
Regulation of G Protein-Coupled Receptors by Ubiquitination

1
Laboratory of GPCR Expression and Signal Transduction (L-GEST), Ghent University, Proeftuinstraat 86, 9000 Ghent, Belgium
2
Center for Medical Genetics Ghent; Ghent University, De Pintelaan 185, 9000 Ghent, Belgium
3
Cancer Research Institute Ghent (CRIG), Ghent University Hospital, Medical Research Building 2, De Pintelaan 185, 9000 Ghent, Belgium
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(5), 923; https://doi.org/10.3390/ijms18050923
Submission received: 6 February 2017
/
Revised: 20 April 2017
/
Accepted: 23 April 2017
/
Published: 27 April 2017
(This article belongs to the Collection G Protein-Coupled Receptor Signaling and Regulation)
Abstract
:G protein-coupled receptors (GPCRs) comprise the largest family of membrane receptors that control many cellular processes and consequently often serve as drug targets. These receptors undergo a strict regulation by mechanisms such as internalization and desensitization, which are strongly influenced by posttranslational modifications. Ubiquitination is a posttranslational modification with a broad range of functions that is currently gaining increased appreciation as a regulator of GPCR activity. The role of ubiquitination in directing GPCRs for lysosomal degradation has already been well-established. Furthermore, this modification can also play a role in targeting membrane and endoplasmic reticulum-associated receptors to the proteasome. Most recently, ubiquitination was also shown to be involved in GPCR signaling. In this review, we present current knowledge on the molecular basis of GPCR regulation by ubiquitination, and highlight the importance of E3 ubiquitin ligases, deubiquitinating enzymes and β-arrestins. Finally, we discuss classical and newly-discovered functions of ubiquitination in controlling GPCR activity.
1. Introduction
1.1. GPCR Signaling
G protein-coupled receptors (GPCRs) represent the largest family of membrane proteins that transduce signals from various stimuli, including photons, pheromones, hormones and neurotransmitters. These receptors regulate numerous physiological processes and are the major pharmacological targets in the treatment of many pathological conditions including neurological and cardiovascular disorders, pain, cancer, endocrine and pulmonary diseases [1,2]. Upon activation by their ligands, GPCRs typically signal via heterotrimeric G proteins. However, GPCRs can also signal independently of G proteins, in this situation signaling is mainly mediated by β-arrestins [3,4]. β-arrestins belong to the arrestin protein family which consists of four members: arrestin 1 (visual arrestin), arrestin 2 (β-arrestin 1), arrestin 3 (β-arrestin 2), and arrestin 4 (cone arrestin). The visual and cone arrestin are localized to retina rods and cones and interact mainly with rhodopsin, while the two β-arrestin isoforms are ubiquitously expressed. All four arrestins share high sequence and structural homology [5,6,7]. β-arrestins were first characterized as key regulators of GPCR desensitization and internalization [8,9]; however, it is now known that β-arrestins can regulate GPCR action by functioning as scaffolds for signaling molecules [4,10,11,12,13]. The phenomenon in which specific ligands preferentially activate G protein-or β-arrestin-mediated signaling upon binding to the same receptor is called biased agonism [14].
1.2. Ubiquitination
Ubiquitin is a 76-amino acid polypeptide that is covalently attached to lysine residues in substrate proteins. The attachment of ubiquitin to substrate proteins is processed by a sequential action of three types of enzymes: ubiquitin activating enzymes (E1), ubiquitin conjugating enzymes (E2), and ubiquitin ligases (E3). First, in an adenosine triphosphate (ATP)-dependent process, ubiquitin is activated by E1. In this step, a thiol-ester linkage is formed between the C-terminal glycine of ubiquitin and a cysteine of E1 at the active site. Next, the activated ubiquitin is transferred to a cysteine residue at the active site of E2. Finally, the E3 ligase directly or indirectly catalyzes the covalent attachment of ubiquitin to the target protein. The human genome encodes two E1s, around 60 E2s, and more than 600 E3 ubiquitin ligases; therefore, it is not surprising that E3s are responsible for providing substrate specificity in the ubiquitination process. E3 ligases are mainly categorized into two families: homologous to E6AP C terminus (HECT), and really interesting new gene (RING), based on the structure of the catalytic domains [19]. In mammals, around 30 HECT domain E3s and around 600 RING-type ligases are expressed [19,20]. The HECT ligases possess catalytic activity, they accept ubiquitin from the E2 enzyme and transfer it to the specific residue in the substrate protein [21]; while RING ligases function by bringing E2 enzymes and substrate proteins in close proximity to each other.
Ubiquitin typically binds to the ε-amino group of a lysine residue; however, recent evidence has shown that ubiquitin can also be attached to other residues, including cysteines, serines, threonines, and the N-terminus of the polypeptide backbone [22,23,24,25,26,27,28].
Ubiquitin can be attached to single or multiple residues in the substrate protein representing monoubiquitination and multimonoubiquitination, respectively. Ubiquitin itself contains seven lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, Lys63) and they can all potentially be ubiquitinated. Attachment of an additional ubiquitin to a previously substrate-bound ubiquitin molecule leads to polyubiquitin chains of different configurations. Different types of ubiquitination are often associated with different functions. Ubiquitination is a reversible modification and ubiquitin moieties can be removed from the substrate protein by a family of deubiquitinating enzymes (DUBs). The human genome encodes almost 100 DUBs classified into five families: the ubiquitin carboxy-terminal hydrolases, ubiquitin–specific proteases (USPs), ovarian tumor-related proteases, Machado-Joseph disease protein domain proteases, and jab1/MPN domain-associated metalloisopeptidases (JAMM). All families are cysteine proteases, except JAMM, which are a family of metalloproteases [29]. USPs represent the largest family of DUBs (58 members) and most of the described DUBs involved in the regulation of GPCR ubiquitination belong to this family [29,30,31]. We have compiled an overview of all published studies on GPCR ubiquitination (including E3 ubiquitin ligases), DUBs, receptor residues which are ubiquitinated, the role of the ubiquitination, and whether the modification is induced by receptor stimulation (Table 1). In this review, we elaborate on some crucial reports and discuss the role of ubiquitination in the regulation of GPCR function.
2. Functional Role of GPCR Ubiquitination
Although an increasing number of in-depth studies about ubiquitination of GPCRs are becoming available, our understanding of the function of this modification in the regulation of these receptors is still very limited. The best characterized role of ubiquitination is the targeting of activated GPCRs for lysosomal degradation [87,88,89]. Additionally, ubiquitin has been shown to serve as a tag in the quality control system, the endoplasmic reticulum-associated degradation (ERAD) pathway, and to direct misfolded proteins for proteasomal degradation. Ubiquitination also regulates cell surface expression of GPCRs via different mechanisms and in this way controls cell responsiveness to different stimuli (Figure 1). Recently, the role of ubiquitin in the regulation of GPCR signaling and its involvement in differential cellular responses upon GPCR activation with biased agonists has been demonstrated. These well-defined and novel roles of ubiquitin in GPCR regulation will all be discussed in this review.
2.1. Regulation of GPCR Cell-Surface Expression by Ubiquitination
In addition to the transcription, translation, and targeting of the receptor to the plasma membrane, down-regulation plays an important role in the regulation of GPCR expression. There are two major pathways involved in the degradation of GPCRs: the ubiquitin proteasome system (UPS), and the endosomal-sorting complex required for the transport (ESCRT) pathway leading to lysosomal degradation. Additionally, deubiquitinating enzymes and β-arrestins play an important role in the regulation of GPCR trafficking. In this chapter, our focus is on single mechanistic studies, but the entire picture is much more dynamic and diverse. Furthermore, very limited studies have focused on the kinetics of these processes, that is the kinetics of ubiquitination, deubiquitination, and the interaction of proteins involved in the regulation of E3 ligases, DUBs, and proteins important for receptor internalization, etc. Extensive knowledge about these processes is of paramount importance for full understanding of the regulation of GPCR cell-surface expression.
2.1.1. Proteasomal Degradation
The initial discovery of the role of ubiquitin was to serve as a tag to mark misfolded or unneeded proteins for degradation in the proteasome. Ubiquitination of GPCRs was shown to be involved in the ERAD, which mainly serves as a quality control system where misfolded receptors are ubiquitinated, and targeted for degradation in the proteasome [90] (Figure 1, pathway 5). During biogenesis, GPCRs and other transmembrane proteins are folded in the endoplasmic reticulum (ER) and adopt specific conformations. This process is assisted by resident molecular chaperons and folding factors. Next, proteins are released from the ER and are transported through the secretory pathway to the Golgi, and upon complete maturation move to their final destination. Misfolded proteins are retrotranslocated from the ER to the cytosol and upon ubiquitination are further directed to the proteasome for proteolytic degradation [64,91]. The 26S proteasome is a protein complex consisting of a 20S protein subunit with a proteolytic function and two 19S regulatory cap subunits. Proteins that are tagged by polyubiquitin chains consisting of at least four ubiquitin moieties are recognized by 19S regulatory subunits. Ubiquitin is then cleaved and recycled while targeted proteins are degraded in the 20S subunit. Lys48-type polyubiquitin chains serve most frequently as signals for proteasomal degradation, but other linkages such as Lys63-type, Lys11-type, linear and non-lysine ubiquitination chains can also be recognized by the proteasome [92,93,94,95].
In this way, only properly folded receptors are delivered to the membrane. The importance of ERAD as a quality control system during biogenesis was shown for the calcium-sensing receptor [83], thyrotropin-releasing hormone receptor [79], μ-opioid receptor (MOR), δ-opioid receptor (DOR) [64,68], and dopamine D4 receptor (D4R) [96].
