
Applying Unconventional Secretion in Ustilago maydis for the Export of Functional Nanobodies

 ,
,
Abstract
:
1. Introduction
2. Results
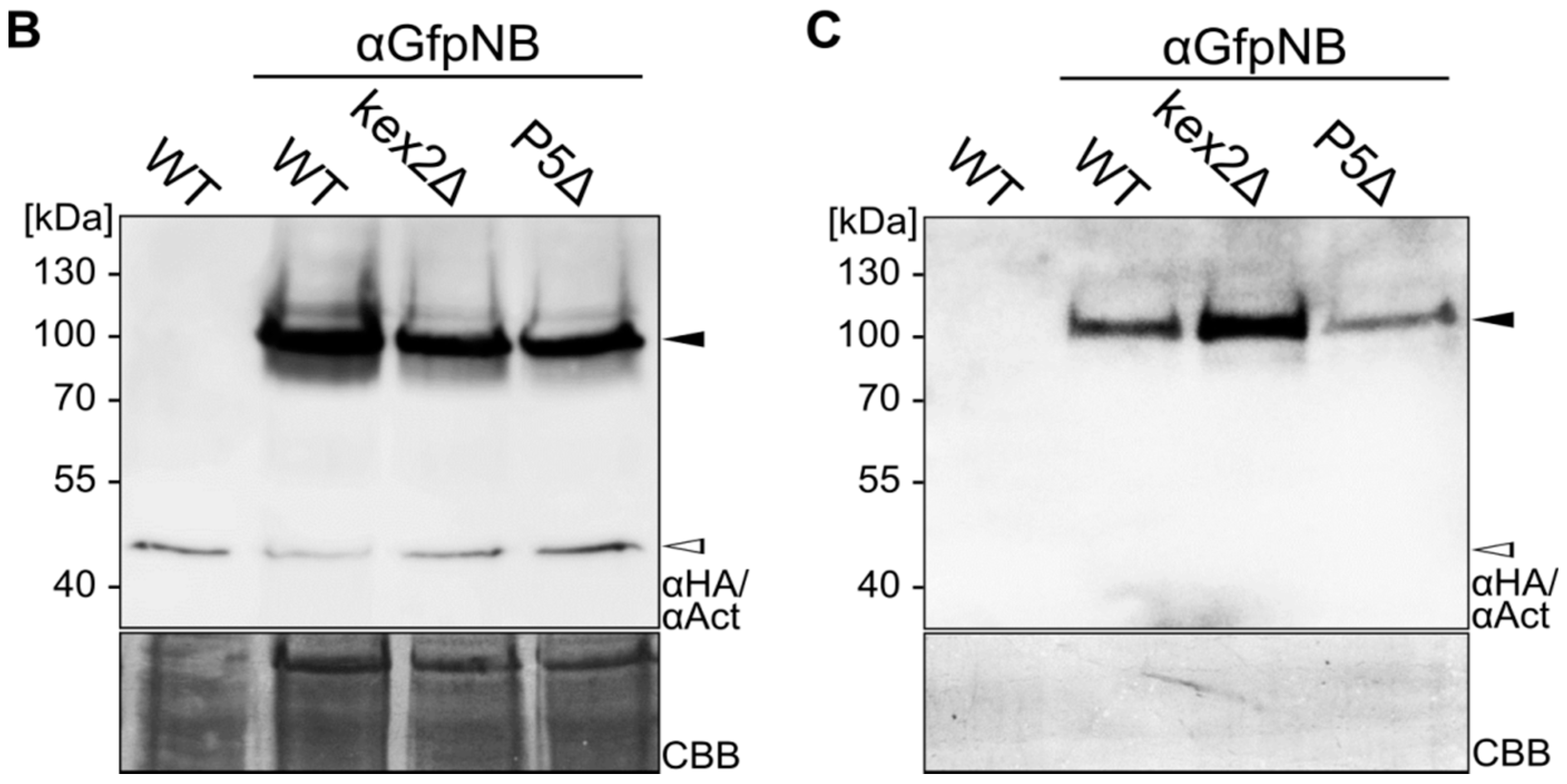
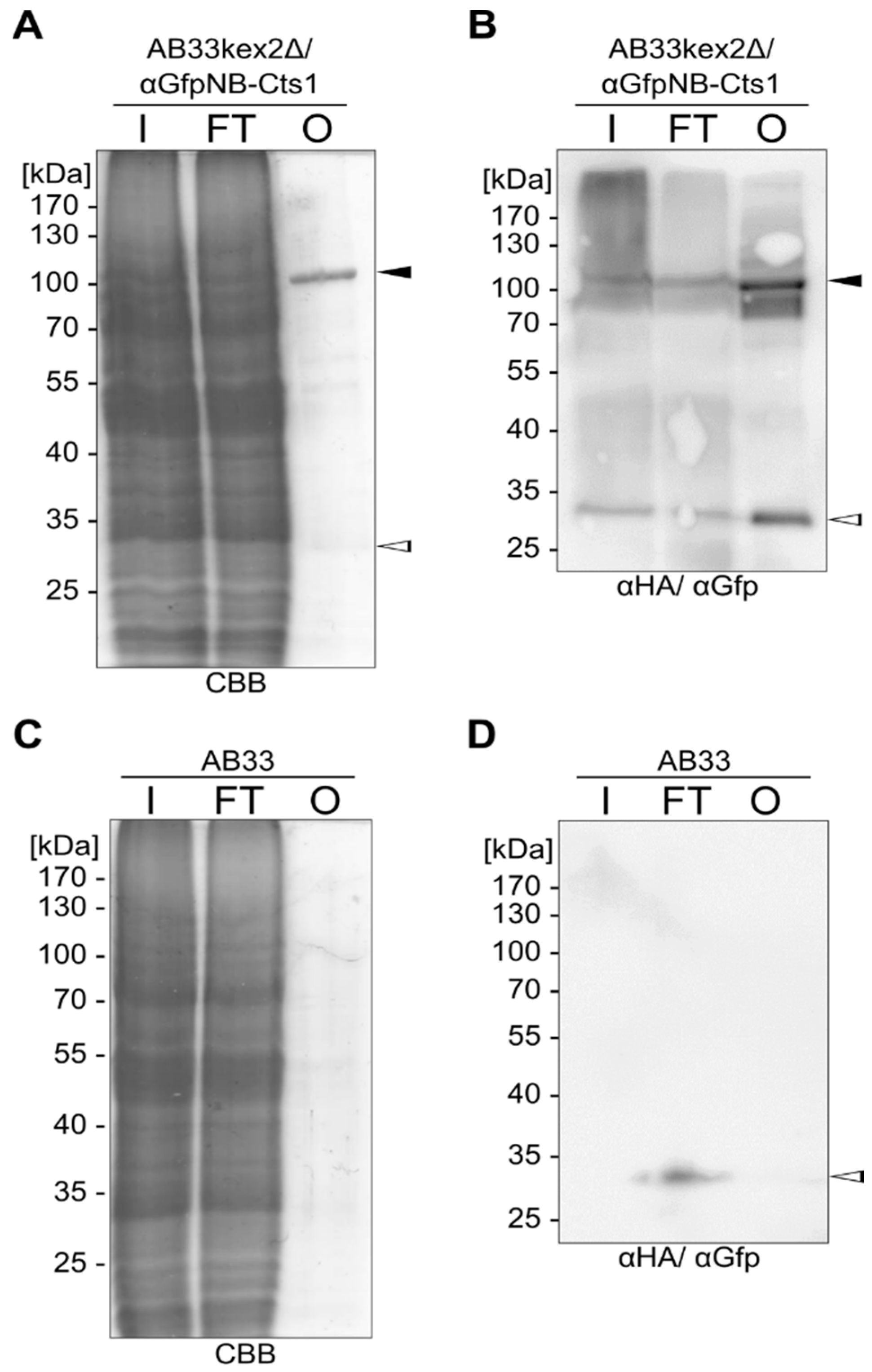
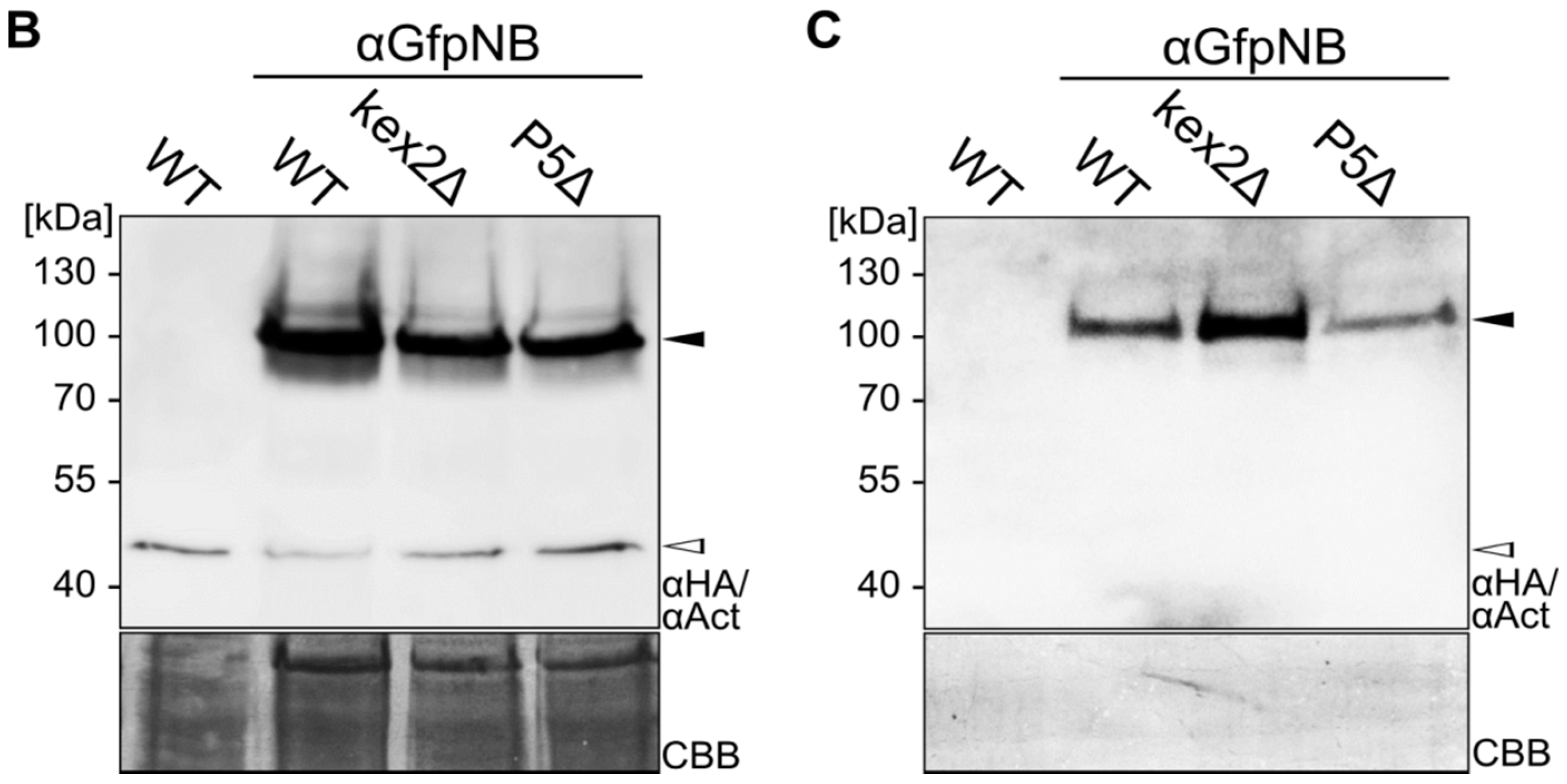
2.1. Expression and Unconventional Secretion of an αGfp Nanobody
2.2. Biochemical Characterization of the αGfp Nanobody
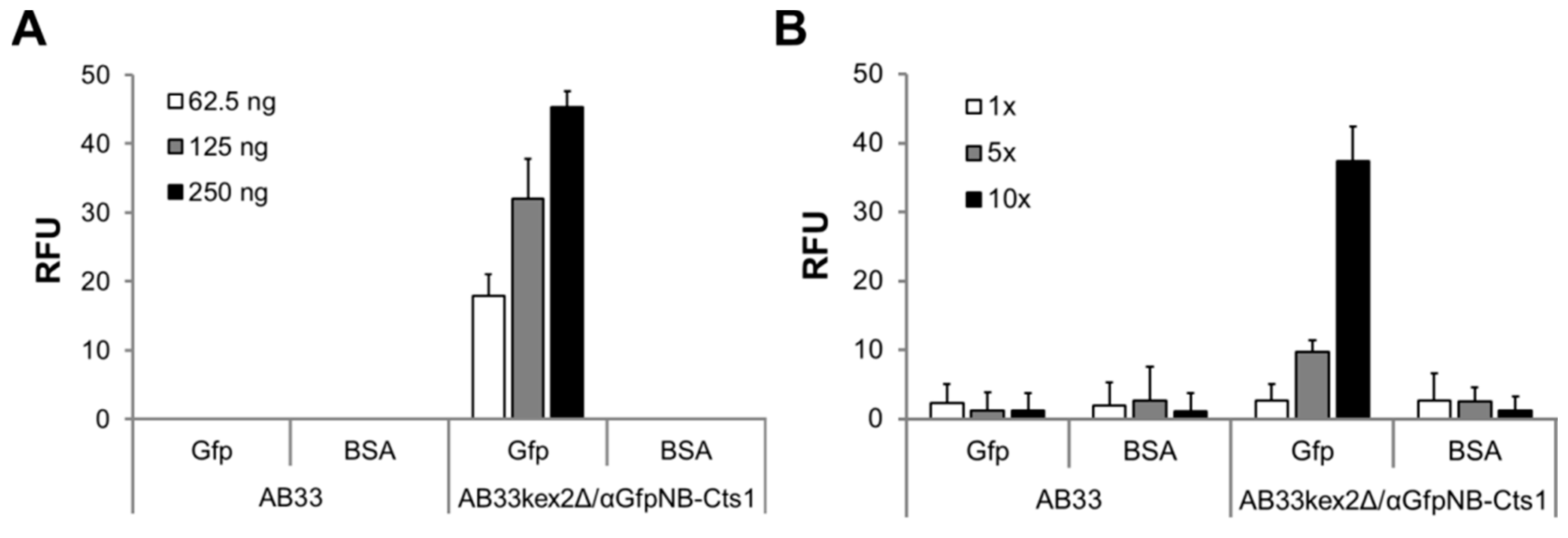
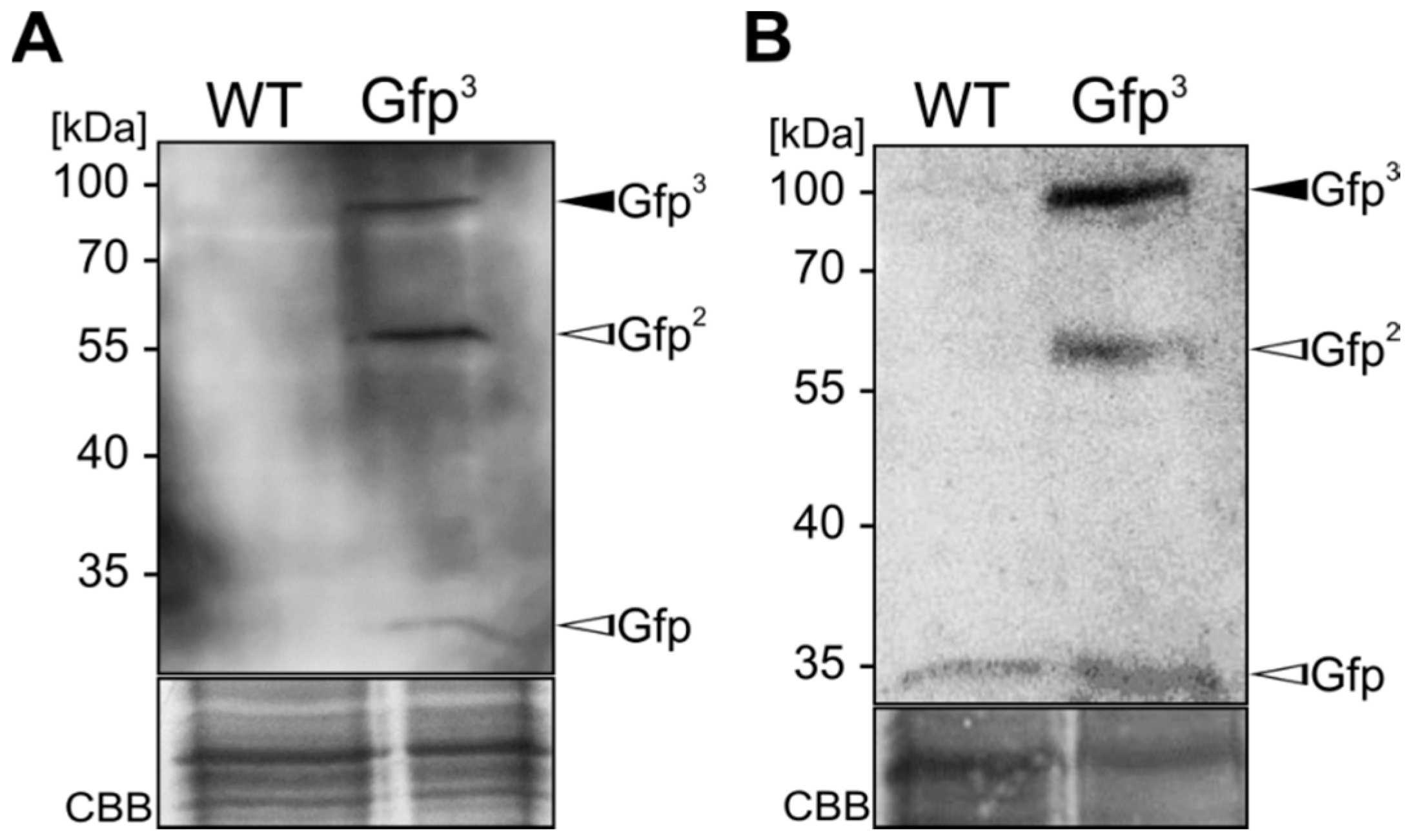
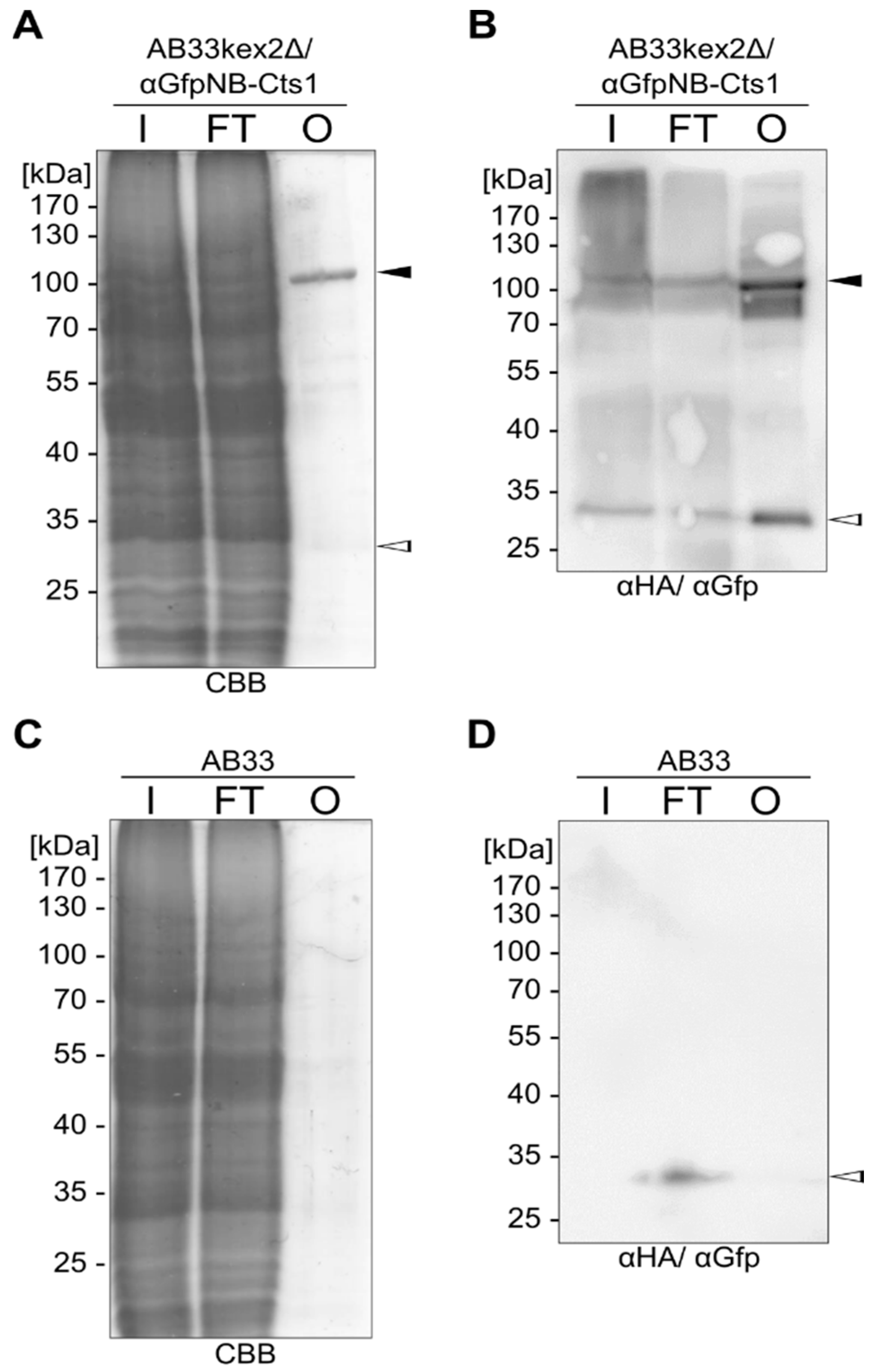
2.3. Exploiting Intrinsic Chitin-Binding Properties to Establish a Gfp-Trap
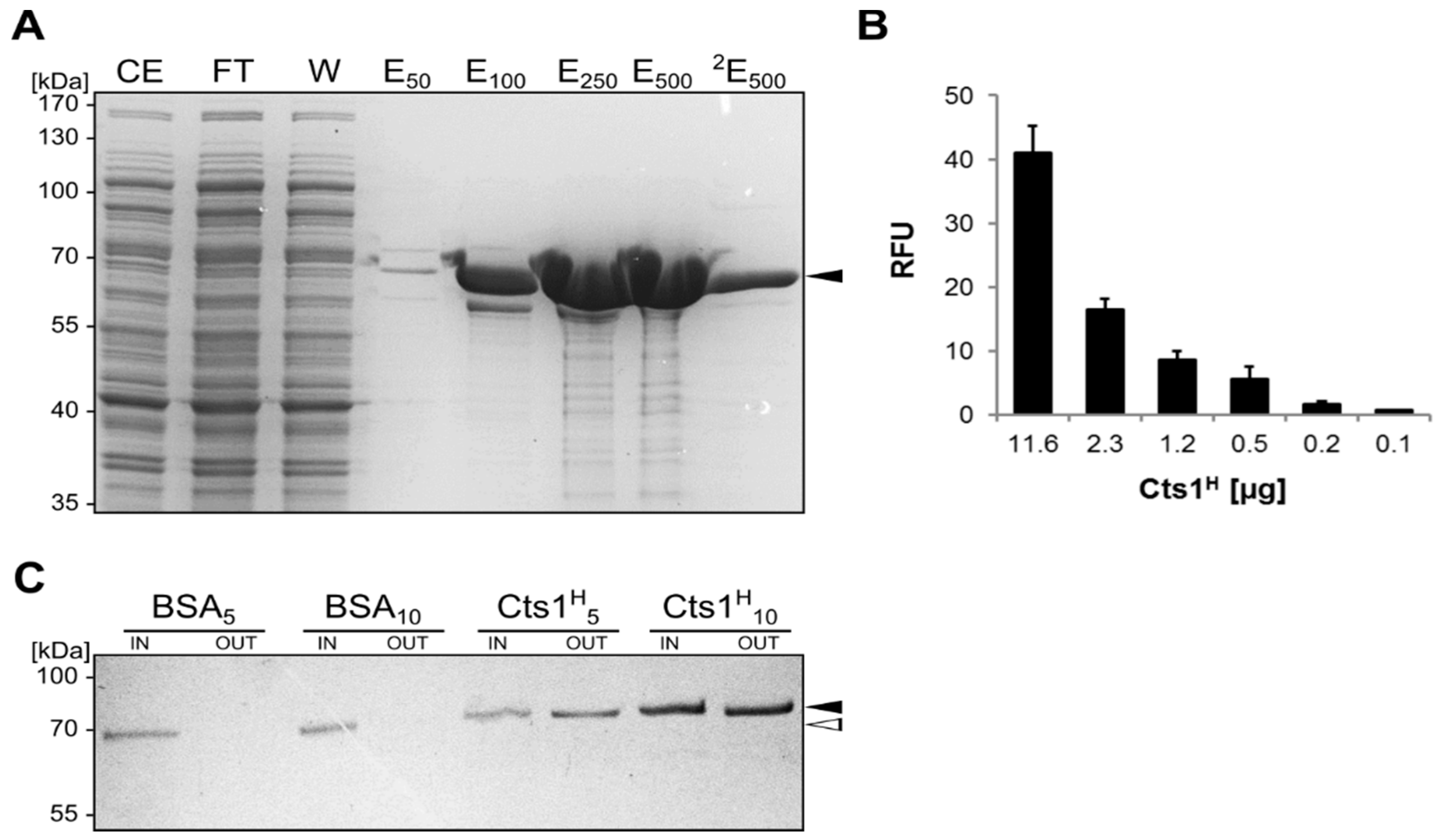
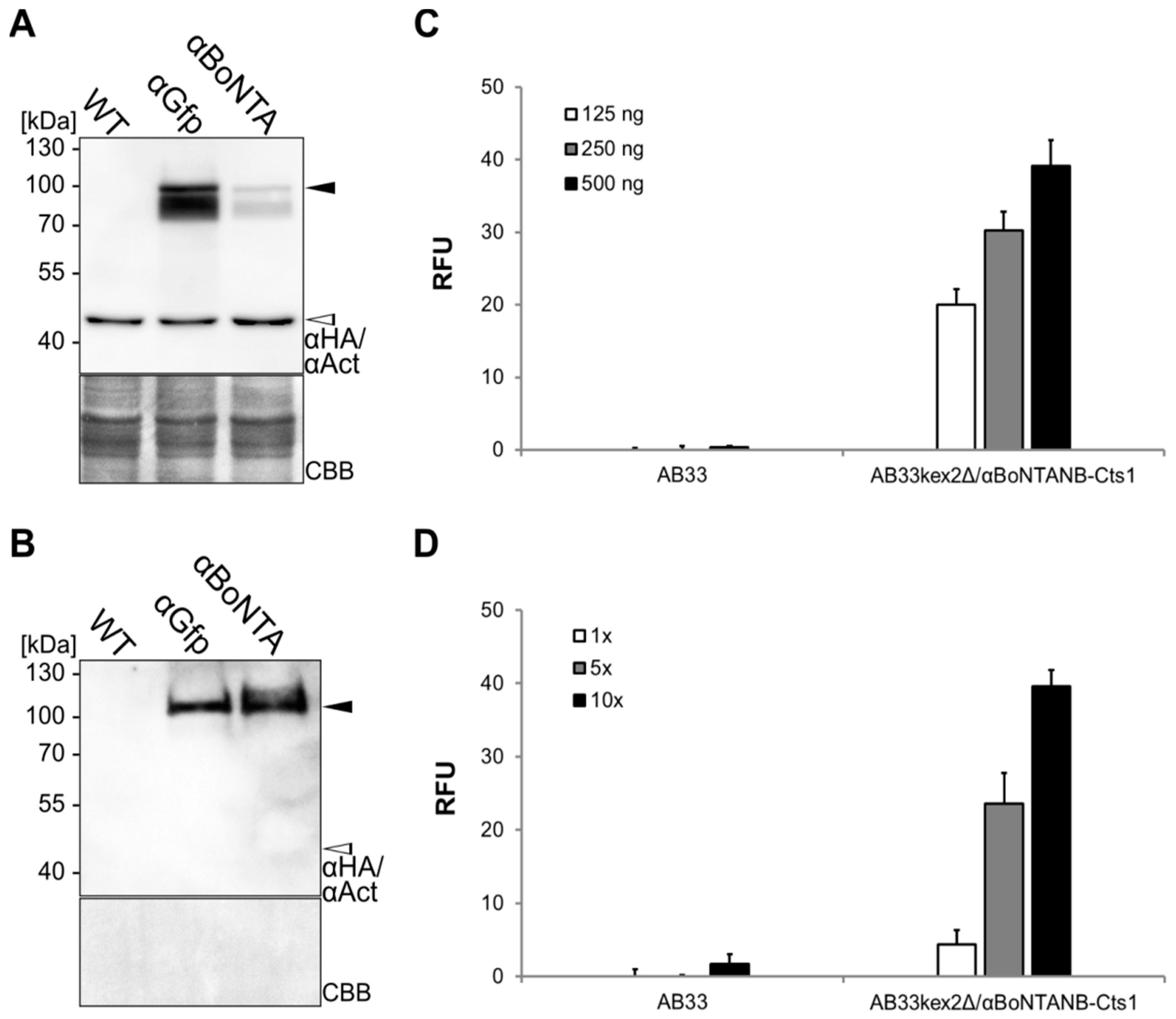
2.4. Expression, Purification and Biochemical Characterization of a Functional Nanobody against Botulinum Toxin A
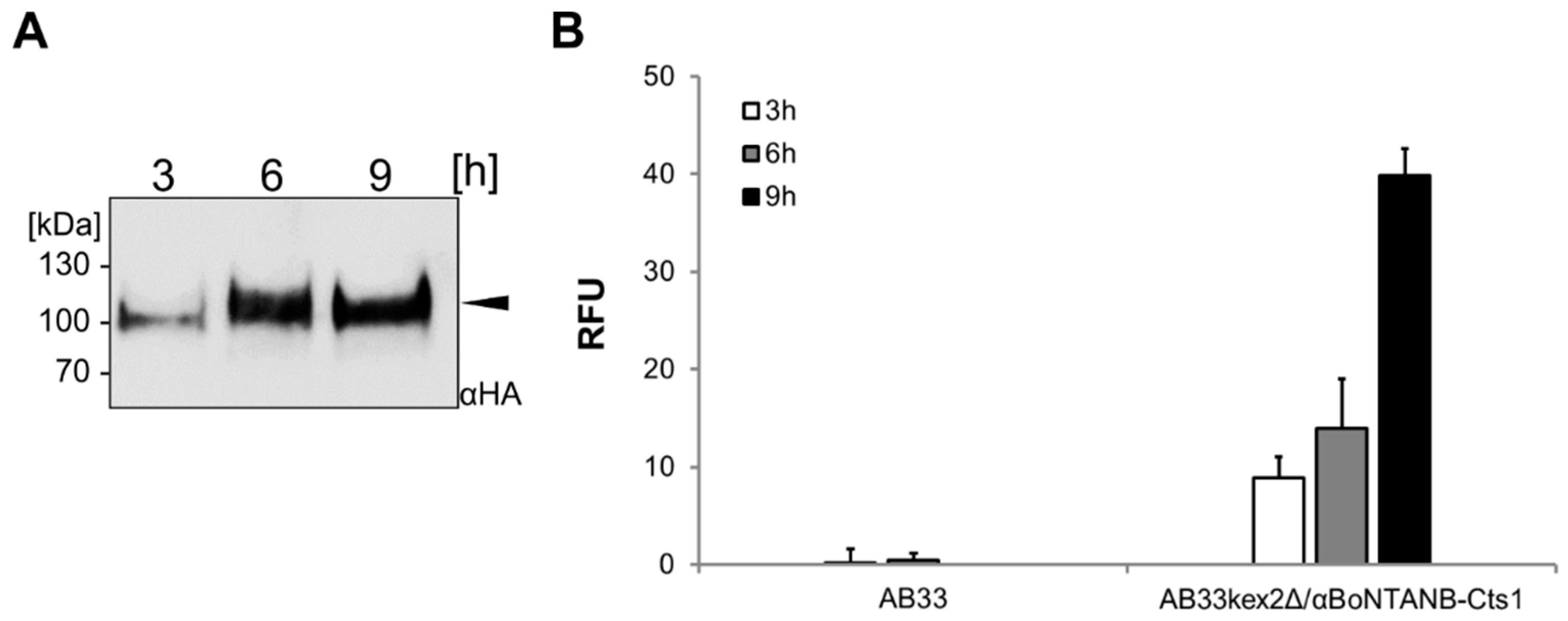
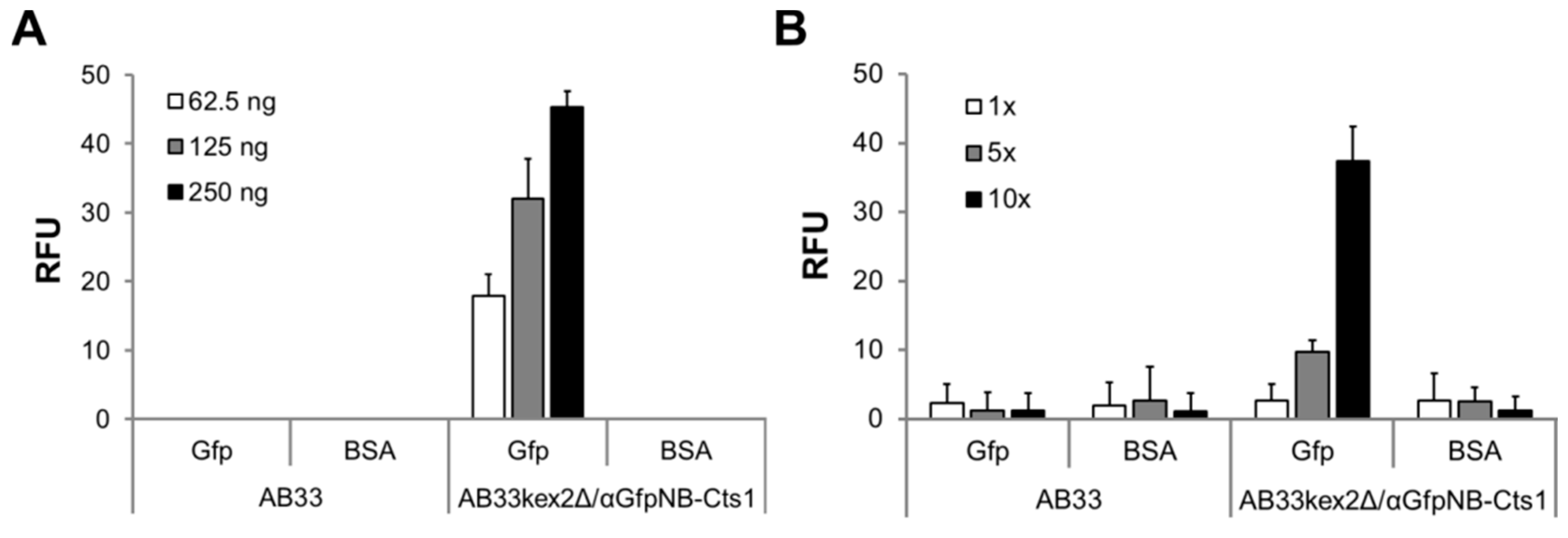
2.5. Optimizing αBoNTA Nanobody Expression
3. Discussion
4. Materials and Methods
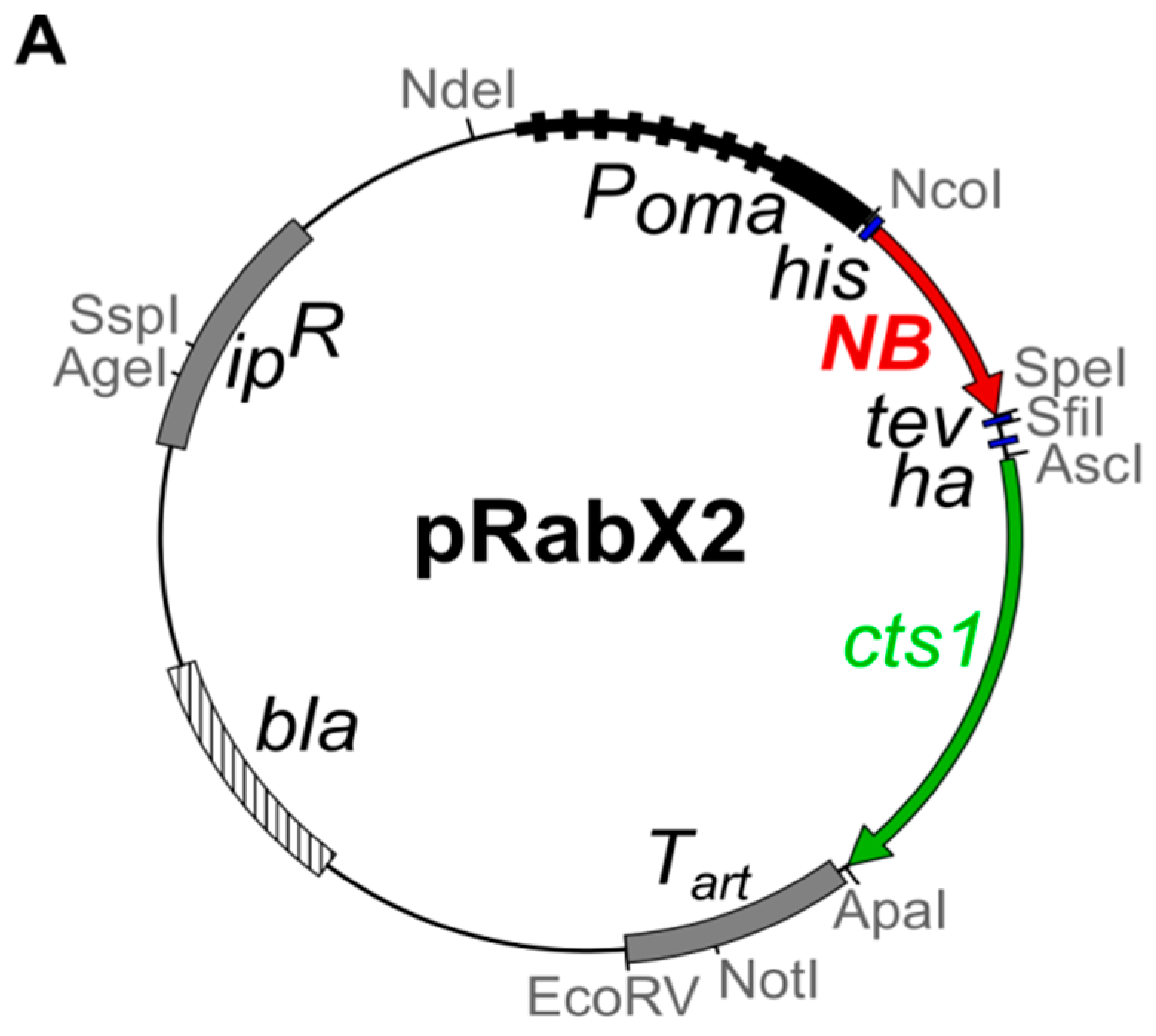
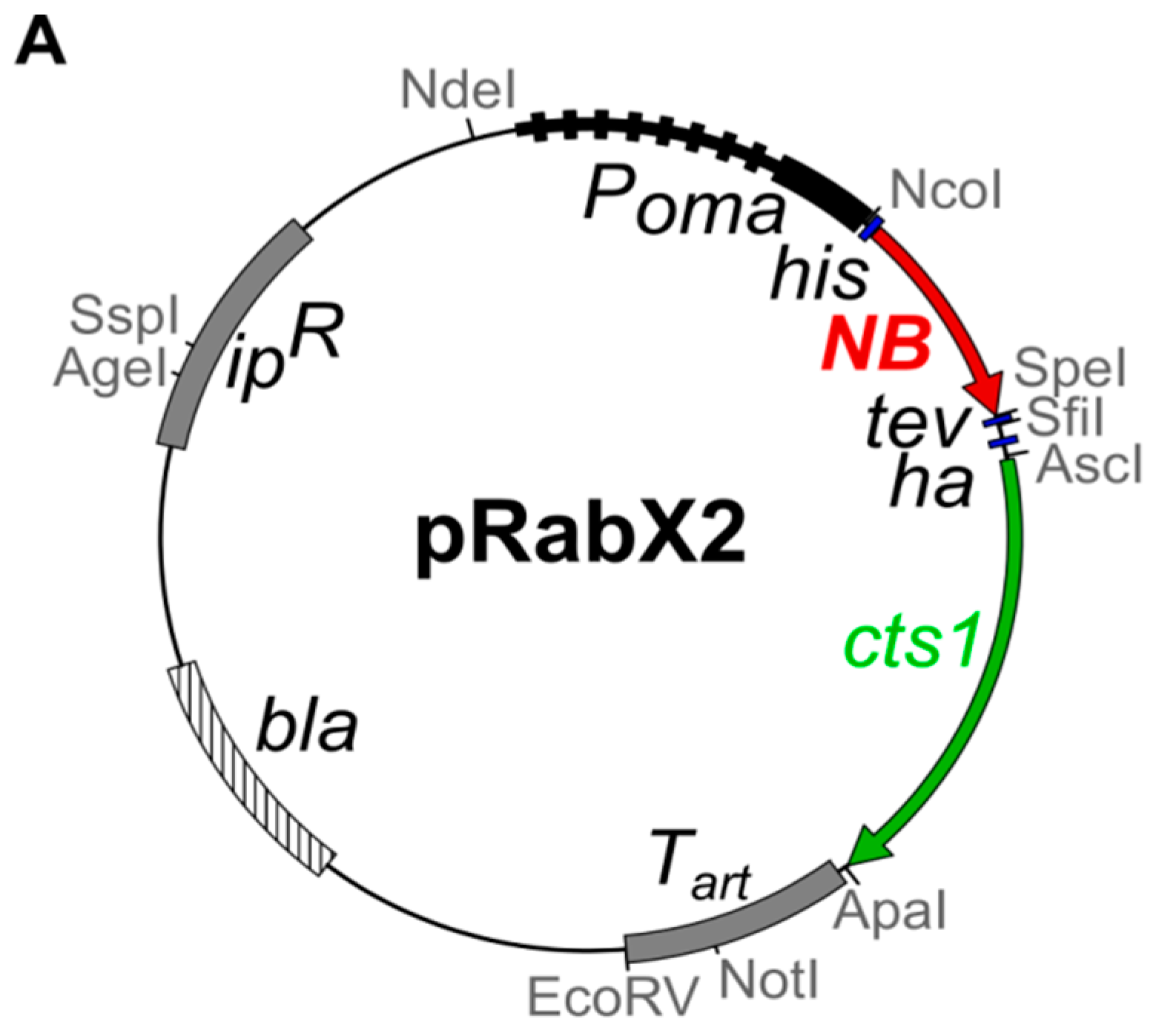
4.1. Microbial Strains, Culture Conditions and Plasmids
4.2. Purification of Cts1-Fusion Proteins from U. maydis
4.3. SDS-PAGE and Western Blot Analysis
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. E. coli Expression and Purification Gfp and Chitinase
4.6. Chitin Binding Assay
4.7. Chitinase Activity Assay
4.8. Gfp Pull-Down with Chitin-Bound Cts1
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| NB | Nanobody; BoNTA, botulinum toxin A |
| α | Anti |
| CBB | Coomassie brilliant blue |
| Gfp | Green fluorescent protein |
| IMAC | Immobilized metal affinity chromatography |
References
- Berlec, A.; Strukelj, B. Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. J. Ind. Microbiol. Biotechnol. 2013, 40, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Lingappa, V.R. Mechanism of protein translocation across the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 1986, 2, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Viotti, C. ER to golgi-dependent protein secretion: The conventional pathway. Methods Mol. Biol. 2016, 1459, 3–29. [Google Scholar] [PubMed]
