
Tubacin, an HDAC6 Selective Inhibitor, Reduces the Replication of the Japanese Encephalitis Virus via the Decrease of Viral RNA Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
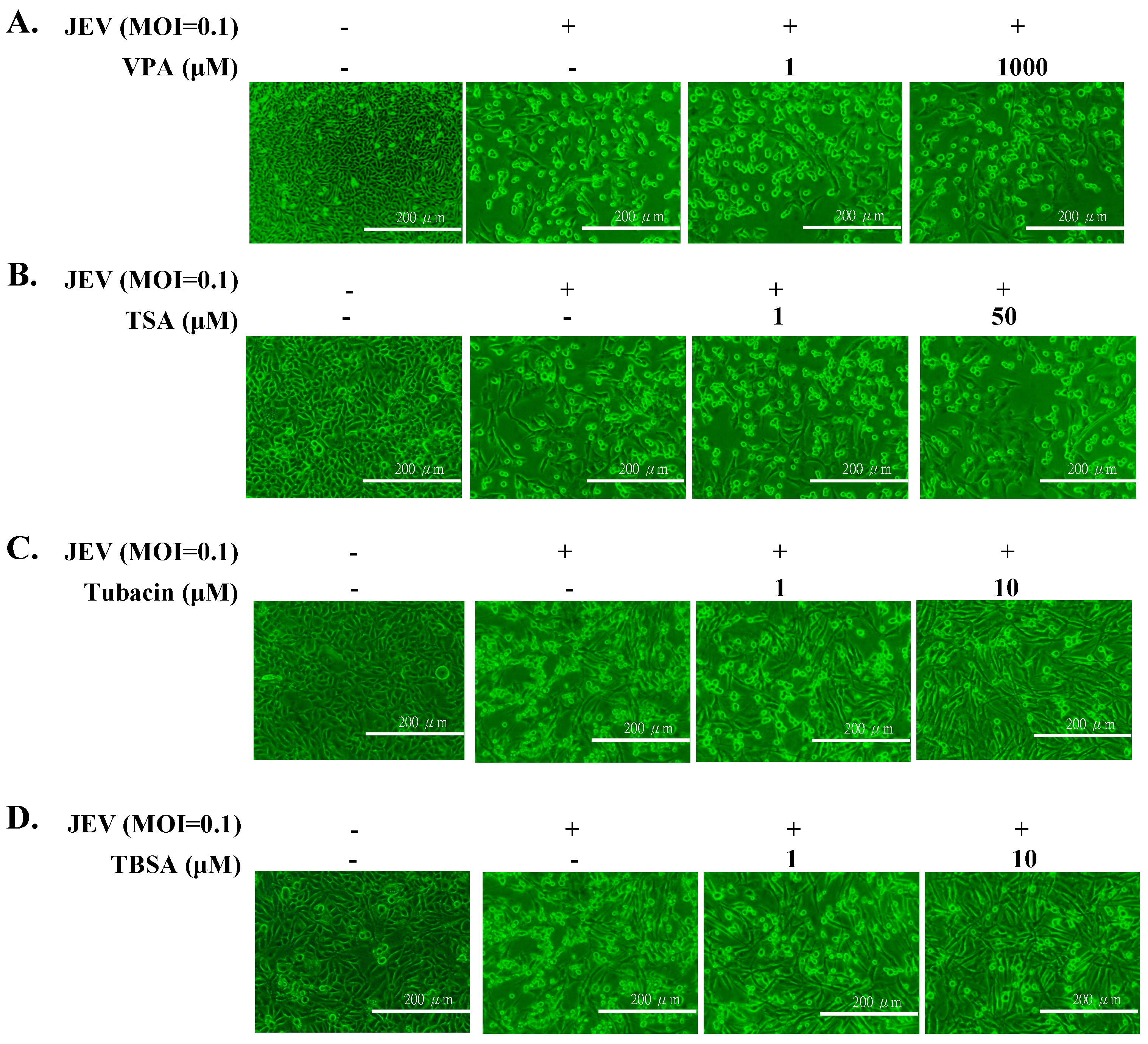
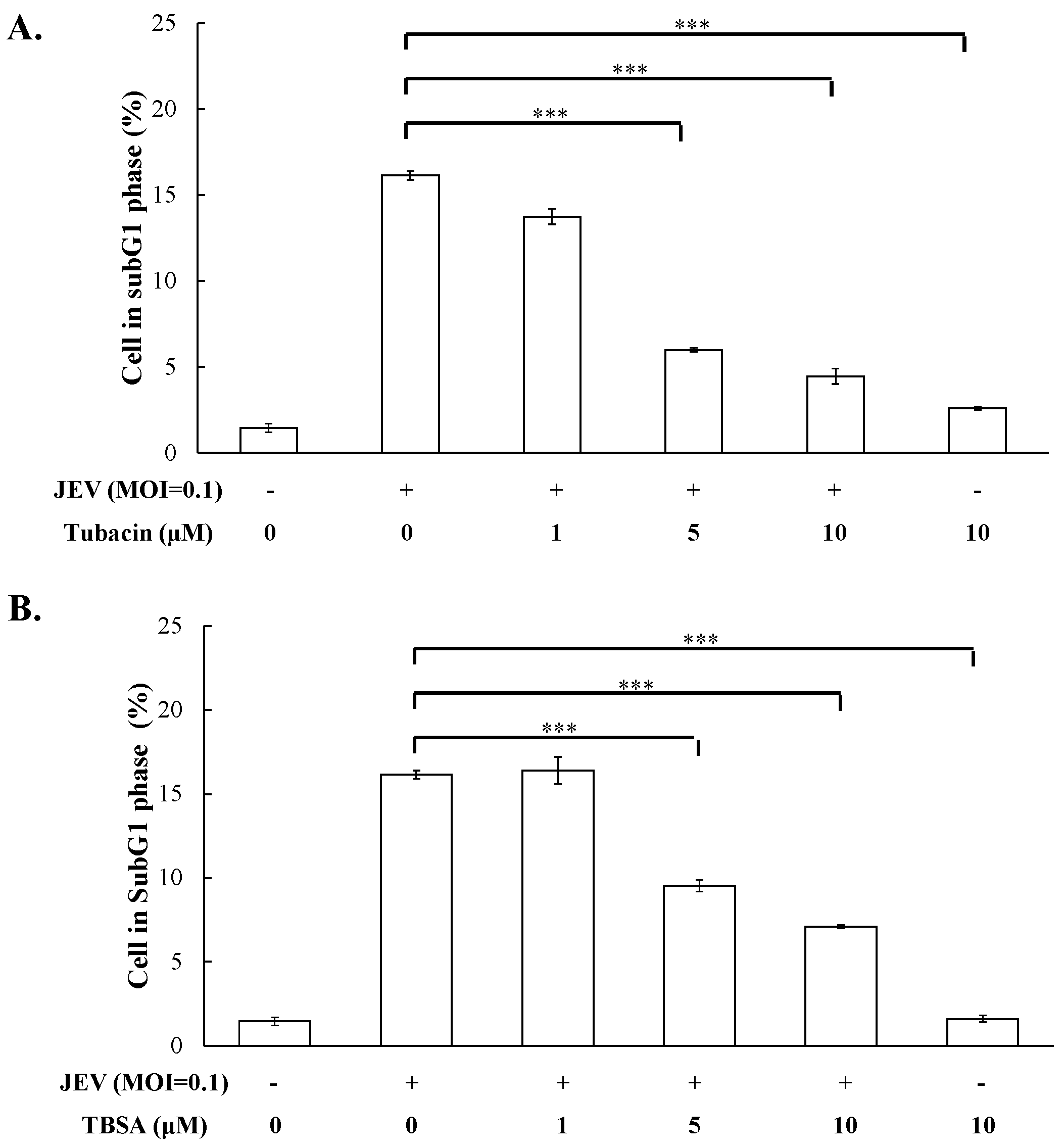
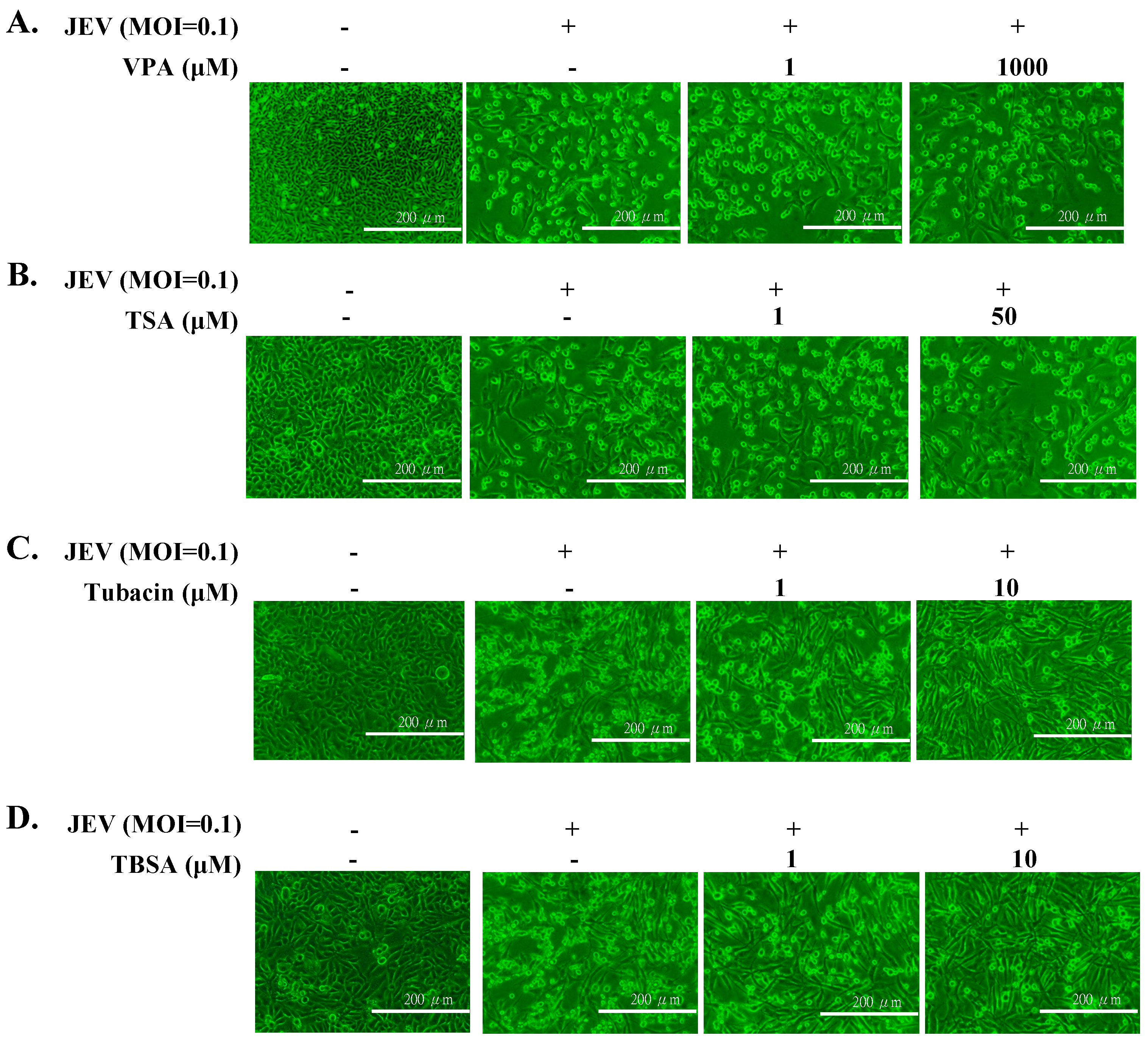
2.1. Antiviral Activity of Pan- and Selective-HDAC Inhibitors Against JEV
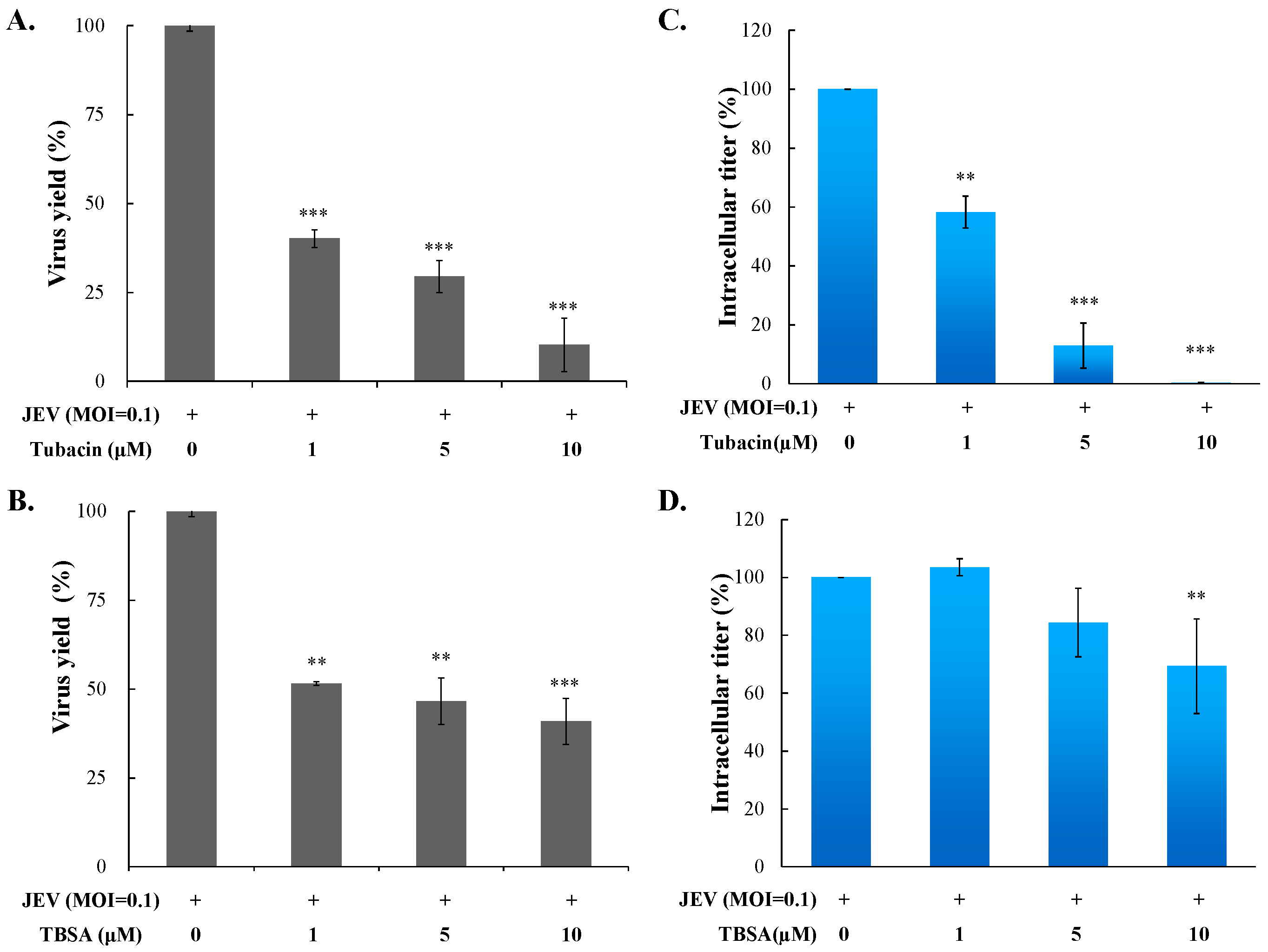
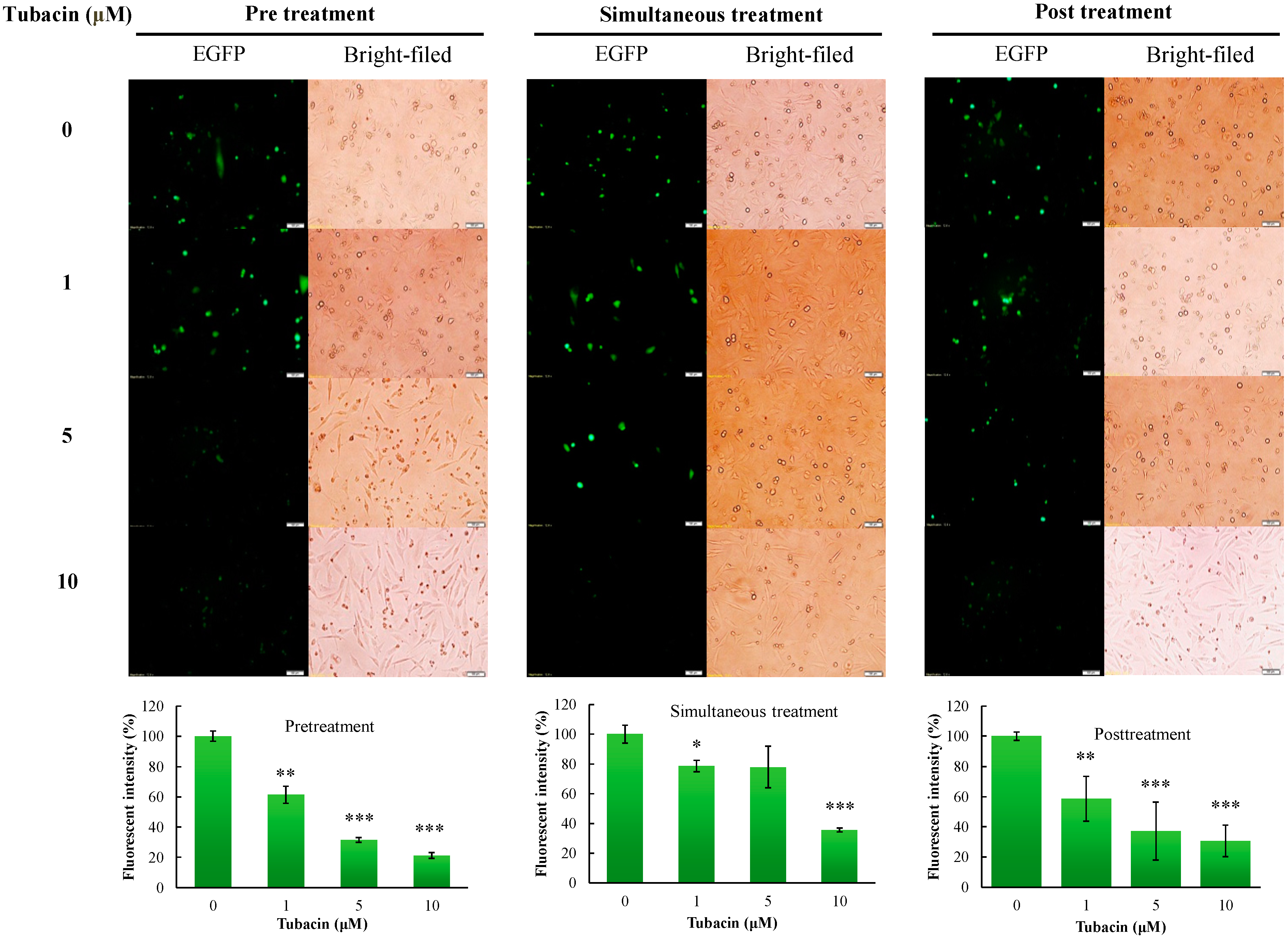
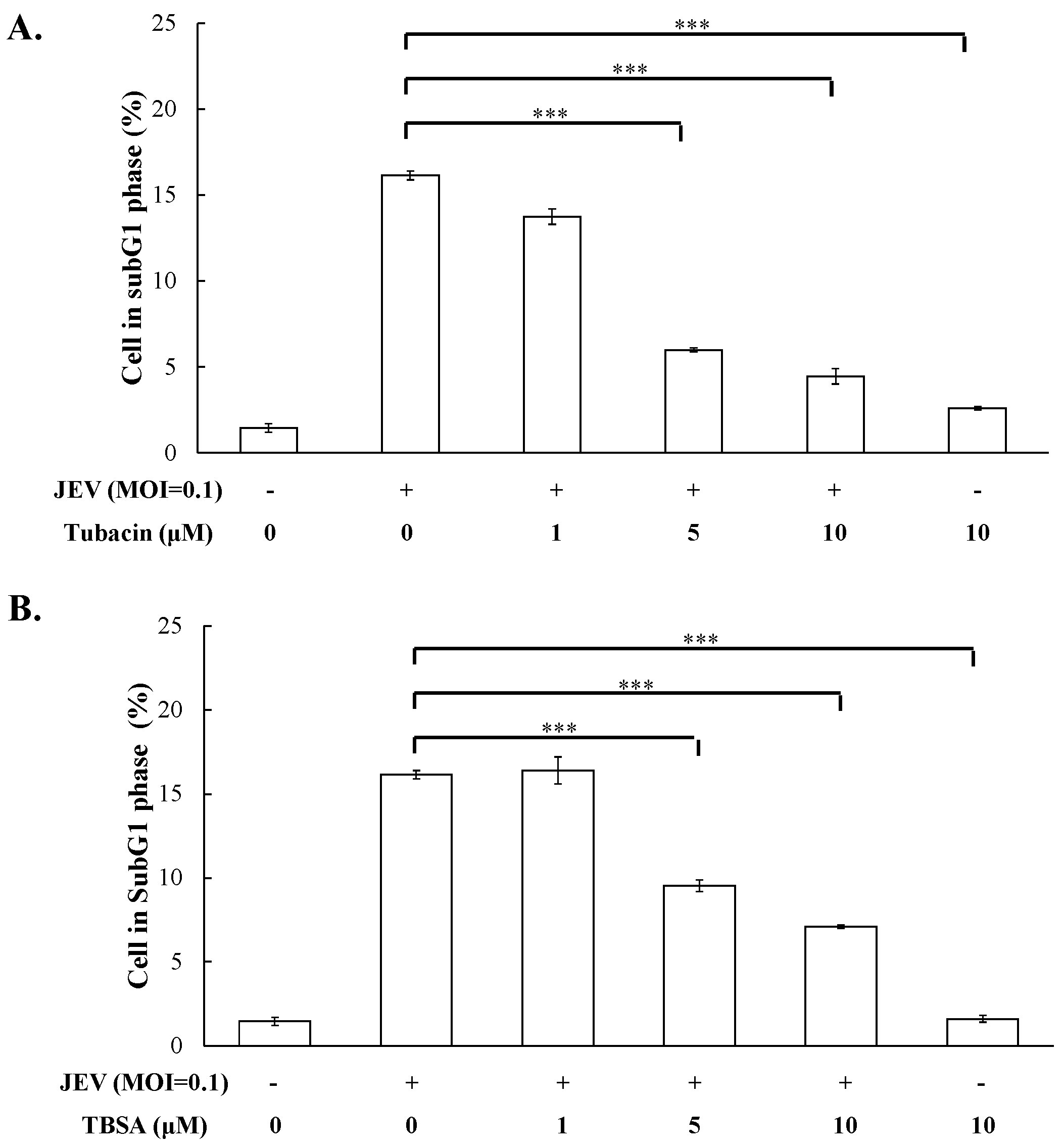
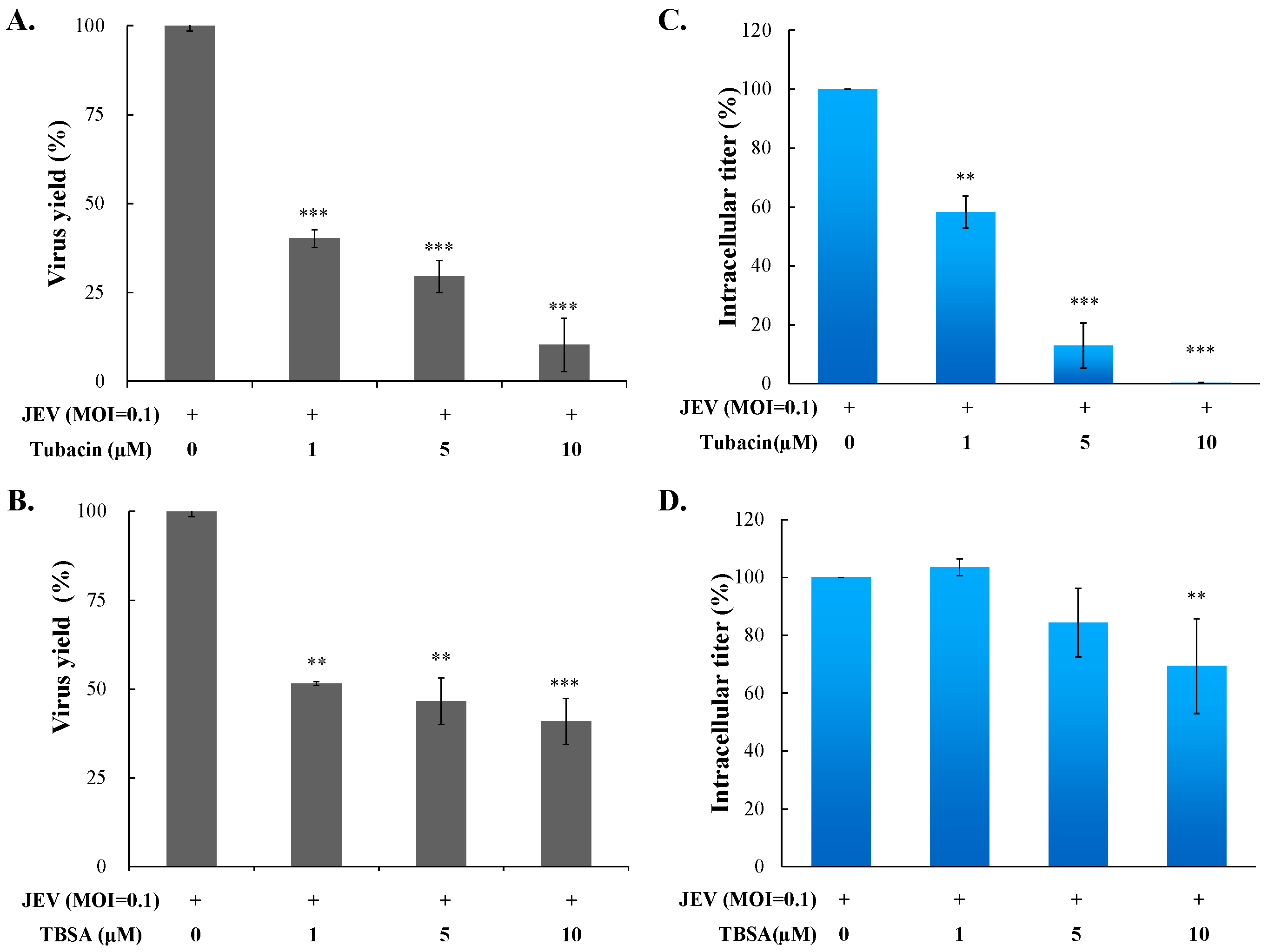
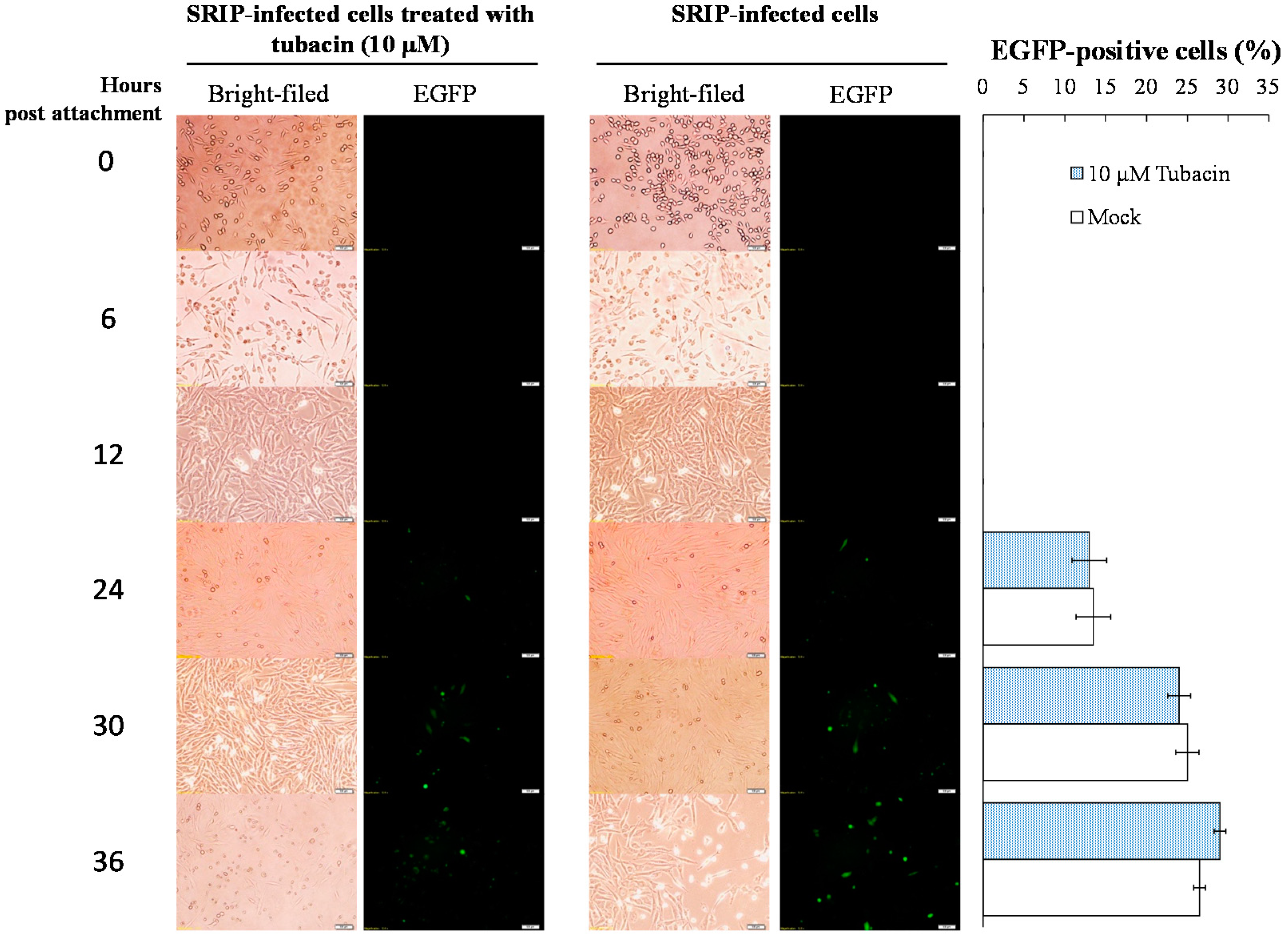
2.2. Preventive and Therapeutic Activities of Tubacin against JEV Infection
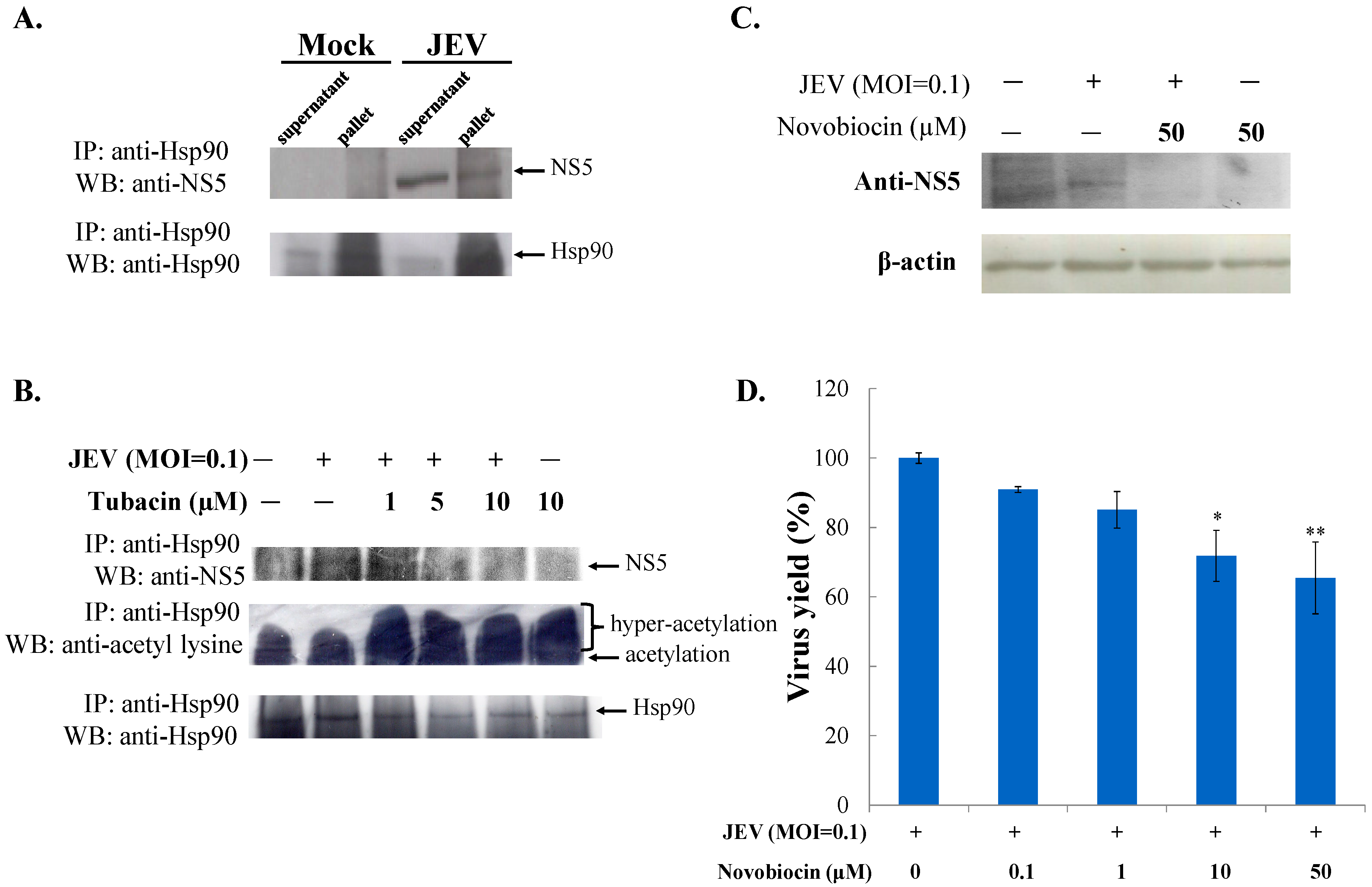
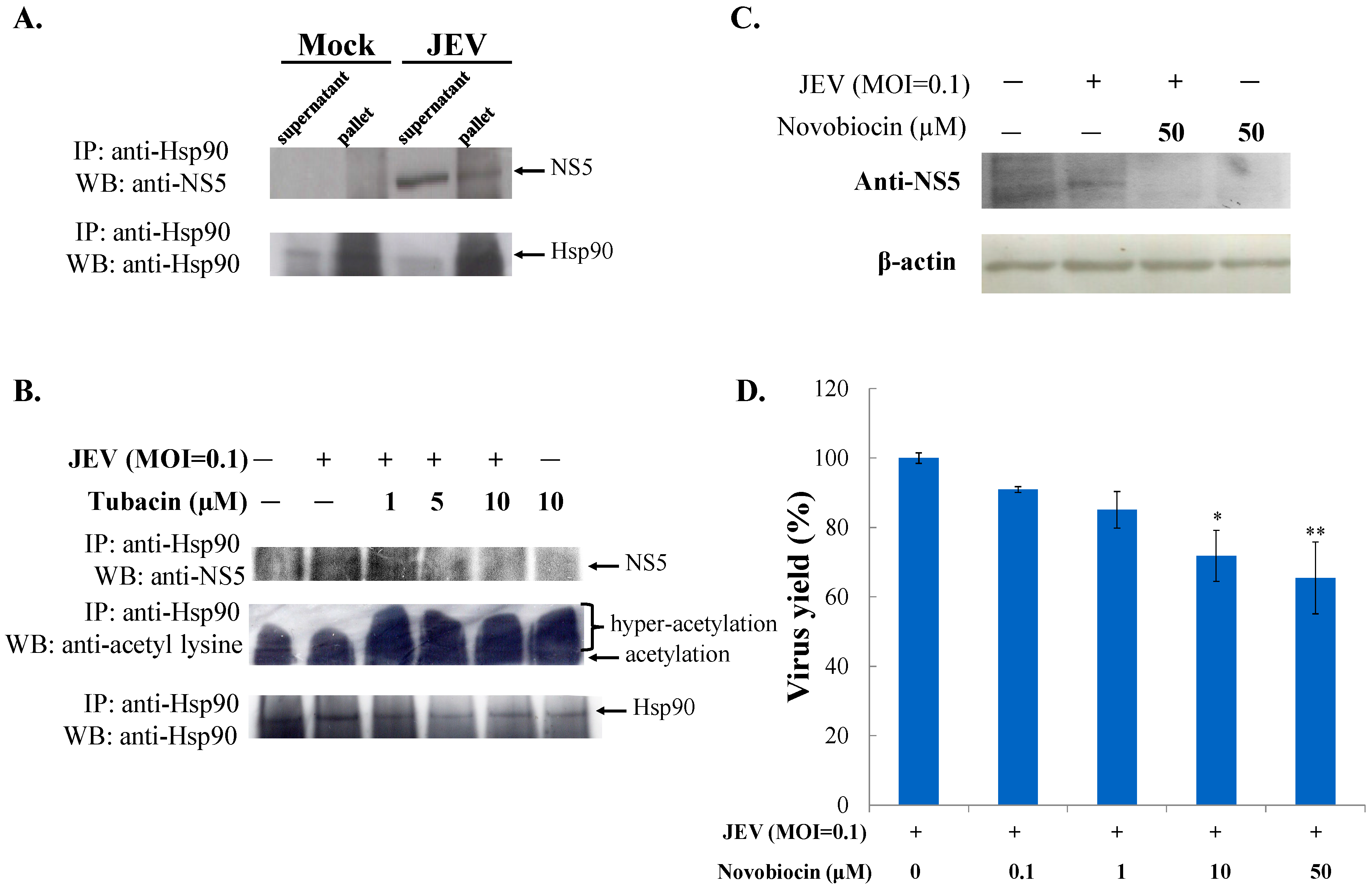
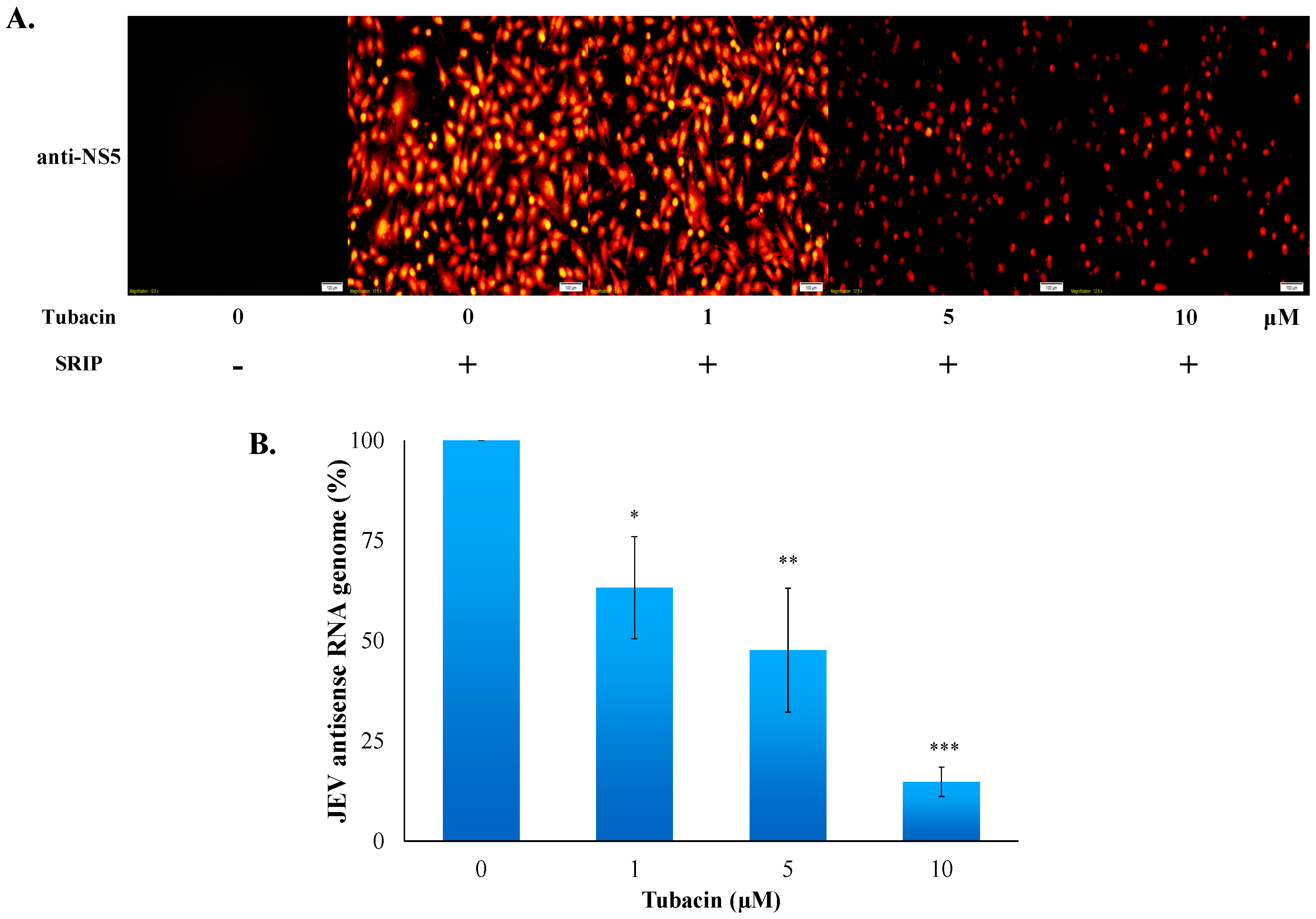
2.3. Tubacin-Induced Hsp90 Hyperacetylation Was Associated with the Reduction of NS5 RNA Polymerase Activity
3. Discussion
4. Methods and Materials
4.1. Cells and Virus
4.2. MTT Cytotoxicity Test
4.3. Inhibitory Assays of HDACi on JEV-Induced Cytopathic Effect and Apoptosis