However, there have been examples of properly folded GPCRs that are deubiquitinated during biosynthesis and transported to the cell surface, for example, the adenosine receptor A2A [32]. Furthermore, several GPCRs have been described to undergo basal and agonist-induced proteasomal degradation (Figure 1, pathway 6). Metabotropic glutamate receptors 1 and 5 (mGluR1 and mGluR5) and the human follitropin receptor are directed to the proteasome, which does not depend on agonist stimulation [54,85]. Additionally, proteasomal degradation has been shown to play an important role in the regulation of different opioid receptors. MOR and DOR agonist-induced down-regulation were attenuated upon pretreatment of the cells with proteasome inhibitors (Z-Leu-Leu-Leu-al, ZLLL; lactacystin; Z-Ile-Glu(OtBu), PSI and Ac-Leu-Leu-nLeu-al, ALLN), while pretreatment with a lysosome inhibitor (E64d) had little effect. In addition, incubation with all tested proteasome inhibitors in the absence of an agonist increased the steady state MOR and DOR levels, indicating that proteasomal degradation also has an important role in the basal turn-over of both receptors [68]. The κ-opioid receptor (KOR), ubiquitinated after phosphorylation and Lys63-type polyubiquitination, was shown as a dominant form of KOR ubiquitination. Agonist-induced ubiquitination of KOR leads to its down-regulation, but surprisingly, the lysine-deficient mutant with a clear decrease in ubiquitination was also down-regulated, suggesting that more than one mechanism may control receptor degradation [66]. Additionally, a combination of both proteasome and lysosome inhibitors was required to completely block KOR degradation, suggesting that both pathways are involved in agonist-promoted down-regulation of the receptor [65]. Although the effect of proteasome inhibitors was observed in the above-mentioned examples, it is not clear how mature receptors can be extracted from the cell membrane and directed to the proteasome. It is possible that the proteasome indirectly influences receptor degradation, that is via proteins involved in the degradative pathway. Such a scenario has been previously proposed by Hicke [97].
2.1.2. Lysosomal Degradation
GPCRs that recognize and integrate signals from various stimuli need to undergo strict regulation to prevent cell overstimulation. One of the main regulatory mechanisms is receptor desensitization, followed by receptor internalization, and subsequent degradation of the receptor in the lysosomes (Figure 1, pathway 1).
The involvement of ubiquitin in the regulation of GPCR trafficking was first demonstrated in 1996 in yeast, and showed that ubiquitination was necessary for the internalization and degradation of the sterile 2 α-factor receptor protein (Ste2p) [98,99]. In mammals, however, ubiquitination is not required for receptor internalization, but seems to regulate, via different mechanisms, lysosomal degradation of the activated receptor via the highly conserved endosomal-sorting complex required for transport (ESCRT) pathway [100,101]. The ESCRT machinery consists of four distinct protein complexes known as ESCRT-0, -I, -II and -III. ESCRT complexes (together with numerous accessory proteins) act sequentially to sort ubiquitinated cargo from early endosomes into intraluminal vesicles (ILVs) of multivesicular bodies (MVBs) before degradation in the lysosome [102].
Ubiquitin-dependent sorting of GPCRs via ESCRT to lysosomes has been shown for many GPCRs (Table 1). The C-X-C chemokine receptor-4 (CXCR4) and proteinase-activated receptor 2 (PAR2) are ubiquitinated and sorted for lysosomal degradation upon agonist stimulation. Alterations in proper ubiquitination of these receptors prevent their lysosomal sorting and degradation. Moreover, disruption of ESCRT components also blocked CXCR4 and PAR2 sorting to lysosomes, confirming the importance of this pathway in receptor degradation [46,47,77,103,104]. Other examples of GPCRs where ubiquitination has been shown to play a role in lysosomal sorting after prolonged agonist treatment are the neurokinin-1 receptor (NK1R) and MOR. When ubiquitination is impaired upon internalization, NK1R is recycled back into the plasma membrane [78]. MOR ubiquitination is necessary for its ESCRT-dependent down-regulation and controls receptor distribution between the limiting endosome membrane and lumen, but is not essential for receptor delivery to the proteolytic compartments. Instead, this is dictated by the MOR C-terminal tail and is independent of receptor ubiquitination [69].
The prototypic β2-adrenergic receptor (β2AR) was shown to be sorted to late endosomes/lysosomes upon agonist (isoproterenol) stimulation, while the mutant receptor (lacking lysines (β2AR-K0)) was not ubiquitinated and showed resistance to lysosomal degradation [34,37]. Treatment of the β2AR with β-arrestin biased agonist (carvedilol) also resulted in ubiquitination and lysosomal sorting of the receptor. This appears to be a distinct type of ubiquitination, rather than the one induced by a balanced agonist in which another E3 ligase is involved and where ubiquitin is probably attached to other non-lysine residues [38]. Upon balanced agonist (isoproterenol) treatment, β2AR is ubiquitinated by the HECT-type E3 ligase neural precursor cell-expressed developmentally downregulated gene 4 (Nedd4) [36]; while upon β-arrestin biased agonist treatment (carvedilol), the RING-type ubiquitin ligase membrane associated ring-CH-type finger 2 (MARCH2) ubiquitinates the wild type receptor as well as the lysine-lacking mutant [38].
Although ubiquitination plays an important role in GPCR lysosomal degradation, there are examples of receptors which can efficiently sort to lysosomes in a ubiquitin-independent manner. The calcitonin-like receptor (which is not ubiquitinated upon activation) and a lysine-deficient mutant of DOR (of which ubiquitination is completely blocked) are still degraded in the lysosomes. These receptors require an ESCRT-0 complex component for their lysosome sorting, confirming that ubiquitination is not obligatory for GPCRs to enter the ESCRT pathway [60,62,103,105]. In contrast, proteinase-activated receptor 1 (PAR1) undergoes agonist-induced ubiquitination and is directed to lysosomes in the next step. However, its degradation is independent of receptor ubiquitination and ubiquitin-binding components of ESCRT-0 and ESCRT-I complexes (hepatocyte growth factor-regulated tyrosine kinase substrate—HRS and Tsg101) [75,106,107]. A later study revealed that ALG-2-interacting protein X (ALIX), an ESCRT-III interacting protein, interacts with the YPX3L motif in the second intracellular loop of PAR1 and purinergic P2Y1 receptor, and is necessary for receptor lysosomal sorting. This conserved motif was also found in the second intracellular loop of several other GPCRs, suggesting that regulation of lysosomal sorting by ALIX could be a common mechanism within the GPCR family [61,74,108,109]. Most recently, it was shown that ALIX is regulated by the arrestin domain-containing protein 3 (ARRDC3), and this regulation is necessary for ALIX to sort PAR1 to the lysosomes. Upon PAR1 activation, ARRDC3 recruits HECT-type E3 ubiquitin ligase WW domain-containing protein 2 (WWP2) which ubiquitinates ALIX. In this way, ARRDC3 facilitates the sorting of PAR1 into ILVs of MVBs [110]. The mechanisms that control ALIX-dependent GPCR sorting at the multivesicular endosomes remain poorly understood; however, it is speculated that ubiquitin may regulate ALIX by facilitating its dimerization, which in turn can promote its interaction with the ESCRT-III complex [110].
This finding demonstrates that direct ubiquitination of the GPCR might not be obligatory for its degradation, but that ubiquitin may still play an important regulatory role in GPCR lysosomal sorting by modifying other proteins involved in this process.
2.1.3. Deubiquitination and GPCR Cell-Surface Expression
Ubiquitination can direct receptors for degradation while also regulating the cell-surface expression of GPCRs. The latter involves the dynamic actions of ubiquitin ligases and DUBs, and in this way modulates cell responsiveness [111].
GPCRs that undergo agonist-induced ubiquitination are usually internalized and targeted for degradation in lysosomes. However, after deubiquitination they can often be redirected to the resensitization pathway and recycled back to the cell surface (Figure 1, pathway 2). Upon stimulation with the agonist isoproterenol, β2AR is internalized and undergoes lysosomal degradation, but its deubiquitination by USP33 and USP20 on late endosomes promotes receptor recycling to the plasma membrane [33]. These USPs are coupled to the receptor in a non-active state and upon agonist stimulation are transferred to β-arrestin 2. This leads to deubiquitination of β-arrestin 2 and its dissociation from the receptor upon internalization [33]. At the late endosomes, USPs can reassociate with the receptor and allow its recycling to the cell surface. It has also been shown that the presence of only one USP is sufficient for receptor recycling. Most recently, it was described that upon physiological stress, USP20 is phosphorylated by protein kinase A and this process regulates trafficking of β2AR to autophagosomes [112].
Ubiquitination of CXCR4 by HECT-type E3 ligase atrophin-1-interacting protein 4 (AIP4) induced by C-X-C motif chemokine ligand 12 (CXCL12) leads to receptor lysosomal degradation [113]. Overexpression of USP14 promotes CXCR4 deubiquitination and allows it to escape degradation [42,48]. Another interesting example represents frizzled-4 receptor (FZD4R) where a balance between ubiquitination and deubiquitination is important for regulating its membrane expression. Constitutive ubiquitination of FZD4R promotes its internalization and lysosomal degradation, while deubiquitination mediated by USP8 leads to recycling and increased cell surface expression [86]. USP8 has also been shown together with another DUB, associated molecule with the SH3-domain of STAM (AMSH), to regulate PAR2 trafficking. These two DUBs are associated with the ESCRT pathway. USP8 and AMSH mediate deubiquitination of PAR2 and its sorting from endosomes to lysosomes [76]. AMSH and USP8 may also regulate trafficking of DOR, but it is unclear whether they do so through direct deubiquitination of DOR, or via regulation of the ESCRT machinery [61]. Furthermore, the importance of these two DUBs has also been shown in the regulation of CXCR4 ubiquitination; however, in this case, they do not work through direct deubiquitination of the receptor. USP8 regulates CXCR4 lysosomal degradation, but does so through the deubiquitination of the ESCRT machinery and not CXCR4 [41]. AMSH was shown to not play a role in the agonist-induced lysosomal degradation of CXCR4, in contrast to its role in PAR2 regulation [44,114]. Additionally, deubiquitination and lysosomal degradation of CXCR4 is regulated directly by USP14 [48]. Overexpression of this DUB has been shown to prevent lysosomal degradation of CXCR4 [42]. These examples highlight that the ubiquitination of a single receptor can be regulated by several DUBs each working with a different mechanism, and that the same DUB can change its mechanism of action depending on the receptor which undergoes regulation.
Additionally, there are GPCRs which require deubiquitination to enter the internalization pathway (Figure 1, pathway 3). The C-X-C chemokine receptor-7 (CXCR7) is ubiquitinated in a basal state on a lysine residue located in the C-terminus. Receptor activation with CXCL12 leads to its reversible deubiquitination in a process that requires phosphorylation and β-arrestin recruitment [49]. When the ligand is removed, the receptor is ubiquitinated again and recycled back to the plasma membrane.
Like CXCR7, PAR1 was described as constitutively ubiquitinated. Agonist stimulation of PAR1 induces its deubiquitination [75]; however, after its internalization, ubiquitination of PAR1 increases once again [74]. Lysine-less PAR1 mutants displayed constitutive endocytosis, suggesting that ubiquitination is necessary for membrane expression of the receptor and negatively regulates its constitutive internalization [75].
In contrast to these observations, the deubiquitination of the adenosine A2A receptor (A2AR) by USP4 is necessary for the cell surface delivery of a functionally active receptor (Figure 1, pathway 4) [32].