- Demain, A.L.; Vaishnav, P. Production of recombinant proteins by microbes and higher organisms. Biotechnol. Adv. 2009, 27, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Wells, E.; Robinson, A.S. Cellular engineering for therapeutic protein production: Product quality, host modification, and process improvement. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Jeon, C.O. High-throughput recombinant protein expression in Escherichia coli: Current status and future perspectives. Open Biol. 2016, 6, 160196. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.; Harwood, C.R. Heterologous protein secretion by bacillus species from the cradle to the grave. Adv. Appl. Microbiol. 2010, 73, 1–25. [Google Scholar] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Idiris, A.; Tohda, H.; Kumagai, H.; Takegawa, K. Engineering of protein secretion in yeast: Strategies and impact on protein production. Appl. Microbiol. Biotechnol. 2010, 86, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, H.; Peterson, R. Making recombinant proteins in filamentous fungi—Are we expecting too much? Front. Microbiol. 2014, 5, 75. [Google Scholar] [PubMed]
- Zhu, J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol. Adv. 2012, 30, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Zemella, A.; Thoring, L.; Hoffmeister, C.; Kubick, S. Cell-free protein synthesis: Pros and cons of prokaryotic and eukaryotic systems. Chembiochem 2015, 16, 2420–2431. [Google Scholar] [CrossRef] [PubMed]
- Gerngross, T.U. Advances in the production of human therapeutic proteins in yeasts and filamentous fungi. Nat. Biotechnol. 2004, 22, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Steringer, J.P.; Müller, H.M.; Nickel, W. Unconventional secretion of fibroblast growth factor 2—A novel type of protein translocation across membranes? J. Mol. Biol. 2015, 427, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Wegehingel, S.; Zehe, C.; Nickel, W. Rerouting of fibroblast growth factor 2 to the classical secretory pathway results in post-translational modifications that block binding to heparan sulfate proteoglycans. FEBS Lett. 2008, 582, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.C. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim. Biophys. Acta 1999, 1473, 172–185. [Google Scholar] [CrossRef]
