inhibits Wnt co-receptor, Lrp6, phosphorylation and β-catenin signalling. Biochem. Biophys. Res. Commun. 2016, 470, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Tada, Y.; Asano, Y. Adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signalling pathway. J. Immunol. 2012, 189, 3231–3241. [Google Scholar] [CrossRef] [PubMed]
- Denzel, M.S.; Scimia, M.C.; Zumstein, P.M. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J. Clin. Investig. 2010, 120, 4342–4352. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Hug, C.; Wang, J.; Ahmad, N.S. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. USA 2004, 101, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Nakerakanti, S.S.; Bujor, A.M.; Trojanowska, M. CCN2 is required for the TGF-β induced activation of Smad1-Erk1/2 signalling network. PLoS ONE 2011, 6, e21911. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Kong, W.; Wang, Z. Increased CCN2 transcription in keloid fibroblasts requires cooperativity between AP-1 and SMAD binding sites. Ann. Surg. 2007, 246, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Cool, B.L.; Laderoute, K.R. AMP-activated protein kinase inhibits transforming growth factor-β-induced Smad3-dependent transcription and myofibroblast transdifferentiation. J. Biol. Chem. 2008, 283, 10461–10469. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qu, M.; Xu, L. Sorafenib exerts an anti-keloid activity by antagonizing TGF-β/Smad and MAPK/ERK signalling pathways. J. Mol. Med. 2016, 94, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Li, G.; Li, S. Aspidin PB, a novel natural anti-fibrotic compound, inhibited fibrogenesis in TGF-β1-stimulated keloid fibroblasts via PI-3K/Akt and Smad signalling pathways. Chem. Biol. Interact. 2015, 238, 66–73. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, X.; Yang, Y. Mechanisms of transforming growth factor β1/Smad signalling mediated by mitogen-activated protein kinase pathways in keloid fibroblasts. Br. J. Dermatol. 2010, 162, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Zhang, Q.; Dong, Y. Adiponectin protects the rats liver against chronic intermittent hypoxia induced injury through AMP-activated protein kinase pathway. Sci. Rep. 2016, 6, 34151. [Google Scholar] [CrossRef] [PubMed]
- Su, C.M.; Lee, W.L.; Hsu, C.J. Adiponectin induces oncostatin M expression in osteoblasts through the PI3K/Akt signalling pathway. Int. J. Mol. Sci. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Gan, L.; Qi, R. Adiponectin decreases lipids deposition by p38 MAPK/ATF2 signalling pathway in muscle of broilers. Mol. Biol. Rep. 2013, 40, 7017–7025. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Cao, Y.; He, Y.R. Adiponectin attenuates lung fibroblasts activation and pulmonary fibrosis induced by paraquat. PLoS ONE 2015, 10, e0125169. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.C.; Li, Y.; Ma, Y. YIA 03–02 adiponectin-mediated epithelial autophagy attenuates hypertensive renal fibrosis. J. Hypertens. 2016, 34, e204. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; Sanz-Garcia, C.; Nagy, L.E. Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr. Pathobiol. Rep. 2015, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Lakota, K.; Wei, J.; Carns, M. Levels of adiponectin, a marker for PPAR-gamma activity, correlate with skin fibrosis in systemic sclerosis: Potential utility as biomarker? Arthritis Res. Ther. 2012, 14, R102. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.; Wanninger, J.; Bauer, S. Adiponectin reduces connective tissue growth factor in human hepatocytes which is already induced in non-fibrotic non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2011, 91, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Mao, S.; Wen, H. Upregulation of proinflammatory genes in skin lesions may be the cause of keloid formation (review). Biomed. Rep. 2013, 1, 833–836. [Google Scholar] [PubMed]
- Clark, R.A. Cutaneous tissue repair: Basic biologic considerations. I. J. Am. Acad. Dermatol. 1985, 13, 701–725. [Google Scholar] [CrossRef]
- Esfahani, M.; Movahedian, A.; Baranchi, M. Adiponectin: An adipokine with protective features against metabolic syndrome. Iran. J. Basic Med. Sci. 2015, 18, 430–442. [Google Scholar] [PubMed]
- Pineiro, R.; Iglesias, M.J.; Gallego, R. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett. 2005, 579, 5163–5169. [Google Scholar] [CrossRef] [PubMed]
- Berner, H.S.; Lyngstadaas, S.P.; Spahr, A. Adiponectin and its receptors are expressed in bone-forming cells. Bone 2004, 35, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Katsiougiannis, S.; Kapsogeorgou, E.K.; Manoussakis, M.N. Salivary gland epithelial cells: A new source of the immunoregulatory hormone adiponectin. Arthritis Rheum. 2006, 54, 2295–2299. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Mamaeghani, M.; Mohammadi, S.; Arefhosseini, S.R. Adiponectin as a potential biomarker of vascular disease. Vasc. Health Risk Manag. 2015, 11, 55–70. [Google Scholar] [PubMed]