The parathyroid hormone 1 receptor (PTH1R) regulates bone growth and extracellular mineral ion homeostasis and represents another interesting example of a GPCR regulated by integrated ubiquitination and deubiquitination mechanisms. This receptor responds to distinct ligands, and stimulation with both the activating ligand PTH (1–34) and the non-activating ligand PTH (7–34) leads to polyubiquitination of PTH1R. The enzymes involved in PTH1R ubiquitination are still unknown, although an enzyme (USP2) involved in deubiquitination of the receptor has been described. Upregulation of USP2 mRNA upon treatment with the activating ligands has been shown in bone [115], and was also detected in rat osteosarcoma cells [82]. The different regulation of USP2 can explain why ubiquitination induced by the activating ligand PTH (1–34) is transient and the non-activating ligand PTH (7–34) is sustained. This difference in receptor ubiquitination has important implications as PTH1R undergoes fast deubiquitination and recycles to the cell membrane upon treatment with the activating ligand PTH (1–34). In contrast, the non-activating ligand PTH (7–34) induces sustained ubiquitination of PTH1R which leads to its proteasomal degradation [82].
2.1.4. β-Arrestins, Ubiquitination and GPCR Trafficking
Our knowledge on the role of β-arrestins in GPCR regulation has increased rapidly. From regulation of GPCR internalization and desensitization, to mediating G protein-independent signaling, β-arrestins appear to be crucial components of the complicated protein network involved in controlling proper GPCR function. β-arrestins function as adaptors for E3 ubiquitin ligases [116,117] or deubiquitinating enzymes [41,118] and as such they play an important role in the regulation of GPCR ubiquitination. However, at times, their mechanism of action is not well defined. The first research on the importance of β-arrestins in the regulation of GPCRs by ubiquitination came from the studies of Shenoy et al. [34] who demonstrated that agonist-induced activation of the β2AR led to the transient ubiquitination of the receptor and of β-arrestin 2. Next, they showed that the mouse double minute 2 homolog (MDM2) ligase was important for the ubiquitination of β-arrestin 2, but that MDM2 depletion had no effect on receptor ubiquitination. Later, it was revealed that another E3 ligase, Nedd4, was responsible for the ubiquitination of β2AR, and that β-arrestin 2 functioned as an adaptor to recruit Nedd4 to activated β2AR [36]. A recent study showed that Nedd4 may be recruited to β2AR independently of β-arrestin 2 through a mechanism mediated by ARRDC3, a member of the α-arrestin protein family [119]. Nabhan et al. [119] showed that ARRDC3 interacted with Nedd4 and β2AR and served as an adaptor for ubiquitination of the receptor mediated by Nedd4. Knockdown of ARRDC3 abolished agonist-induced ubiquitination and lysosomal degradation of β2AR. However, another group [120] recently presented a report that β-arrestin 2, and not ARRDC3, was the adaptor protein necessary for the ubiquitination of β2AR. They also concluded that β-arrestin 2 and ARRDC3 functioned sequentially. Upon β2AR activation, β-arrestin 2 mediated the ubiquitination of the receptor by recruiting Nedd4 and promoted receptor endocytosis. ARRDC3 (as well as other ARRDC proteins (2 and 4)) functioned as secondary adaptors recruited to internalized β2AR-Nedd4 complexes on endosomes [120]. β-arrestin 1 was shown to bind and co-localize with HECT-type AIP4 ubiquitin ligases on early endosomes upon CXCR4 activation [42]. Depletion of β-arrestin 1 blocked agonist promoted degradation of CXCR4 by preventing its trafficking from early endosomes to lysosomes. Interestingly, ubiquitination and internalization of CXCR4 were not affected, suggesting that the interaction between β-arrestin 1 and AIP4 was not required for ubiquitination of the receptor at the plasma membrane, but was probably important for subsequent post-internalization events. One possible explanation is the adaptor function of β-arrestin 1 for AIP4-mediated ubiquitination of ESCRT-0 components. β-arrestin 1 was shown to interact with signal transducing adapter molecule 1 (STAM-1) (a subunit of ESCRT-0) [44]. Disruption of this interaction reduced ubiquitination of the second ESCRT-0 subunit, HRS, which in turn enhanced CXCR4 degradation. Involvement of β-arrestins in the regulation of ubiquitination of other GPCRs (such as the MOR and vasopressin V2 receptor (V2R)) was also demonstrated, but the precise role of β-arrestins in these processes was unclear. Using β-arrestin1/2 knockout mouse embryonic fibroblast (MEF) cells, Groer et al. [67] showed that β-arrestin1 was crucial for [d-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) mediated ubiquitination while morphine (the other agonist) could not induce MOR ubiquitination, nor when G protein-coupled receptor kinase 2 (GRK2) was overexpressed. As the scaffolding machinery has not been investigated, we do not know which E3 ligase is involved. However, another study [59] showed that stimulation of MOR with the non-selective agonist [D-Ala2, D-Leu5]-Enkephalin (DADLE) induced ubiquitination of MOR by the HECT-type E3 ligase Smurf2.
In case of V2R, β-arrestin 2 was shown to be the critical component in the rapid ubiquitination and degradation of the receptor upon agonist stimulation [80].
Although β-arrestins are mainly adaptors for E3 ubiquitin ligases, one study [49] showed that constitutively ubiquitinated CXCR7, upon stimulation, underwent deubiquitination in a process that required phosphorylation and β-arrestin recruitment. The authors proposed a model in which β-arrestins may function as scaffolds for deubiquitinating enzymes, but the exact function of these proteins in the regulation of CXCR7 ubiquitination still needs to be investigated.
All the studies presented clearly show that β-arrestins are very important regulators of GPCR ubiquitination, although their precise role in this process is still poorly understood.
2.2. Importance of Ubiquitination in GPCR Signaling and Biased Agonism
2.2.1. GPCR Signaling
Although most studies have highlighted the role of ubiquitination as a mechanism controlling GPCR down-regulation, there is also evidence that ubiquitin influences GPCR signaling. Many GPCRs signal via activation of the mitogen-activated protein kinase (MAPK) pathway, also known as the extracellular-signal regulated kinase (ERK) pathway. The MAPK pathway is a chain of proteins in a cell that transfer the signal from the receptor on the cell surface to the DNA in the nucleus, or to other subcellular targets causing cellular responses including proliferation, gene expression, differentiation, mitosis, cell survival, and apoptosis. Signaling molecules in this pathway communicate between each other by adding a phosphate group to the neighboring proteins. This phosphorylation event functions as an “on/off” signal, leading to the activation or inhibition of the next signaling molecule in the chain [121]. The most commonly studied MAPK pathway initiated by GPCR activation is the p44/42 MAPK (ERK1/2) pathway. This pathway can be activated by G protein-dependent and -independent mechanisms [122,123,124]. The G protein-independent MAPK signaling often requires β-arrestins as scaffolds for the signaling complexes [10,125,126]. We can assume that the scaffold function of β-arrestins is related to their ubiquitination status and their ability to co-internalize with certain GPCRs. The ubiquitination of β-arrestin 2 is controlled by the ubiquitin ligase Mdm2 and the deubiquitinating enzyme USP33, and is regulated by the activation of GPCRs, e.g., β2AR [35]. Based on the affinity of β-arrestin and trafficking patterns, GPCRs are divided into two classes. Receptors from class A (e.g., β2AR) form a transient complex with β-arrestin 2 and internalize without them, which promotes fast recycling. In contrast, receptors from class B (e.g., V2R) form more stable complexes with β-arrestin, internalize into endosomes together with β-arrestin 2 and are slowly recycled. Stimulation of the β2AR and V2R leads to transient or stable β-arrestin ubiquitination, respectively. Thus, ubiquitinated β-arrestin internalizes (together with the receptor) towards endosomes with activated MAPK. Such a situation was demonstrated for AT1aR [127,128]. The precise mechanism demonstrating how ubiquitination of β-arrestin 2 favors sustained p44/42 MAPK activation is unclear. It is possible that β-arrestin 2-bound ubiquitin allows a more stable interaction between β-arrestin 2 and the receptor which promotes their co-internalization and sustained MAPK activation on endosomes. Furthermore, ubiquitinated β-arrestin 2 may work as an adaptor for ubiquitin binding domain (UBD) proteins which mediate sustained MAPK signaling [129].
Another study indicated that ubiquitination may play a role in the regulation of G protein-dependent signaling. CXCR4-mediated p44/42 MAPK phosphorylation is G protein-dependent and does not require β-arrestin. Malik et al. [45] showed that two proteins crucial for lysosomal sorting of CXCR4 (the ubiquitin E3 ligase AIP4 and the ESCRT-0 subunit STAM-1) were also involved in the regulation of CXCR4-mediated p44/42 MAPK activation. Depletion of either AIP4 or STAM-1, as well as inhibition of the interaction between these two proteins, blocked phosphorylation of p44/42 MAPK upon CXCR4 activation. Moreover, it was observed that there was a discrete subpopulation of AIP4 and STAM-1 in caveolar microdomains with CXCR4 which appeared to mediate MAPK activation. To explain this observation, the authors proposed a mechanism in which ubiquitination of STAM-1 by AIP4 in caveolae regulated the activation of p44/42 MAPK signaling initiated by CXCR4, but further research is required for clarification [45]. Interestingly, extracellular ubiquitin can bind to the CXCR4 receptor and functions as a natural agonist of this receptor. Binding of the extracellular ubiquitin promotes intracellular Ca2+ flux and reduces cAMP levels via the Gαi/o protein [130]. These studies present a novel idea on the importance of ubiquitin in the regulation of GPCR signaling. Ubiquitin does not only influence receptor signaling by modifying the receptor itself or various receptor interacting proteins, but can also serve as a direct activator of the receptor on the extracellular site of the cell.
Recently, signaling via another chemokine receptor, CXCR2, was shown to be strictly dependent on the ubiquitination status of the receptor. A single lysine residue (Lys327) in the C-terminal tail of CXCR2 was identified as a site of ubiquitination. Moreover, this ubiquitination seemed to play a key role in receptor internalization and signaling. CXCR2, where Lys327 was mutated to arginine, displayed decreased polyubiquitination, failed to recruit β-arrestin, and did not internalize upon stimulation of the receptor. Furthermore, the intracellular signaling—including both early events, such as MAPK phosphorylation and the increase in calcium flux, and the later activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1 (AP1) transcription factors—was blunted. The precise mechanism of how the ubiquitination of a single lysine can regulate so many aspects of CXCR2 signaling still needs to be elucidated [40]. In addition, a study where all lysines in the C-terminal tail of the CXCR2 were mutated [131] showed that these residues were not crucial for CXCR2 degradation in the lysosomes.