- Cooper, D.N.; Barondes, S.H. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J. Cell Biol. 1990, 110, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W. Pathways of unconventional protein secretion. Curr. Opin. Biotechnol. 2010, 21, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, P.; Feldbrügge, M.; Schipper, K. The corn smut fungus Ustilago maydis as an alternative expression system for biopharmaceuticals. In Fungal Biology. Gene Expression Systems in Fungi: Advancements and Applications; Schmoll, M., Dattenböck, C., Eds.; Springer Book Series; Springer International Publishing Switzerland: Cham, Switzerland, 2016; pp. 183–200. [Google Scholar]
- Feldbrügge, M.; Kellner, R.; Schipper, K. The biotechnological use and potential of plant pathogenic smut fungi. Appl. Microbiol. Biotechnol. 2013, 97, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Koepke, J.; Kaffarnik, F.; Haag, C.; Zarnack, K.; Luscombe, N.M.; König, J.; Ule, J.; Kellner, R.; Begerow, D.; Feldbrügge, M. The RNA-binding protein RRM4 is essential for efficient secretion of endochitinase Cts1. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.; Sarkari, P.; Kreibich, S.; Brefort, T.; Feldbrügge, M.; Schipper, K. Applying unconventional secretion of the endochitinase Cts1 to export heterologous proteins in Ustilago maydis. J. Biotechnol. 2012, 161, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, P.; Reindl, M.; Stock, J.; Müller, O.; Kahmann, R.; Feldbrügge, M.; Schipper, K. Improved expression of single-chain antibodies in Ustilago maydis. J. Biotechnol. 2014, 191, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Langner, T.; Göhre, V. Fungal chitinases: Function, regulation, and potential roles in plant/pathogen interactions. Curr. Genet. 2016, 62, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Langner, T.; Özturk, M.; Hartmann, S.; Cord-Landwehr, S.; Moerschbacher, B.; Walton, J.D.; Göhre, V. Chitinases are essential for cell separation in Ustilago maydis. Eukaryot. Cell 2015, 14, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.; Terfrüchte, M.; Schipper, K. A reporter system to study unconventional secretion of proteins avoiding N-glycosylation in Ustilago maydis. In Unconventional Protein Secretion: Methods and Protocols; Pompa, A., de Marchis, F., Eds.; Springer Protocols; Springer: New York, NY, USA, 2016; Volume 1459, pp. 149–160. [Google Scholar]