4.4. Quantitative Assays of Virus Yield and Intracellular Viral Titer
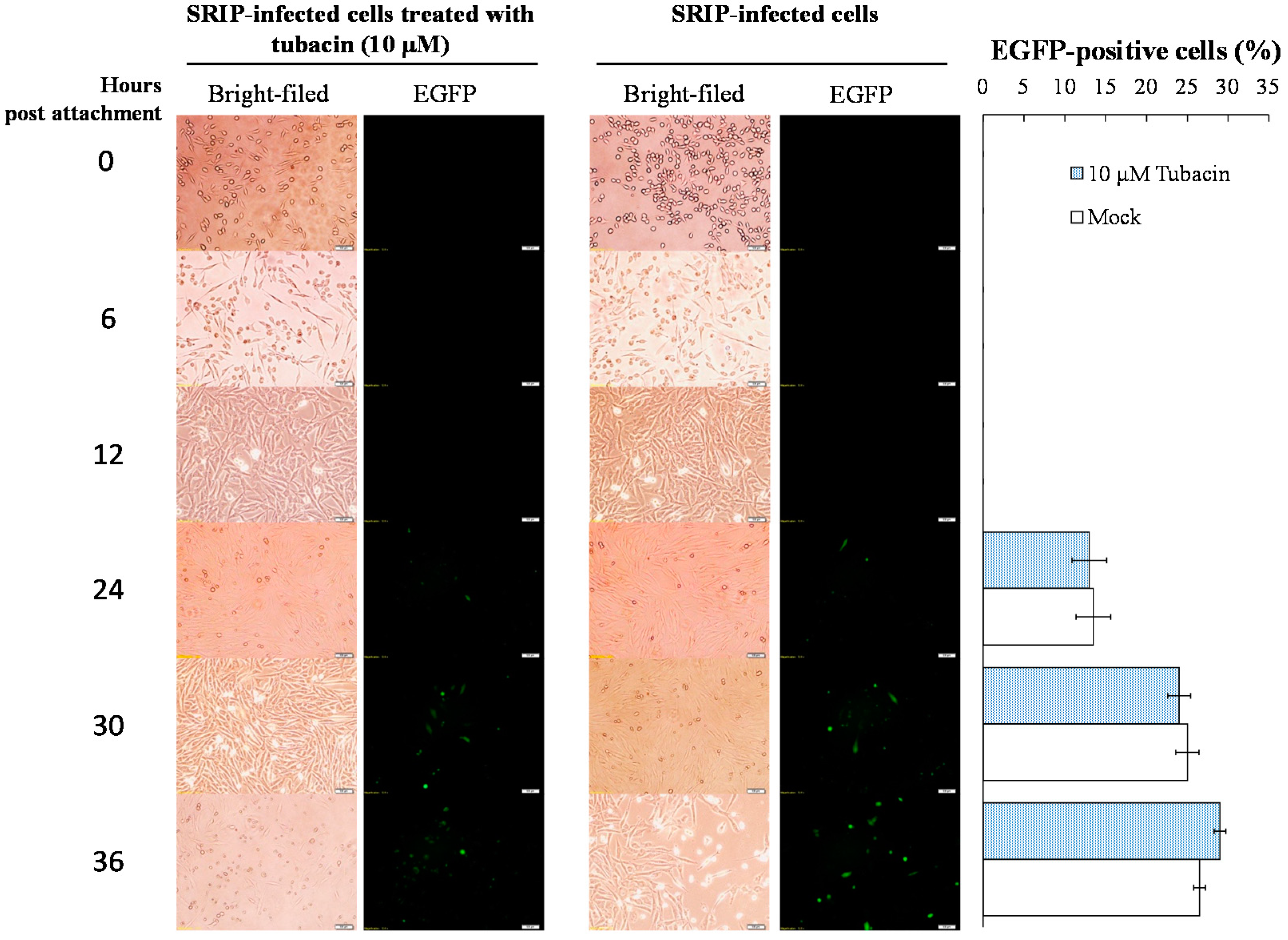
4.5. Virus Attachment Assays with Single Run Infectious Particles (SRIPs) and Virions
4.6. Time-of-Addition Assay
4.7. Detection of Viral NS5 Expression Using Immnunofluorescence
4.8. Quantification of Replicon RNA Expression Using RT-PCR
4.9. Co-Immunoprecipitation and Western Blotting Assays
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Umenai, T.; Krzysko, R.; Bektimirov, T.A.; Assaad, F.A. Japanese encephalitis: Current worldwide status. Bull. World Health Organ. 1985, 63, 625–631. [Google Scholar] [PubMed]
- Van den Hurk, A.F.; Ritchie, S.A.; Mackenzie, J.S. Ecology and geographical expansion of Japanese encephalitis virus. Annu. Rev. Entomol. 2009, 54, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Reisen, W.K. Present and future arboviral threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar]