Ubiquitination is also important for p38 MAPK activation. It was revealed that activation of PAR1 with α-thrombin led to its Lys63-type polyubiquitination mediated by the Nedd4-2 E3 ubiquitin ligase [71]. Ubiquitinated PAR1 recruits the transforming growth factor-β-activated protein kinase-1 binding protein-2 (TAB2) which binds to the receptor via its ubiquitin binding motif and functions as an adaptor for TAB1 protein, resulting in TAB2-TAB1-p38 signaling complex formation on endosomes. The phosphorylation of p38 occurred in a non-canonical way as the two kinases typically necessary for this process, mitogen-activated protein kinase kinase 3 (MKK3) and MKK6, were not obligatory in the case of PAR1 initiated p38 phosphorylation. Ubiquitin and TAB-dependent activation of p38 was required for thrombin-induced endothelial barrier permeability in vitro, and signaling by p38 MAPK was essential for PAR1-stimulated vascular leakage in vivo. Interestingly, ubiquitination of the purinergic GPCR P2Y was also required for the TAB-dependent activation of p38 MAPK upon receptor activation [71]. This finding suggests that non-canonical p38 signaling is not limited to PAR1 and can be a more general phenomenon with an important role in GPCR ubiquitination.
2.2.2. Biased Agonism
Biased agonism is a phenomenon where different ligands elicit distinct responses at the same receptor [14]. Understanding the molecular basis of biased agonism is of great importance in pharmacology as it will allow the development of drugs which produce beneficial responses with limited side effects [3,132,133,134]. Recent studies have demonstrated that ubiquitination is important in determining differential GPCR regulation induced by biased agonists. Here we provide examples that show that various ligands induce distinct ubiquitination patterns of the same receptor.
MOR, a major target for opiate drugs, is a receptor that displays various responses when activated with different ligands [135]. Morphine and DAMGO (two MOR-specific agonists) cause a difference in the recruitment of β-arrestins (1/2) and in MOR ubiquitination. Activation of MOR with DAMGO leads to MOR internalization mediated by β-arrestin 1 and β-arrestin 2. In contrast, morphine induces only β-arrestin 2 recruitment, followed by receptor internalization [67]. Next, there was a huge difference in the ubiquitination of MOR upon incubation with morphine or DAMGO (as DAMGO induces MOR ubiquitination), while morphine treatment had no effect on the ubiquitination of the receptor. Further experiments proved that β-arrestin 1 (and not β-arrestin 2) was important in MOR ubiquitination. Another study [62] showed that stimulation of MOR with the non-selective agonist DADLE also induced ubiquitination of the receptor, which controlled its internalization as ubiquitin-deficient MOR did not internalize through clathrin-coated pits as efficiently as wild-type receptors.
Another interesting example (previously mentioned in Section 2.1.2) is β2AR, which can be ubiquitinated by different E3 ligases upon treatment with different ligands. Upon activation of β2AR with its balanced agonist isoproterenol, the receptor undergoes rapid ubiquitination by the HECT-type ubiquitin ligase Nedd4, followed by lysosomal degradation [36]. The β-arrestin biased agonist carvedilol stimulates β-arrestin2-dependent signaling, but is also an antagonist for G protein-dependent signalling [136]. Stimulation of β2AR with carvedilol also leads to ubiquitination and lysosomal degradation, but ubiquitination is now added to the receptor via the RING-type E3 ligase MARCH2 [38].
All examples presented above show that distinct ligands can differentially modulate the activity of the ubiquitin machinery that plays a role in the regulation of biased signaling.
2.3. Transubiquitination
GPCRs are known to transactivate other receptors such as growth factor tyrosine kinase receptors via release of membrane-anchored ligands, or through the modulation of receptor cytoplasmic domains [137]. Recent studies have revealed that some receptors and antagonists can stimulate GPCR transubiquitination and degradation. The orexin receptor (OX2), involved in the regulation of the sleep/wake cycle, is ubiquitinated and degraded in response to signaling by the proinflammatory cytokine tumor necrosis factor α (TNF-α) [70]. An example of an antagonist which promotes ubiquitination of a GPCR is FTY720. This inhibitor of sphingosine-1-phosphate receptor (S1PR) signaling induces phosphorylation of the C-terminal domain of the receptor followed by receptor internalization, polyubiquitination, and proteasomal degradation, and finally results in a functional antagonism of S1P signaling [56,57].
Moreover, certain GPCRs promote transubiquitination of other GPCRs which can dramatically change cell responsiveness. This phenomenon was first described for the angiotensin-II type 1 receptor (AT1R) in response to dopamine D5 receptor (D5R) activation. Both receptors have opposite influences on cellular signal transduction, where signaling of AT1R is prohypertensive while D5R signaling is antihypertensive. Li et al. [39] demonstrated that activation of D5R promoted ubiquitination and proteasomal degradation of AT1R, but it was unclear whether the two receptors interacted directly with each other, or if it was an indirect effect mediated by downstream signaling molecules (Figure 2(1)).
Accumulating evidence suggests that GPCRs can also form functional homo- and heteromers as well as higher order oligomers which exhibit different signaling properties when compared to monomers [138,139,140,141,142,143,144,145,146,147]. DOR and MOR were shown to form heteromers, and depending on the ligand used, the heteromers were recycled or directed for lysosomal degradation [63]. Stimulation of DOR with its specific agonist (Delt I) promoted ubiquitination, but not phosphorylation of MOR and led to degradation of both receptors (Figure 2(2)). Disruption of the heteromer by an interfering peptide containing the first transmembrane domain of MOR rescued MOR cell-surface expression and increased cell sensitivity to opiate agonists [63], while DOR could still undergo internalization.
3. Conclusions
It is widely accepted that ubiquitination plays a vital role in the regulation of many cellular processes. However, our knowledge about the exact functions of ubiquitination in the coordination of GPCR action is still limited. The increasing amount of research in this field highlights the huge importance of E3 ubiquitin ligases, DUBs and other GPCR-interacting proteins (like β-arrestins) in coordinating complicated ubiquitin-mediated processes in a very flexible way. The role of ubiquitination in regulating lysosomal degradation of activated GPCRs is already well-established. Recent findings have provided evidence for the additional involvement of ubiquitin in many other regulatory mechanisms like receptor trafficking, β-arrestin-, and G protein-mediated signaling, and even in the modulation of differential responses initiated by biased agonists. The incredible diversity of the mechanisms directly or indirectly controlled by ubiquitination warrants future studies to uncover additional, yet unknown functions of this posttranslational modification in GPCR regulation. Amongst others, these studies should focus on identifying proteins which are involved in or are targets of ubiquitination initiated by the activation of specific GPCRs. Finally, understanding the molecular basis of the regulatory processes controlled by ubiquitin will be of great value in the development of new drugs and therapies.
Acknowledgments
This work was financially supported by Fonds voor Wetenschappelijk Onderzoek-Research Foundation Flanders (FWO). Kamila Skieterska is a recipient of a pre-doctoral FWO fellowship. Pieter Rondou has a post-doctoral FWO fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Congreve, M.; Langmead, C.J.; Mason, J.S.; Marshall, F.H. Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 2011, 54, 4283–4311. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.S.; Bortolato, A.; Congreve, M.; Marshall, F.H. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of β-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane translocation. Structure 2001, 9, 869–880. [Google Scholar] [CrossRef]
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. The 2.8 A crystal structure of visual arrestin: A model for arrestin’s regulation. Cell 1999, 97, 257–270. [Google Scholar] [CrossRef]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Lefkowitz, R.J. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2001, 2, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.S.; Tian, X.; Benovic, J.L. Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 2014, 27, 63–71. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A. Beta-arrestins as regulators of signal termination and transduction: How do they determine what to scaffold? Cell. Signal. 2011, 23, 621–629. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 2010, 62, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Clarke, W.P.; Von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Dunn, H.; Ferguson, S.S.G. Regulation of G protein-coupled receptor activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 2012, 165, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Jean-Alphonse, F.; Hanyaloglu, A. Regulation of GPCR signal networks via membrane trafficking. Mol. Cell. Endocrinol. 2011, 331, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hanyaloglu, A.C.; Zastrow, M.v. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 537–568. [Google Scholar] [CrossRef] [PubMed]
- Chini, B.; Parenti, M. G-protein-coupled receptors, cholesterol and palmitoylation: Facts about fats. J. Mol. Endocrinol. 2009, 42, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. Ring domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT family of E3 ubiquitin ligases: Multiple players in cancer development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Coscoy, L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Sci. STKE 2005, 309, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Herr, R.A.; Hansen, T.H. Ubiquitination of substrates by esterification. Traffic 2012, 13, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Ben-Saadon, R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004, 14, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Okuda-Shimizu, Y.; Hendershot, L.M. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol. Cell. 2010, 40, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Herr, R.A.; Chua, W.J.; Lybarger, L.; Wiertz, E.J.H.J.; Hansen, T.H. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase MK3. J. Cell Biol. 2007, 177, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Skieterska, K.; Rondou, P.; Lintermans, B.; Van Craenenbroeck, K. KLHL12 promotes non-lysine ubiquitination of the dopamine receptors D4.2 and D4.4, but not of the ADHD-associated D4.7 variant. PLoS ONE 2015, 10, e014565. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Turcu, F.E.R.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Milojevic, T.; Reiterer, V.; Stefan, E.; Korkhov, V.M.; Dorostkar, M.M.; Ducza, E.; Ogris, E.; Boehm, S.; Freissmuth, M.; Nanoff, C. The ubiquitin-specific protease USP4 regulates the cell surface level of the A2A receptor. Mol. Pharmacol. 2006, 69, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Modi, A.S.; Shukla, A.K.; Xiao, K.; Berthouze, M.; Ahn, S.; Wilkinson, K.D.; Miller, W.E.; Lefkowitz, R.J. β-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase MDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6650–6655. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Xiao, K.; Venkataramanan, V.; Snyder, P.M.; Freedman, N.J.; Weissman, A.M. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 2008, 283, 22166–22176. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Shenoy, S.K. β2-adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J. Biol. Chem. 2011, 286, 12785–12795. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-o.; Xiao, K.; Kim, J.; Wu, J.-H.; Wisler, J.W.; Nakamura, N.; Freedman, N.J.; Shenoy, S.K. MARCH2 promotes endocytosis and lysosomal sorting of carvedilol-bound β2-adrenergic receptors. J. Cell Biol. 2012, 199, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Armando, I.; Yu, P.; Escano, C.; Mueller, S.C.; Asico, L.; Pascua, A.; Lu, Q.; Wang, X.; Villar, V.A.M. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J. Clin. Investig. 2008, 118, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Leclair, H.M.; Dubois, S.M.; Azzi, S.; Dwyer, J.; Bidere, N.; Gavard, J. Control of CXCR2 activity through its ubiquitination on k327 residue. BMC Cell Biol. 2014, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Berlin, I.; Higginbotham, K.M.; Dise, R.S.; Sierra, M.I.; Nash, P.D. The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 2010, 285, 37895–37908. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Trejo, J.; Benovic, J.L.; Marchese, A. Arrestin-2 interacts with the E3 ubiquitin ligase AIP4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 2007, 282, 36971–36979. [Google Scholar] [CrossRef] [PubMed]