- Muyldermans, S. Single domain camel antibodies: Current status. J. Biotechnol. 2001, 74, 277–302. [Google Scholar] [CrossRef]
- Joosten, V.; Lokman, C.; van den Hondel, C.A.; Punt, P.J. The production of antibody fragments and antibody fusion proteins by yeasts and filamentous fungi. Microb. Cell Factories 2003, 2, 1. [Google Scholar] [CrossRef]
- Desmyter, A.; Spinelli, S.; Roussel, A.; Cambillau, C. Camelid nanobodies: Killing two birds with one stone. Curr. Opin. Struct. Biol. 2015, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Maurer, S.; Kaffarnik, F.; Ladendorf, O.; Brachmann, A.; Kämper, J.; Feldbrügge, M. Tetracycline-regulated gene expression in the pathogen Ustilago maydis. Fungal Genet. Biol. 2006, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.; Saxonov, S.; Gilbert, W. Regularities of context-dependent codon bias in eukaryotic genes. Nucleic Acids Res. 2002, 30, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Doehlemann, G.; Reissmann, S.; Assmann, D.; Fleckenstein, M.; Kahmann, R. Two linked genes encoding a secreted effector and a membrane protein are essential for Ustilago maydis-induced tumour formation. Mol. Microbiol. 2011, 81, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Funkhouser, J.D.; Aronson, N.N., Jr. Chitinase family GH18: Evolutionary insights from the genomic history of a diverse protein family. BMC Evolut. Biol. 2007, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, S. Charakterisierung der Unkonventionell Sekretierten Endochitinase Cts1 in Ustilago maydis. Master’s Thesis, Heinrich Heine University Düsseldorf, Düsseldorf, Germany, 2013. [Google Scholar]
- Mukherjee, J.; Tremblay, J.M.; Leysath, C.E.; Ofori, K.; Baldwin, K.; Feng, X.; Bedenice, D.; Webb, R.P.; Wright, P.M.; Smith, L.A.; et al. A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PLoS ONE 2012, 7, e29941. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, U.; Zolghadr, K.; Muyldermans, S.; Schepers, A.; Cardoso, M.C.; Leonhardt, H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell Proteom. 2008, 7, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Arnon, S.S.; Schechter, R.; Maslanka, S.E.; Jewell, N.P.; Hatheway, C.L. Human botulism immune globulin for the treatment of infant botulism. N. Engl. J. Med. 2006, 354, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control. Investigational heptavalent botulinum antitoxin (HBAT) to replace licensed botulinum antitoxin AB and investigational botulinum antitoxin E. Morb. Mortal. Wkly. Rep. 2010, 59, 299. [Google Scholar]