- Misra, U.K.; Kalita, J.; Goel, D.; Mathur, A. Clinical, radiological and neurophysiological spectrum of JEV encephalitis and other non-specific encephalitis during post-monsoon period in India. Neurol. India 2003, 51, 55–59. [Google Scholar] [PubMed]
- Liu, T.H.; Liang, L.C.; Wang, C.C.; Liu, H.C.; Chen, W.J. The blood-brain barrier in the cerebrum is the initial site for the Japanese encephalitis virus entering the central nervous system. J. Neurovirol. 2008, 14, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Tsai, T.H.; Chang, T.J.; Wong, M.L. Cloning and Sequencing of Complete cDNA of Japanese Encephalitis Virus YL Strain in Taiwan. Virus Genes. 2003, 26, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Kalita, J. Overview: Japanese encephalitis. Prog. Neurobiol. 2010, 91, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Pastorino, B.A.; Peyrefitte, C.N.; Rolland, D.; Grandadam, M.; Tolou, H.J. Functional characterization of the NS2B/NS3 protease complex from seven viruses belonging to different groups inside the genus Flavivirus. Virus Res. 2006, 120, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Park, G.S.; Morris, K.L.; Hallett, R.G.; Bloom, M.E.; Best, S.M. Identification of residues critical for the interferon antagonist function of Langat virus NS5 reveals a role for the RNA-dependent RNA polymerase domain. J. Virol. 2007, 81, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.K.; Růžek, D.; Chhatbar, C.; Mishra, R.; Johri, M.K.; Singh, S.K. Japanese encephalitis virus: From genome to infectome. Microbes Infect. 2011, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Nishimura, Y.; Ichimura, T.; Suzuki, K.; Miyamura, T.; Suzuki, T.; Moriishi, K.; Matsuura, Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006, 25, 5015–5025. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Krämer, O.H. HDAC2: A critical factor in health and disease. Trends Pharmacol. Sci. 2009, 30, 647–655. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Gregoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Jiang, F.; Loo, Y.M.; Hsu, S.; Hsiang, T.Y.; Marcotrigiano, J.; Gale, M., Jr. Regulation of Retinoic Acid Inducible Gene-I (RIG-I) Activation by the Histone Deacetylase 