- Holleman, J.; Marchese, A. The ubiquitin ligase deltex-3l regulates endosomal sorting of the G protein-coupled receptor CXCR4. Mol. Biol. Cell 2014, 25, 1892–1904. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Marchese, A. Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 2010, 21, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Soh, U.J.K.; Trejo, J.A.; Marchese, A. Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J. Biol. Chem. 2012, 287, 9013–9027. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Benovic, J.L. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR44 mediates lysosomal sorting. J. Biol. Chem. 2001, 276, 45509–45512. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Raiborg, C.; Santini, F.; Keen, J.H.; Stenmark, H.; Benovic, J.L. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 2003, 5, 709–722. [Google Scholar] [CrossRef]
- Mines, M.A.; Goodwin, J.S.; Limbird, L.E.; Cui, F.-F.; Fan, G.-H. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK activation. J. Biol. Chem. 2009, 284, 5742–5752. [Google Scholar] [CrossRef] [PubMed]
- Canals, M.; Scholten, D.J.; de Munnik, S.; Han, M.K.L.; Smit, M.J.; Leurs, R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS ONE 2012, 7, e34192. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Orba, Y.; Nagashima, K.; Takahashi, R.; Fujitani, N.; Matsumura, S.; Hata, A.; Kubota, K. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of parkin. J. Neurochem. 2006, 99, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Rondou, P.; Haegeman, G.; Vanhoenacker, P.; Van Craenenbroeck, K. BTB protein KLHL1212 targets the dopamine D4 receptor for ubiquitination by a CUL3-based E3 ligase. J. Biol. Chem. 2008, 283, 11083–11096. [Google Scholar] [CrossRef] [PubMed]
- Rondou, P.; Skieterska, K.; Packeu, A.; Lintermans, B.; Vanhoenacker, P.; Vauquelin, G.; Haegeman, G.; Van Craenenbroeck, K. KLHL12-mediated ubiquitination of the dopamine D4 receptor does not target the receptor for degradation. Cell. Signal. 2010, 22, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.D.; Bariteau, J.T.; Magenis, L.M.; Dias, J.A. Regulation of follitropin receptor cell surface residency by the ubiquitin-proteasome pathway. Endocrinology 2003, 144, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-T.; Lai, Y.-J.; Makarova, N.; Tigyi, G.; Lin, W.-C. The lysophosphatidic acid 2 receptor mediates down-regulation of SIVA-1 to promote cell survival. J. Biol. Chem. 2007, 282, 37759–37769. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.L.; Chang, S.-H.; Thangada, S.; Wu, M.-T.; Rezaul, K.; Blaho, V.; Hwang, S.-I.; Han, D.K.; Hla, T. Engagement of s1p1-degradative mechanisms leads to vascular leak in mice. J. Clin. Investig. 2011, 121, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.L.; Thangada, S.; Wu, M.-T.; Liu, C.H.; Macdonald, T.L.; Lynch, K.R.; Lin, C.-Y.; Hla, T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J. Biol. Chem. 2007, 282, 9082–9089. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.N.; Guasti, L.; Clark, A.J. The E3 ubiquitin ligase mahogunin ubiquitinates the melanocortin 2 receptor. Endocrinology 2011, 152, 4224–4231. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; Hislop, J.N.; Grove, J.; Thorn, K.; Marsh, M.; von Zastrow, M. Regulation of endocytic clathrin dynamics by cargo ubiquitination. Dev. Cell 2012, 23, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Tanowitz, M.; von Zastrow, M. Ubiquitination-independent trafficking of G protein-coupled receptors to lysosomes. J. Biol. Chem. 2002, 277, 50219–50222. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; Henry, A.G.; Marchese, A.; von Zastrow, M. Ubiquitination regulates proteolytic processing of G protein-coupled receptors after their sorting to lysosomes. J. Biol. Chem. 2009, 284, 19361–19370. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; White, I.J.; Marsh, M.; von Zastrow, M.; Hislop, J.N. The role of ubiquitination in lysosomal trafficking of δ-opioid receptors. Traffic 2011, 12, 170–184. [Google Scholar] [CrossRef] [PubMed]
- He, S.Q.; Zhang, Z.N.; Guan, J.S.; Liu, H.R.; Zhao, B.; Wang, H.B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.Y. Facilitation of µ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron 2010, 69, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Petaja-Repo, U.E.; Hogue, M.; Laperriere, A.; Bhalla, S.; Walker, P.; Bouvier, M. Newly synthesized human δ-opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J. Biol. Chem. 2001, 276, 4416–4423. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-G.; Benovic, J.L.; Liu-Chen, L.-Y. Mechanisms of agonist-induced down-regulation of the human κ-opioid receptor: Internalization is required for down-regulation. Mol. Pharmacol. 2000, 58, 795–801. [Google Scholar] [PubMed]
- Li, J.-G.; Haines, D.S.; Liu-Chen, L.-Y. Agonist-promoted lys63-linked polyubiquitination of the human κ-opioid receptor is involved in receptor down-regulation. Mol. Pharmacol. 2008, 73, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Groer, C.E.; Schmid, C.L.; Jaeger, A.M.; Bohn, L.M. Agonist-directed interactions with specific β-arrestins determine µ-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 2011, 286, 31731–31741. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, K.; Bandari, P.; Chinen, N.; Howells, R.D. Proteasome involvement in agonist-induced down-regulation of µ and δ opioid receptors. J. Biol. Chem. 2001, 276, 12345–12355. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; Henry, A.G.; von Zastrow, M. Ubiquitination in the first cytoplasmic loop of µ-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J. Biol. Chem. 2011, 286, 40193–40204. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Cai, G.-Q.; Zheng, A.; Wang, Y.; Jia, J.; Fang, H.; Yang, Y.; Hu, M.; Ding, Q. Tumor necrosis factor-alpha regulates the hypocretin system via mRNA degradation and ubiquitination. Biochim. Biophys. Acta 2011, 1812, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, N.J.; Aguilar, B.; Smith, T.H.; Le, P.; Soohoo, A.L.; Puthenveedu, M.A.; Nizet, V.; Trejo, J. Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J. Cell Biol. 2015, 210, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Dupre, D.J.; Chen, Z.; Le Gouill, C.; Theriault, C.; Parent, J.-L.; Rola-Pleszczynski, M.; Stankova, J. Trafficking, ubiquitination, and down-regulation of the human platelet-activating factor receptor. J. Biol. Chem. 2003, 278, 48228–48235. [Google Scholar] [CrossRef] [PubMed]
- Donnellan, P.D.; Kinsella, B.T. Immature and mature species of the human prostacyclin receptor are ubiquitinated and targeted to the 26s proteasomal or lysosomal degradation pathways, respectively. J. Mol. Signal. 2009, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dores, M.R.; Grimsey, N.; Canto, I.; Barker, B.L.; Trejo, J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation-and ubiquitination-dependent sorting signals. J. Biol. Chem. 2011, 286, 40760–40770. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.L.; Marchese, A.; Trejo, J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J. Cell Biol. 2007, 177, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, B.; Murphy, J.E.; Cottrell, G.S.; Bunnett, N.W. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J. Biol. Chem. 2009, 284, 28453–28466. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Cottrell, G.S.; Gehringer, D.; Schmidlin, F.; Grady, E.F.; Bunnett, N.W. C-cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J. Biol. Chem. 2005, 280, 16076–16087. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.S.; Padilla, B.; Pikios, S.; Roosterman, D.; Steinhoff, M.; Gehringer, D.; Grady, E.F.; Bunnett, N.W. Ubiquitin-dependent down-regulation of the neurokinin-1 receptor. J. Biol. Chem. 2006, 281, 27773–27783. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Zhu, C.-C.; Hinkle, P.M. Thyrotropin-releasing hormone receptor processing: Role of ubiquitination and proteasomal degradation. Mol. Endocrinol. 2003, 17, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.P.; Lefkowitz, R.J.; Shenoy, S.K. Regulation of v2 vasopressin receptor degradation by agonist-promoted ubiquitination. J. Biol. Chem. 2003, 278, 45954–45959. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Livak, M.F.; Bernier, M.; Muller, D.C.; Carlson, O.D.; Elahi, D.; Maudsley, S.; Egan, J.M. Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E538–E547. [Google Scholar] [CrossRef] [PubMed]
- Alonso, V.; Magyar, C.E.; Wang, B.; Bisello, A.; Friedman, P.A. Ubiquitination-deubiquitination balance dictates ligand-stimulated PTHR sorting. J. Bone Miner. Res. 2011, 26, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niwa, J.-I.; Sobue, G.; Breitwieser, G.E. Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase Dorfin. J. Biol. Chem. 2006, 281, 11610–11617. [Google Scholar] [CrossRef] [PubMed]
- Lahaie, N.; Kralikova, M.; Prezeau, L.; Blahos, J.; Bouvier, M. Post-endocytotic deubiquitination and degradation of the metabotropic g-aminobutyric acid receptor by the ubiquitin-specific protease 14. J. Biol. Chem. 2016, 291, 7156–7170. [Google Scholar] [CrossRef] [PubMed]
- Moriyoshi, K.; Iijima, K.; Fujii, H.; Ito, H.; Cho, Y.; Nakanishi, S. Seven in absentia homolog 1a mediates ubiquitination and degradation of group 1 metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 8614–8619. [Google Scholar] [CrossRef] [PubMed]
- Mukai, A.; Yamamoto-Hino, M.; Awano, W.; Watanabe, W.; Komada, M.; Goto, S. Balanced ubiquitylation and deubiquitylation of frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 2010, 29, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Trejo, J.A. Ubiquitination of G protein-coupled receptors: Functional implications and drug discovery. Mol. Pharmacol. 2012, 82, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K. Seven-transmembrane receptors and ubiquitination. Circ. Res. 2007, 100, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Alonso, V.; Friedman, P.A. Minireview: Ubiquitination-regulated G protein-coupled receptor signaling and trafficking. Mol. Endocrinol. 2013, 27, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.A.; Lawson, T.G.; Velayutham, M.; Zweier, J.L.; Pickart, C.M. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 2002, 416, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Van Craenenbroeck, K.; Clark, S.D.; Cox, M.J.; Oak, J.N.; Liu, F.; Van Tol, H.H. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J. Biol. Chem. 2005, 280, 19350–19357. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. Gettin’down with ubiquitin: Turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 1999, 9, 107–112. [Google Scholar] [CrossRef]