- Ardekani, L.S.; Gargari, S.L.; Rasooli, I.; Bazl, M.R.; Mohammadi, M.; Ebrahimizadeh, W.; Bakherad, H.; Zare, H. A novel nanobody against urease activity of Helicobacter pylori. Int. J. Infect. Dis. 2013, 17, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Nozaki, S.; Hartanto, D.; Miyano, R.; Nakayama, K. Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J. Cell Sci. 2015, 128, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, Z.; Shao, L.; Kong, X.; Hou, X.; Tian, D.; Sun, Y.; Xiao, Y.; Yu, L. Nanobody-derived nanobiotechnology tool kits for diverse biomedical and biotechnology applications. Int. J. Nanomed. 2016, 11, 3287–3303. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Muyldermans, S.; Depicker, A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014, 32, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Frenken, L.G.; van der Linden, R.H.; Hermans, P.W.; Bos, J.W.; Ruuls, R.C.; de Geus, B.; Verrips, C.T. Isolation of antigen specific llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J. Biotechnol. 2000, 78, 11–21. [Google Scholar] [CrossRef]
- Okazaki, F.; Aoki, J.; Tabuchi, S.; Tanaka, T.; Ogino, C.; Kondo, A. Efficient heterologous expression and secretion in Aspergillus oryzae of a llama variable heavy-chain antibody fragment V(HH) against EGFR. Appl. Microbiol. Biotechnol. 2012, 96, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Lustigman, S.; Montero, E.; Oksov, Y.; Lobo, C.A. PfRH5: A novel reticulocyte-binding family homolog of Plasmodium falciparum that binds to the erythrocyte, and an investigation of its receptor. PLoS ONE 2008, 3, e3300. [Google Scholar] [CrossRef]
- Douglas, A.D.; Baldeviano, G.C.; Lucas, C.M.; Lugo-Roman, L.A.; Crosnier, C.; Bartholdson, S.J.; Diouf, A.; Miura, K.; Lambert, L.E.; Ventocilla, J.A.; et al. A PfRH5-based vaccine is efficacious against heterologous strain blood-stage Plasmodium falciparum infection in aotus monkeys. Cell Host Microbe 2015, 17, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, L.Y.; Bartholdson, S.J.; Crosnier, C.; Campos, M.G.; Wanaguru, M.; Nguon, C.; Kwiatkowski, D.P.; Wright, G.J.; Rayner, J.C. A