- Neuman, M.G.; Cohen, L.B.; Nanau, R.M. Biomarkers in nonalcoholic fatty liver disease. Can. J. Gastroenterol. Hepatol. 2014, 28, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Ye, P.; Peng, X. Circulating adiponectin levels in various malignancies: An updated meta-analysis of 107 studies. Oncotarget 2016, 7, 48671–48691. [Google Scholar]
- Witt, E.; Maliri, A.; McGrouther, D.A. RAC activity in keloid disease: Comparative analysis of fibroblasts from margin of keloid to its surrounding normal skin. Eplasty 2008, 8, e19. [Google Scholar] [PubMed]
- Profyris, C.; Tziotzios, C.; Do Vale, I. Cutaneous scarring: Pathophysiology, molecular mechanisms, and scar reduction therapeutics Part I. The molecular basis of scar formation. J. Am. Acad. Dermatol. 2012, 66, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Muragaki, Y.; Ooshima, A. Keloid-derived fibroblasts show increased secretion of factors involved in collagen turnover and depend on matrix metalloproteinase for migration. Br. J. Dermatol. 2005, 153, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.; Diegelmann, R.; Oliver, N. Fibronectin is overproduced by keloid fibroblasts during abnormal wound healing. Mol. Cell Biol. 1989, 9, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Chipev, C.C.; Simman, R.; Hatch, G. Myofibroblast phenotype and apoptosis in keloid and palmar fibroblasts in vitro. Cell Death Differ. 2000, 7, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Smith, T.; Rahman, K. Adiponectin agonist ADP355 attenuates CCl4-induced liver fibrosis in mice. PLoS ONE 2014, 9, e110405. [Google Scholar] [CrossRef] [PubMed]
- Brochu-Gaudreau, K.; Rehfeldt, C.; Blouin, R. Adiponectin action from head to toe. Endocrine 2010, 37, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Fisman, E.Z.; Tenenbaum, A. Adiponectin: A manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc. Diabetol. 2014, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Weiyuan, M. The effect of Metformin on the proliferation and collagen synthesis of human keloids fibroblasts. Zhonghua Zheng Xing Wai Ke Za Zhi 2015, 31, 291–295. [Google Scholar] [PubMed]
- Park, G.; Yoon, B.S.; Moon, J.H. Green tea polyphenol epigallocatechin-3-gallate suppresses collagen production and proliferation in keloid fibroblasts via inhibition of the STAT3-signalling pathway. J. Investig. Dermatol. 2008, 128, 2429–2441. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.J.; Chen, L.; Li, L. Adiponectin inhibits lipopolysaccharide-induced adventitial fibroblast migration and transition to myofibroblasts via AdipoR1-AMPK-iNOS pathway. Mol. Endocrinol. 2010, 24, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, S.C.; Chen, G. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1644–1659. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Tsou, H.K.; Chen, J.C. Adiponectin enhances intercellular adhesion molecule-1 expression and promotes monocyte adhesion in human synovial fibroblasts. PLoS ONE 2014, 9, e92741. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Negative Control | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT |
| AdipoR1 | GGCUAAAGGACAACGACUATT | UAGUCGUUGUCCUUGAGCCTT |
| AdipoR2 | CCUGGCAAAUGUGACAUCUTT | AGAUGUCACAUUUGCCAGGTT |
| T-cadherin | CAGCGAUGGCGGCUUAGUUTT | AACUAAGCCGCCAUCGCUGTT |
| Calreticulin | GGCAUACGCUGAGGAGUUUTT | AAACUCCUCAGCGUAUGCCTT |
| Gene | Forward (5′–3′) | Reverse (5′–3′) | Product Size (bp) |
|---|---|---|---|
| Adiponectin | GGAGAACCTGGAGAAGGTGC | GTACAGCCCAGGAATGTTGC | 171bp |
| AdipoR1 | AATTCCTGAGCGCTTCTTTCCT | CATAGAAGTGGACAAAGGCTGC | 101bp |
| AdipoR2 | TGCAGCCATTATAGTCTCCCAG | GAATGATTCCACTCAGGCCTAG | 101bp |
| T-cadherin | GACAAGCCATCTCCCAACAT | CAACATCCAGTCCAGCCATA | 151bp |
| Calreticulin | GGCAUACGCUGAGGAGUUUTT | AAACUCCUCAGCGUAUGCCTT | 150bp |
| Collagen I | AGCCAGCAGATCGAGAACAT | TCCTTGGGGTTCTTGCTGAT | 245bp |
| FN | GCCAGATGATGAGCTGCAC | GAGCAAATGGCACCGAGATA | 142bp |
| α-SMA | CAGGGCTGTTTTCCCATCCAT | GCCATGTTCTATCGGGTACTTC | 142bp |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC | 226bp |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Li, J.; Liu, H.; Jian, X.; Zou, Q.; Zhao, Q.; Le, Q.; Chen, H.; Gao, X.; He, C. Adiponectin Is Involved in Connective Tissue Growth Factor-Induced Proliferation, Migration and Overproduction of the Extracellular Matrix in Keloid Fibroblasts. Int. J. Mol. Sci. 2017, 18, 1044. https://doi.org/10.3390/ijms18051044
Luo L, Li J, Liu H, Jian X, Zou Q, Zhao Q, Le Q, Chen H, Gao X, He C. Adiponectin Is Involved in Connective Tissue Growth Factor-Induced Proliferation, Migration and Overproduction of the Extracellular Matrix in Keloid Fibroblasts. International Journal of Molecular Sciences. 2017; 18(5):1044. https://doi.org/10.3390/ijms18051044
Chicago/Turabian StyleLuo, Limin, Jun Li, Han Liu, Xiaoqing Jian, Qianlei Zou, Qing Zhao, Qu Le, Hongdou Chen, Xinghua Gao, and Chundi He. 2017. "Adiponectin Is Involved in Connective Tissue Growth Factor-Induced Proliferation, Migration and Overproduction of the Extracellular Matrix in Keloid Fibroblasts" International Journal of Molecular Sciences 18, no. 5: 1044. https://doi.org/10.3390/ijms18051044