- Hicke, L.; Riezman, H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 1996, 84, 277–287. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef]
- Marchese, A.; Paing, M.M.; Temple, B.R.S.; Trejo, J.A. G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; von Zastrow, M. Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic 2011, 12, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, B.; Bunnett, N.W.; Cottrell, G.S. Hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J. Biol. Chem. 2007, 282, 29646–29657. [Google Scholar] [CrossRef] [PubMed]
- Malerod, L.; Stuffers, S.; Brech, A.; Stenmark, H. Vps22/eap30 in ESCRT-II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic 2007, 8, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.S.; Padilla, B.; Pikios, S.; Roosterman, D.; Steinhoff, M.; Grady, E.F.; Bunnett, N.W. Post-endocytic sorting of calcitonin receptor-like receptor and receptor activity-modifying protein 1. J. Biol. Chem. 2007, 282, 12260–12271. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Paing, M.M.; Lin, H.; Montagne, W.A.; Marchese, A.; Trejo, J. Ap-3 regulates PAR1 ubiquitin-independent MVB/lysosomal sorting via an ALIX-mediated pathway. Mol. Biol. Cell 2012, 23, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, A.; Wolfe, B.L.; Griffin, C.T.; Magnuson, T.; Trejo, J. An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: Evidence for retromer-, Hrs-, and Tsg101-independent functions of sorting nexins. Mol. Biol. Cell 2006, 17, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Chen, B.; Lin, H.; Soh, U.J.; Paing, M.M.; Montagne, W.A.; Meerloo, T.; Trejo, J. Alix binds a YPX3L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell Biol. 2012, 197, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Grimsey, N.J.; Mendez, F.; Trejo, J. ALIX regulates the ubiquitin-independent lysosomal sorting of the P2y 1 purinergic receptor via a YPX3Lmotif. PLoS ONE 2016, 11, e0157587. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Lin, H.; Grimsey, N.; Mendez, F.; Trejo, J. The α-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 2015, 26, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Mukai, A.; Yamamoto-Hino, M.; Komada, M.; Okano, H.; Goto, S. Balanced ubiquitination determines cellular responsiveness to extracellular stimuli. Cell. Mol. Life Sci. 2012, 69, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Jean-Charles, P.-Y.; Shenoy, S.K. Phosphorylation of the deubiquitinase USP20 by protein kinase a regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 2015, 290, 8888–8903. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Robia, S.L.; Marchese, A. The e3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol. Biol. Cell 2009, 20, 1324–1339. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.I.; Wright, M.H.; Nash, P.D. Amsh interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J. Biol. Chem. 2010, 285, 13990–14004. [Google Scholar] [CrossRef] [PubMed]
- Miles, R.; Sluka, J.; Halladay, D.; Santerre, R.; Hale, L.; Bloem, L.; Patanjali, S.; Galvin, R.; Ma, L.; Hock, J. Parathyroid hormone (HPTH 1–38) stimulates the expression of UBP41, an ubiquitin-specific protease, in bone. J. Cell. Biochem. 2002, 85, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, M.; Herrador, A.; Haguenauer-Tsapis, R.; Vincent, O.; Leon, S. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem. Res. Int. 2012, 2012, 242764. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K. Arrestin interaction with E3 ubiquitin ligases and deubiquitinases: Functional and therapeutic implications. Handb. Exp. Pharmacol. 2014, 219, 187–203. [Google Scholar] [PubMed]
- Jean-Charles, P.-Y.; Rajiv, V.; Shenoy, S.K. Ubiquitin-related roles of b-arrestins in endocytic trafficking and signal transduction. J. Cell. Physiol. 2016, 231, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Pan, H.; Lu, Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the β2-adrenergic receptor. EMBO Rep. 2010, 11, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-O.; Kommaddi, R.P.; Shenoy, S.K. Distinct roles for b-arrestin2 and arrestin-domain-containing proteins in β2 adrenergic receptor trafficking. EMBO Rep. 2013, 14, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.-E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions 1. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Shenoy, S.K.; Wei, H.; Lefkowitz, R.J. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 2004, 279, 35518–35525. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, D.K.; Luttrell, L.M. Signaling in time and space: G protein-coupled receptors and mitogen-activated protein kinases. Assay Drug Dev. Technol. 2003, 1, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Strungs, E.G.; Luttrell, L.M. Arrestin-dependent activation of ERK and SRC family kinases. Handb. Exp. Pharmacol. 2014, 219, 225–257. [Google Scholar] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. Trafficking patterns of b-arrestin and G protein-coupled receptors determined by the kinetics of β-arrestin deubiquitination. J. Biol. Chem. 2003, 278, 14498–14506. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. Receptor-specific ubiquitination of β-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J. Biol. Chem. 2005, 280, 15315–15324. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Trejo, J.A. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell. Signal. 2013, 25, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Marchese, A.; Majetschak, M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J. Biol. Chem. 2010, 285, 15566–15576. [Google Scholar] [CrossRef] [PubMed]
- Baugher, P.J.; Richmond, A. The carboxyl-terminal PDZ ligand motif of chemokine receptor CXCR2 modulates post-endocytic sorting and cellular chemotaxis. J. Biol. Chem. 2008, 283, 30868–30878. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M. Minireview: More than just a hammer: Ligand “Bias” And pharmaceutical discovery. Mol. Endocrinol. 2014, 28, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Whalen, E.; Rajagopal, S.; Lefkowitz, R. Therapeutic potential of β-arrestin-and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the µ-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of β-blocker action: Carvedilol stimulates β-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Wetzker, R.; Bohmer, F.-D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 2003, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, M. Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2001, 2, 274–286. [Google Scholar] [CrossRef] [PubMed]
- George, S.R.; O’Dowd, B.F. A novel dopamine receptor signaling unit in brain: Heterooligomers of D1 and D2 dopamine receptors. Sci. World J. 2007, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J. Dimerization in GPCR mobility and signaling. Curr. Opin. Pharmacol. 2010, 10, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Prinster, S.C.; Hague, C.; Hall, R.A. Heterodimerization of G protein-coupled receptors: Specificity and functional significance. Pharmacol. Rev. 2005, 57, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, M.; Novi, F.; Schallmach, E.; Lin, R.; Baragli, A.; Colzi, A.; Griffon, N.; Corsini, G.U.; Sokoloff, P.; Levenson, R. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J. Biol. Chem. 2001, 276, 30308–30314. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Guidolin, D.; Agnati, L.F.; Borroto-Escuela, D.O. Dopamine heteroreceptor complexes as therapeutic targets in parkinson’s disease. Expert Opin. Ther. Targets 2015, 19, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Frankowska, M.; Ferraro, L.; Guidolin, D.; Ciruela, F.; Agnati, L.F. The changing world of G protein-coupled receptors: From monomers to dimers and receptor mosaics with allosteric receptor-receptor interactions. J. Recept. Signal Transduct. 2010, 30, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Marcellino, D.; Guidolin, D.; Woods, A.S.; Agnati, L.F. Heterodimers and receptor mosaics of different types of G-protein-coupled receptors. Physiology 2008, 23, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Borroto-Escuela, D.O.; Marcellino, D.; Romero-Fernandez, W.; Frankowska, M.; Guidolin, D.; Filip, M.; Ferraro, L.; Woods, A.; Tarakanov, A. GPCR heteromers and their allosteric receptor-receptor interactions. Curr. Med. Chem. 2012, 19, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Tarakanov, A.; Fernandez, W.R.; Ferraro, L.; Tanganelli, S.; Filip, M.; Agnati, L.F.; Garriga, P.; Diaz-Cabiale, Z.; Borroto-Escuela, D.O. Diversity and bias through receptor-receptor interactions in GPCR heteroreceptor complexes. Focus on examples from dopamine D2 receptor heteromerization. Front. Endocrinol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Role of ubiquitin in GPCR trafficking. (1) Many GPCRs have been described to undergo agonist-induced ubiquitination and down-regulation. Upon endocytosis, they are often directed for lysosomal degradation via the conserved endosomal-sorting complex required for the transport (ESCRT) machinery (1a); however, some of them can be deubiquitinated and directed to the resensitization pathway (1b). (2) Constitutive ubiquitination of frizzled-4 receptor (FZD4R) promotes its internalization and lysosomal degradation, while deubiquitination leads to its recycling and increased cell surface expression. (3) Some GPCRs are basally ubiquitinated (steady-state) and upon agonist binding are deubiquitinated and internalized. After subsequent ubiquitination, they can recycle back to the cell surface. (4) Some properly folded GPCRs (e.g., A2AR) require deubiquitination to be delivered to the cell surface. (5) Ubiquitination also functions as a quality control system in which misfolded, polyubiquitinated receptors are directed for proteasomal degradation via the endoplasmic reticulum-associated degradation (ERAD) pathway. (6) Some GPCRs are ubiquitinated at the plasma membrane and are directed to the proteasome via a poorly understood process.
Figure 1.
Role of ubiquitin in GPCR trafficking. (1) Many GPCRs have been described to undergo agonist-induced ubiquitination and down-regulation. Upon endocytosis, they are often directed for lysosomal degradation via the conserved endosomal-sorting complex required for the transport (ESCRT) machinery (1a); however, some of them can be deubiquitinated and directed to the resensitization pathway (1b). (2) Constitutive ubiquitination of frizzled-4 receptor (FZD4R) promotes its internalization and lysosomal degradation, while deubiquitination leads to its recycling and increased cell surface expression. (3) Some GPCRs are basally ubiquitinated (steady-state) and upon agonist binding are deubiquitinated and internalized. After subsequent ubiquitination, they can recycle back to the cell surface. (4) Some properly folded GPCRs (e.g., A2AR) require deubiquitination to be delivered to the cell surface. (5) Ubiquitination also functions as a quality control system in which misfolded, polyubiquitinated receptors are directed for proteasomal degradation via the endoplasmic reticulum-associated degradation (ERAD) pathway. (6) Some GPCRs are ubiquitinated at the plasma membrane and are directed to the proteasome via a poorly understood process.