full-length recombinant Plasmodium falciparum PfRH5 protein induces inhibitory antibodies that are effective across common PfRH5 genetic variants. Vaccine 2013, 31, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hjerrild, K.A.; Jin, J.; Wright, K.E.; Brown, R.E.; Marshall, J.M.; Labbe, G.M.; Silk, S.E.; Cherry, C.J.; Clemmensen, S.B.; Jorgensen, T.; et al. Production of full-length soluble Plasmodium falciparum RH5 protein vaccine using a Drosophila melanogaster Schneider 2 stable cell line system. Sci. Rep. 2016, 6, e30357. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Chen, L.; Healer, J.; Lopaticki, S.; Boyle, M.; Triglia, T.; Ehlgen, F.; Ralph, S.A.; Beeson, J.G.; Cowman, A.F. Reticulocyte-binding protein homologue 5—An essential adhesin involved in invasion of human erythrocytes by Plasmodium falciparum. Int. J. Parasitol. 2009, 39, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Ord, R.L.; Rodriguez, M.; Yamasaki, T.; Takeo, S.; Tsuboi, T.; Lobo, C.A. Targeting sialic acid dependent and independent pathways of invasion in Plasmodium falciparum. PLoS ONE 2012, 7, e30251. [Google Scholar] [CrossRef] [PubMed]
- Crosnier, C.; Bustamante, L.Y.; Bartholdson, S.J.; Bei, A.K.; Theron, M.; Uchikawa, M.; Mboup, S.; Ndir, O.; Kwiatkowski, D.P.; Duraisingh, M.T.; et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 2011, 480, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.S.; Pandey, A.K.; Singh, H.; Sahar, T.; Emmanuel, A.; Chitnis, C.E.; Chauhan, V.S.; Gaur, D. Bacterially expressed full-length recombinant Plasmodium falciparum RH5 protein binds erythrocytes and elicits potent strain-transcending parasite-neutralizing antibodies. Infect. Immun. 2014, 82, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Clare, J.J.; Rayment, F.B.; Ballantine, S.P.; Sreekrishna, K.; Romanos, M.A. High-level expression of tetanus toxin fragment C in Pichia pastoris strains containing multiple tandem integrations of the gene. Nat. Biotechnol. 1991, 9, 455–460. [Google Scholar] [CrossRef]