6. EBioMedicine 2016, 9, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone Deacetylase Inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Shapira, L.; Ralph, M.; Tomer, E.; Cohen, S.; Kobiler, O. Histone Deacetylase Inhibitors Reduce the Number of Herpes Simplex Virus-1 Genomes Initiating Expression in Individual Cells. Front. Microbiol. 2016, 7, 1970. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Gélinas, C.; Dougherty, J.P. HDAC Inhibitors Containing a Benzamide Functional Group and a Pyridyl Cap are Preferentially Effective HIV-1 Latency Reversing Agents in Primary Resting CD4+ T Cells. J Gen. Virol. 2017. [Google Scholar] [CrossRef]
- Kozlov, M.V.; Kleymenova, A.A.; Konduktorov, K.A.; Malikova, A.Z.; Kochetkov, S.N. Selective inhibitor of histone deacetylase 6 (tubastatin A) suppresses proliferation of hepatitis Cvirus replicon in culture of human hepatocytes. Biochemistry (Mosc.) 2014, 79, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.H.; Tao, Y.; Zhang, Z.Z.; Chen, W.X.; Cai, X.F.; Chen, K.; Ko, B.C.; Song, C.L.; Ran, L.K.; Li, W.Y.; et al. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor AP-1. J. Virol. 2014, 88, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Taguwa, S.; Frydman, J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 2012, 1823, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Ren, Y.; Mohapatra, A.; Bali, P.; Mandawat, A.; Rao, R.; Herger, B.; Yang, Y.; Atadja, P.; Wu, J.; et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-α levels and transcriptional activity: A result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 2007, 13, 4882–4890. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV andsensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Su, Z.; Song, S.; Χu, H.; Zhang, B.; Yi, L.; Tian, M.; Wang, H. Histone deacetylase inhibitors suppress RSV infection and alleviate virus-induced airway inflammation. Int. J. Mol. Med. 2016, 38, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Cheung, C.Y. Histone deacetylase 6 inhibits influenza A virus release by down-regulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J. Virol. 