Figure 2.
Transubiquitination between GPCRs: Activation of one GPCR can promote ubiquitination of another GPCR. (1) Agonist stimulation of dopamine D5 receptor (D5R) leads to the ubiquitination of angiotensin-II type 1 receptor (AT1R), disruption of the interaction between the two receptors, and subsequent proteasomal degradation of AT1R. The precise mechanism is poorly understood; (2) Activation of δ-opioid receptor (DOR) with its specific agonist leads to the ubiquitination of μ-opioid receptor (MOR) and co-internalization of both receptors which are in the next steps targeted for lysosomal degradation. This causes a decrease in cell responsiveness to opiate agonists.
Figure 2.
Transubiquitination between GPCRs: Activation of one GPCR can promote ubiquitination of another GPCR. (1) Agonist stimulation of dopamine D5 receptor (D5R) leads to the ubiquitination of angiotensin-II type 1 receptor (AT1R), disruption of the interaction between the two receptors, and subsequent proteasomal degradation of AT1R. The precise mechanism is poorly understood; (2) Activation of δ-opioid receptor (DOR) with its specific agonist leads to the ubiquitination of μ-opioid receptor (MOR) and co-internalization of both receptors which are in the next steps targeted for lysosomal degradation. This causes a decrease in cell responsiveness to opiate agonists.


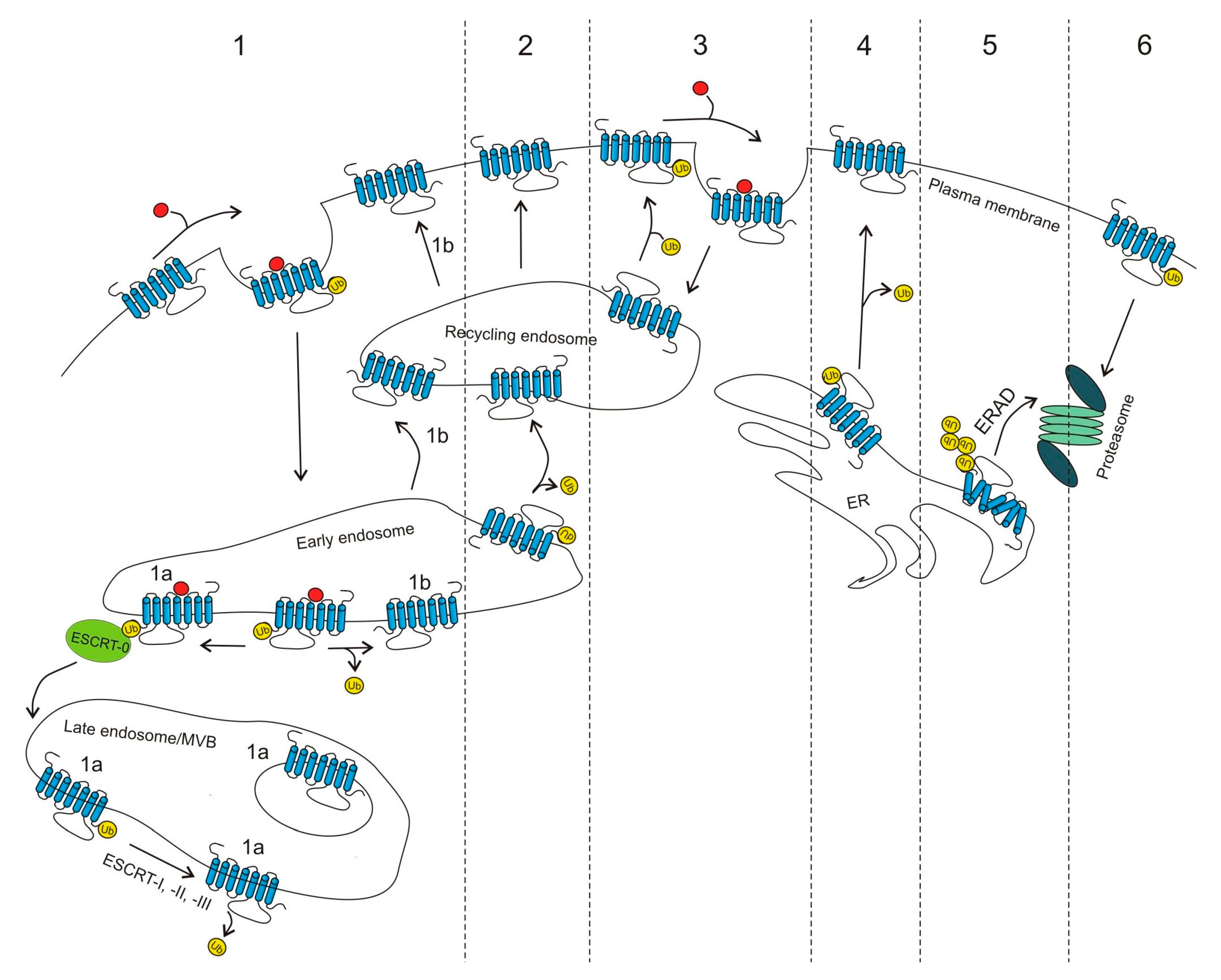{kind=link}
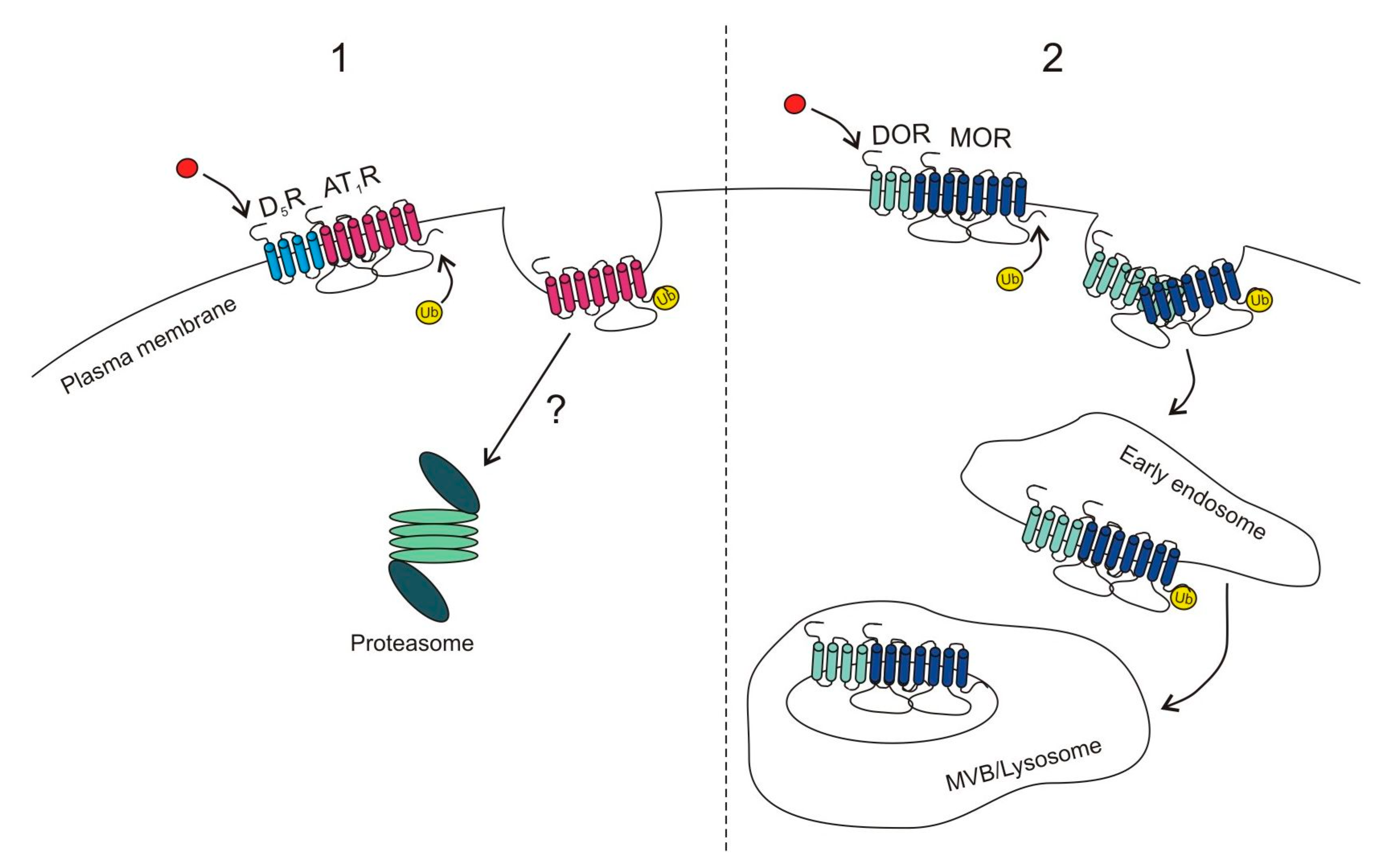{kind=link}
Table 1.
G protein-coupled receptors (GPCR) ubiquitination. The table presents a summary of current knowledge regarding GPCR ubiquitination. The list includes the GPCR family, receptor name and available information on receptor ubiquitination, including E3 ligase, and deubiquitinating enzymes (DUB) and ubiquitin-binding sites. Next, we mention whether ubiquitination is agonist induced or constitutive. Finally, the reported effect on GPCR function is stated.
Table 1.