- Wang, G.; Xia, Y.; Gu, Z.; Zhang, H.; Chen, Y.Q.; Chen, H.; Ai, L.; Chen, W. A new potential secretion pathway for recombinant proteins in Bacillus subtilis. Microb. Cell Factories 2015, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, L.; Fu, G.; Zhou, W.; Sun, Y.; Zheng, P.; Sun, J.; Zhang, D. A novel strategy for protein production using non-classical secretion pathway in Bacillus subtilis. Microb. Cell Factories 2016, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. Ustilago maydis. In Handbook of Genetics; Plenum Press: New York, NY, USA, 1974; Volume 1, pp. 575–595. [Google Scholar]
- Bösch, K.; Frantzeskakis, L.; Vranes, M.; Kämper, J.; Schipper, K.; Göhre, V. Genetic manipulation of the plant pathogen Ustilago maydis to study fungal biology and plant microbe interactions. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.; König, J.; Julius, C.; Feldbrügge, M. A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol. Genet. Genom. 2004, 272, 216–226. [Google Scholar] [CrossRef]
- Loubradou, G.; Brachmann, A.; Feldbrügge, M.; Kahmann, R. A homologue of the transcriptional repressor Ssn6p antagonizes cAMP signalling in Ustilago maydis. Mol. Microbiol. 2001, 40, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.; Weinzierl, G.; Kämper, J.; Kahmann, R. Identification of genes in the bW/bE regulatory cascade in Ustilago maydis. Mol. Microbiol. 2001, 42, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Relevant Genotype/Resistance | Uma 1 | Plasmid Transformed | Progenitor | Reference |
|---|---|---|---|---|---|
| E. coli Rosetta2 (DE3) pLysS | F− ompT hsdSB (rB− mB−) gal dcm (DE3) pLysSRARE2 (CamR) | 791 | - | - | Novagen (Merck-Millipore) |
| E. coli Rosetta2 (DE3) pLysS pET15b_His-Gfp | F− ompT hsdSB (rB− mB−) gal dcm pLysSRARE2 (CamR) pET15b_His-Gfp (AmpR) | 1464 | pET15b_His-Gfp (pUMa2156) (expression of a 6xHis-Gfp fusion protein) | UMa791 | this study |
| E. coli Rosetta2 (DE3) pLysS pET15b_His-Cts1 | F− ompT hsdSB (rB− mB−) gal dcm pLysSRARE2 (CamR) pET15b_His-Cts1 (AmpR) | 1170 | pET15b_His-Cts1 (pUMa1951) (expression of a 6xHis-Cts1 fusion protein) | UMa791 | this study |
| Strains | Relevant Genotype/Resistance | UMa 1 | Reference | Plasmid Integrated | Manipulated Locus | Progenitor Strain |
|---|---|---|---|---|---|---|
| AB33 | a2 PnarbW2bE1 | 133 | [65] | - | b | FB2 |
| PhleoR | ||||||
| AB33 αGfp-Cts1 | ipr[Pomahis:agpf:tev:hacts1:ubi1 3′UTR] ips | 1396 | this study | pRabX2 PomaHis-αGfp-NB-TH-Cts1 (pUMa2240) | cbx | AB33 |