2014, 88, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Panella, S.; Marcocci, M.E.; Celestino, I.; Valente, S.; Zwergel, C.; Li Puma, D.D.; Nencioni, L.; Mai, A.; Palamara, A.T.; Simonetti, G. MC1568 inhibits HDAC6/8 activity and influenza A virus replication in lung epithelial cells: Role ofHsp90 acetylation. Future Med. Chem. 2016, 8, 2017–2031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ogden, A.; Aneja, R.; Zhou, J. Diverse roles of HDAC6 in viral infection: Implications for antiviral therapy. Pharmacol. Ther. 2016, 164, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.Y.; Hour, M.-J.; Wang, C.-Y.; Huang, S.-H.; Mu, W.-X.; Chang, Y.-C.; Lin, C.-W. Single-Round Infectious Particle Antiviral Screening Assays for the Japanese Encephalitis Virus. Viruses 2017, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Lien, J.C.; Chen, C.J.; Liu, Y.C.; Wang, C.Y.; Ping, C.F.; Lin, Y.F.; Huang, A.C.; Lin, C.W. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. Int. J. Mol. Sci. 2016, 17, 1386. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-Y.; Chang, Y.-C.; Hua, C.-H.; Chuang, C.; Huang, S.-H.; Kung, S.-H.; Hour, M.-J.; Lin, C.-W. Tubacin, an HDAC6 Selective Inhibitor, Reduces the Replication of the Japanese Encephalitis Virus via the Decrease of Viral RNA Synthesis. Int. J. Mol. Sci. 2017, 18, 954. https://doi.org/10.3390/ijms18050954
Lu C-Y, Chang Y-C, Hua C-H, Chuang C, Huang S-H, Kung S-H, Hour M-J, Lin C-W. Tubacin, an HDAC6 Selective Inhibitor, Reduces the Replication of the Japanese Encephalitis Virus via the Decrease of Viral RNA Synthesis. International Journal of Molecular Sciences. 2017; 18(5):954. https://doi.org/10.3390/ijms18050954
Chicago/Turabian StyleLu, Chien-Yi, Yi-Chih Chang, Chun-Hung Hua, Chieh Chuang, Su-Hua Huang, Szu-Hao Kung, Mann-Jen Hour, and Cheng-Wen Lin. 2017. "Tubacin, an HDAC6 Selective Inhibitor, Reduces the Replication of the Japanese Encephalitis Virus via the Decrease of Viral RNA Synthesis" International Journal of Molecular Sciences 18, no. 5: 954. https://doi.org/10.3390/ijms18050954