G protein-coupled receptors (GPCR) ubiquitination. The table presents a summary of current knowledge regarding GPCR ubiquitination. The list includes the GPCR family, receptor name and available information on receptor ubiquitination, including E3 ligase, and deubiquitinating enzymes (DUB) and ubiquitin-binding sites. Next, we mention whether ubiquitination is agonist induced or constitutive. Finally, the reported effect on GPCR function is stated.
| GPCR | E3 Ligase | DUB | Residues | Induced/Constitutive | Role | Comment | Reference |
|---|---|---|---|---|---|---|---|
| Class A GPCRs | |||||||
| Adenosine receptors | |||||||
| A2A | N.D. | USP4 | N.D. | Constitutive | N.D. | Deubiquitination necessary for surface expression | [32] |
| Adrenoceptors | |||||||
| β2 | Nedd4 | USP20, USP33 | Lys in IC3 and C-term. | Agonist (isoproterenol) | Lysosomal degradation, regulation of arrestin-mediated signaling | β-arrestin 2 involved | [33,34,35,36,37] |
| MARCH2 | N.D. | Non-Lys | β-arrestin biased agonist (carvedilol) | Lysosomal degradation | N.D. | [38] | |
| Angiotensin receptors | |||||||
| AT1 | N.D. | N.D. | N.D. | Activation of D5R (Fenoldopam) | Proteasomal degradation of glycosylated receptor | Polyubiquitination | [39] |
| Chemokine receptors | |||||||
| CXCR2 | N.D. | N.D. | Lys327 | Agonist (IL-8) | Internalization, signaling | Polyubiquitination | [40] |
| CXCR4 | AIP4 | USP14, USP8 (indirectly) | Three Lys in C-term | Agonist (SDF-1α = CXCL12) | Lysosomal degradation; together with STAM-1 role in p44/42 MAPK activation | β-arrestin 1 involved; DTX3L–controls sorting to lysosomes by blocking activity of AIP4 | [41,42,43,44,45,46,47,48] |
| CXCR7 | N.D. | Upon stimulation | Lys in C-term | Constitutive | Ubiquitination required for membrane expression of the receptor | β-arrestin involved | [49] |
| Class A Orphans | |||||||
| GPR37 | Parkin | N.D. | C-term | Constitutive | ERAD | N.D. | [50] |
| HRD1 | N.D. | N.D. | Induced by overexpression of ATF6 | ERAD | Degradation of GPR37 reduces ER stress induced apoptosis | [51] | |
| Dopamine Receptors | |||||||
| D1R, D2R | N.D. | N.D. | N.D. | Constitutive | N.D. | N.D. | [52] |
| D4R | Cullin3 | N.D. | Non-Lys ubiquitination | Constitutive | Does not influence degradation | Polyubiquitination | [28,52,53] |
| D5R | N.D. | N.D. | N.D. | Constitutive | N.D. | N.D. | [52] |
| Glycoprotein hormone receptors | |||||||
| FSH | N.D. | N.D. | Mainly in IC3 | Constitutive | Cell-surface expression | Other residues can also be ubiquitinated | [54] |
| Lysophospholipid receptors | |||||||
| LPA2 | N.D. | N.D. | N.D. | Agonist (LPA) | Proteasomal degradation, cell survival | N.D. | [55] |
| S1P1 | WWP2 | N.D. | N.D. | Functional antagonist (FTY720P) | Proteasomal degradation | Polyubiquitination | [56,57] |
| Melanocortin receptors | |||||||
| MC2 | Mahogunin | N.D. | N.D. | Agonist (ACTH) | N.D. | Multi-monoubiquitination | [58] |
| Opioid receptors | |||||||
| δ (DOR) | AIP4 | N.D. | Lys | Agonist (DADLE) | Proteasomal degradation | Polyubiquitination, stimulates transport to ILVs | [59,60,61,62] |
| N.D. | N.D. | N.D. | Select. agonist (Deltropin I) | Lysosomal degradation | Co-degradation with MOR | [63] | |
| N.D. | N.D. | N.D. | Constitutive | Proteasomal degradation | ER-retained receptor | [64] | |
| κ (KOR) | N.D. | N.D. | Lys338, Lys 349, Lys 378 in C-term | Constitutive but enhanced by agonists | Lysosomal and proteasomal degradation | Lys63 polyubiquitination; β-arrestin involved; enhanced by receptor phosphorylation | [65,66] |
| µ (MOR) | N.D. | N.D. | Residue in IC1 | Agonist (DAMGO, DADLE) | Lysosomal and proteasomal degradation | β-arrestin 1 involved | [67,68,69] |
| Smurf2 | N.D. | Lys94 and Lys96 in IC1 | Non-selective agonist (DADLE) | Internalization by controlling maturation of the receptor-containing CCPs | Polyubiquitination; β-arrestin 2 involved | [59] | |
| N.D. | N.D. | N.D. | DOR activation (Deltropin) | Co-degradation with DOR in lysosomes | N.D. | [63] | |
| Orexin receptors | |||||||
| OX2 | cIAP-1 and -2 are important | N.D. | N.D. | TNF-α | Degradation | N.D. | [70] |
| P2Y receptors | |||||||
| P2Y1 | Nedd4-2 | N.D. | Lys in C-term | Agonist (ADP) | p38 MAPK activation | N.D. | [71] |
| Platelet-activating receptors | |||||||
| PAF receptor | Cbl is important | N.D. | N.D. | Constitutive | Agonist (PAF)-dependent down-regulation in proteasome and lysosome | Monoubiquitination | [72] |
| Prostanoid receptors | |||||||
| IP | N.D. | N.D. | N.D. | Agonist (cicaprost-mature receptor) | Lysosomal degradation of mature receptor; proteasomal degradation of immature receptor | Polyubiquitination | [73] |
| Proteinase-activated receptors | |||||||
| PAR1 | N.D. | Upon stimulation | Lys421, Lys422 in C-term | Constitutive and agonist-induced (SFLLRN-NH2) | Basal ubiquitination blocks constitutive internalization; agonist-dependent ubiquitination is involved in internalization | N.D. | [74,75] |
| Nedd4-2 | Lys | Agonist (α-thrombin) | p38 MAPK activation | Lys63-type polyubiquitination | [71] | ||
| PAR2 | Cbl | AMSH and USP8 | Lys | Agonist (peptide SLIGR-NH2) | Lysosomal degradation | Monoubiquitination; DUBs are essential for lysosomal trafficking | [76,77] |
| Tachykinin receptors | |||||||
| NK1 | N.D. | N.D. | Lys | Agonist (Substance P) | Down-regulation and degradation | N.D. | [78] |
| Thyrotropin-releasing hormone receptor | |||||||
| TRH1 | N.D. | N.D. | N.D. | Constitutive | ERAD | N.D. | [79] |
| Vasopressin and oxytocin receptors | |||||||
| V2 | N.D. | N.D. | Lys268 in IC3 | Agonist (Arg-vasopr.) | Degradation | β-arrestin 2 involved | [80] |
| Class B GPCRs | |||||||
| Glucagon receptors | |||||||
| GIP | N.D. | N.D. | N.D. | Agonist (GIP) | Proteasomal degradation | N.D. | [81] |
| Parathyroid hormone receptors | |||||||
| PTH1 | N.D. | USP2 | N.D. | Activating PTH [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34] and non-activating PTH [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34] ligands | PTH [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]-induced proteasomal degradation | Lys48-type polyubiquitination | [82] |
| Class C GPCRs | |||||||
| Calcium-sensing receptors | |||||||
| CaS | Dorphin | N.D. | Lys | Constitutive | ERAD | N.D. | [83] |
| GABAB receptors | |||||||
| GABAB1 | N.D. | USP14 | Lys | Constitutive and induced by PMA | Internalization and lysosomal degradation | N.D. | [84] |
| Metabotropic glutamate receptors | |||||||
| mGlu1a mGlu5 | Siah1A | N.D. | Lys | Constitutive | Proteasomal degradation | N.D. | [85] |
| Class Frizzled GPCRs | |||||||
| FZD4 | N.D. | USP8 | N.D. | Constitutive | Internalization; lysosomal degradation | N.D. | [86] |
N.D., non-defined; A2A, Adenosine A2A receptor; Nedd4, Neural precursor cell-expressed developmentally downregulated gene 4; IC1 or 3, Intracellular loop 1 or 3; MARCH2, Membrane associated ring-CH-type finger 2; AT1, Angiotensin-II receptor type 1; D1R-D5R, Dopamine D1-D5 receptor; CXCR2-CXCR7, C-X-C chemokine receptor-2- 7; IL-8, Interleukine 8; AIP4, Atrophin-1-interacting protein 4; USP, Ubiquitin-specific protease; CXCL12, C-X-C motif chemokine ligand 12; STAM-1, Signal transducing adapter molecule 1; MAPK; Mitogen-activated protein kinase; DTX3L, Deltex E3 ubiquitin Ligase 3L; HRD1, ERAD-associated E3 ubiquitin-protein ligase HRD1; ATF6, Activating transcription factor 6; ERAD, Endoplasmic reticulum-associated degradation; ER, Endoplasmic reticulum; FSH, Follicle-stimulating hormone receptor; LPA2, Lysophosphatidic acid receptor 2; S1P1, Sphingosine-1-phosphate receptor 1; WWP2, WW domain-containing protein 2; ACTH, Adrenocorticotropic hormone; DOR, δ-opioid receptor; KOR, κ-opioid receptor; MOR, µ-opioid receptor; ILV, Intraluminal vesicle; DADLE, [D-Ala2, D-Leu5]-Enkephalin; DAMGO, [d-Ala2, N-MePhe4, Gly-ol]-enkephalin; Smurf2, SMAD specific E3 ubiquitin protein ligase 2; CCPs, Clathrin-coated pits; OX2, Orexin receptor 2; cIAP-1, Cellular inhibitor of apoptosis protein-1; TNF-α, Tumor necrosis factor α; P2Y1, P2Y purinoceptor 1; PAF, Platelet-activating factor receptor; IP, Prostanoid IP receptor; PAR1/PAR2, Proteinase-activated receptor 1 and 2; Cbl, E3 ubiquitin-protein ligase Cbl; AMSH, Associated molecule with the SH3-domain of STAM; NK1, Neurokinin-1 receptor; TRH1, Thyrotropin-releasing hormone receptor; GIP, Gastric inhibitory polypeptide; PTH1, Parathyroid hormone 1 receptor; CaS, Calcium-sensing receptor 1; GABAB1, Gamma-aminobutyric acid B1; PMA, phorbol 12-myristate 13-acetate; mGlu1a/mGlu5, Metabotropic glutamate receptor 1a or 5; Siah1A, Seven in absentia 1A; FZD4, Frizzled-4 receptor.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Skieterska, K.; Rondou, P.; Van Craenenbroeck, K. Regulation of G Protein-Coupled Receptors by Ubiquitination. Int. J. Mol. Sci. 2017, 18, 923. https://doi.org/10.3390/ijms18050923
AMA Style
Skieterska K, Rondou P, Van Craenenbroeck K. Regulation of G Protein-Coupled Receptors by Ubiquitination. International Journal of Molecular Sciences. 2017; 18(5):923. https://doi.org/10.3390/ijms18050923
Chicago/Turabian StyleSkieterska, Kamila, Pieter Rondou, and Kathleen Van Craenenbroeck. 2017. "Regulation of G Protein-Coupled Receptors by Ubiquitination" International Journal of Molecular Sciences 18, no. 5: 923. https://doi.org/10.3390/ijms18050923
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.