| CbxR | ||||||
| AB33kex2Δ/αGfp-Cts1 | FRTwt[um02843Δ::hyg]FRTwt | 1397 | this study | pRabX2 PomaHis-αGfp-NB-TH-Cts1 (pUMa2240) | cbx | UMa803 [25] |
| ipr[Pomahis:agpf:tev:ha:cts1:ubi1 3′UTR] ips | ||||||
| CbxR, HygR | ||||||
| AB33P5Δ/αGfp-Cts1 | FRT5[um04400Δ::hyg]FRT5 | 1465 | this study | pRabX2 PomaHis-αGfp-NB-TH-Cts1 (pUMa2240) | cbx | UMa1391 [25] |
| FRT3[um11908Δ] | ||||||
| FRT2[um00064 Δ] | ||||||
| FRTwt[um02178Δ] | ||||||
| FRT1[um04926Δ] | ||||||
| ipr[Pomahis:agpf:tev:ha:cts1:ubi1 3′UTR] ips | ||||||
| PhleoR, CbxR | ||||||
| SG200 | a1:mfa2, bE1, bW2 | 67 | [35] | - | - | - |
| PhleoR | ||||||
| SG200 Gfp3 | ipr[Potefegfp:egfp:egfp]ips | 587 | [36] | - | cbx | SG200 [35] |
| CbxR | ||||||
| AB33kex2Δ/αBoNTA-Cts1 | FRTwt[um02843Δ::hyg] | 1870 | this study | pRabX2 PomaHis-αBoNTA-NB-TH-Cts1 (pUMa2863) | cbx | UMa803 [25] |
| FRTwt ipr[Pomahis:abonta:tev:ha:cts1:ubi1 3′UTR] ips | ||||||
| PhleoR, HygR, CbxR |
| Designation | Nucleotide Sequence (5′–3′) |
|---|---|
| oMF502 | ACGACGTTGTAAAACGACGGCCAG |
| oMF503 | TTCACACAGGAAACAGCTATGACC |
| oRL1085 | CACCATATGTTTGGACGTCTTAAGCACAGGATGTCTCGCGCTCGACTAGACG |
| oRL1086 | GTGGGATCCTTACTTGAGGCCGTTCTTGACATTGTCCC |
| oRL1384 | GTGGGATCCTTACTTGTACAGCTCGTCCATGCCG |
| oRL1385 | CACCATATGGTGAGCAAGGGCGAGGAGC |
| oRL1577 | GCCATGGCGGCCCATCACCACCATCACCACCATCACCACCATCATATGGCCGACGTCCAGCT |
| oRL1578 | GACTAGTCGACGAGACGGTGA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terfrüchte, M.; Reindl, M.; Jankowski, S.; Sarkari, P.; Feldbrügge, M.; Schipper, K. Applying Unconventional Secretion in Ustilago maydis for the Export of Functional Nanobodies. Int. J. Mol. Sci. 2017, 18, 937. https://doi.org/10.3390/ijms18050937
Terfrüchte M, Reindl M, Jankowski S, Sarkari P, Feldbrügge M, Schipper K. Applying Unconventional Secretion in Ustilago maydis for the Export of Functional Nanobodies. International Journal of Molecular Sciences. 2017; 18(5):937. https://doi.org/10.3390/ijms18050937
Chicago/Turabian StyleTerfrüchte, Marius, Michèle Reindl, Silke Jankowski, Parveen Sarkari, Michael Feldbrügge, and Kerstin Schipper. 2017. "Applying Unconventional Secretion in Ustilago maydis for the Export of Functional Nanobodies" International Journal of Molecular Sciences 18, no. 5: 937. https://doi.org/10.3390/ijms18050937