A Quantitative Acetylomic Analysis of Early Seed Development in Rice (Oryza sativa L.)


Abstract
:1. Introduction
2. Results
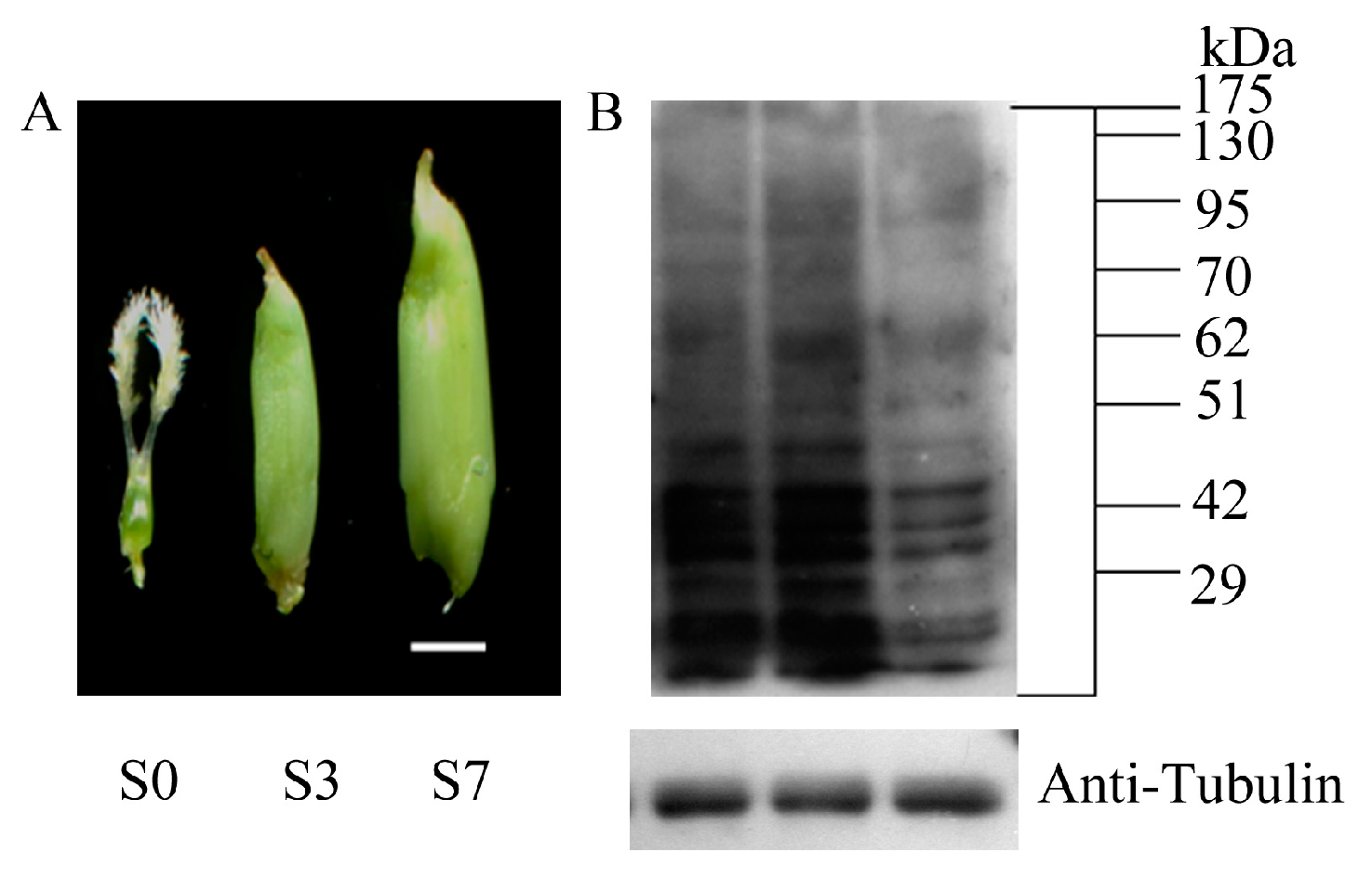
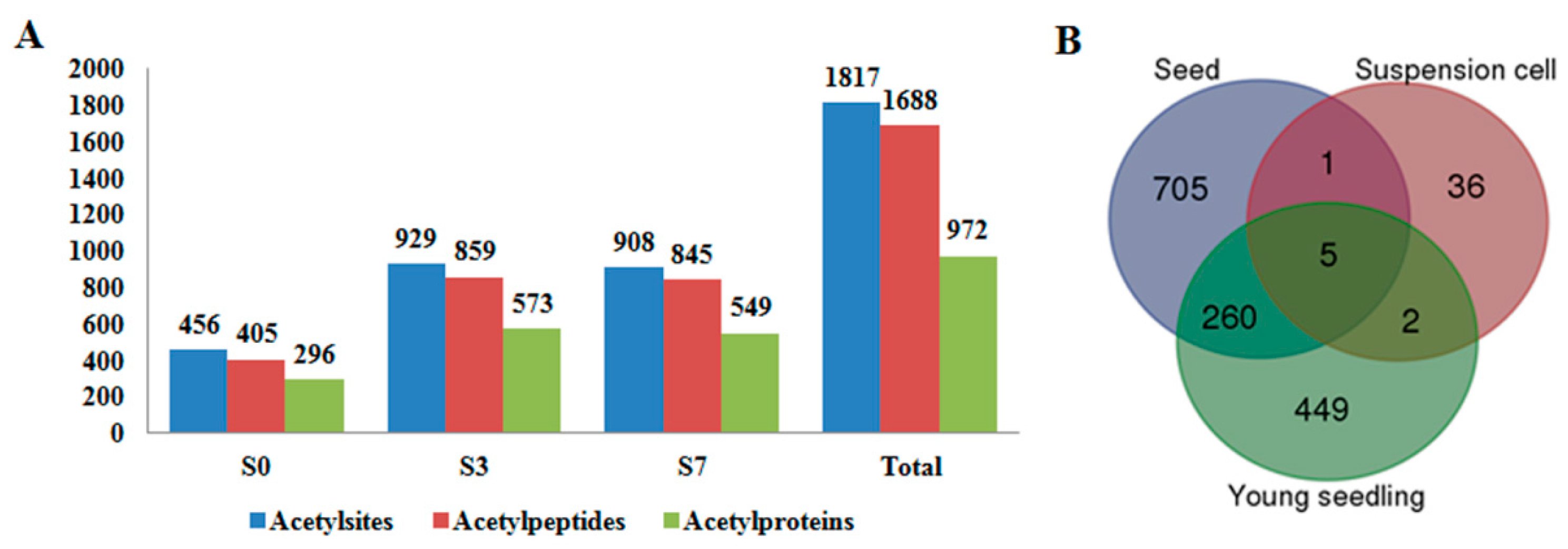
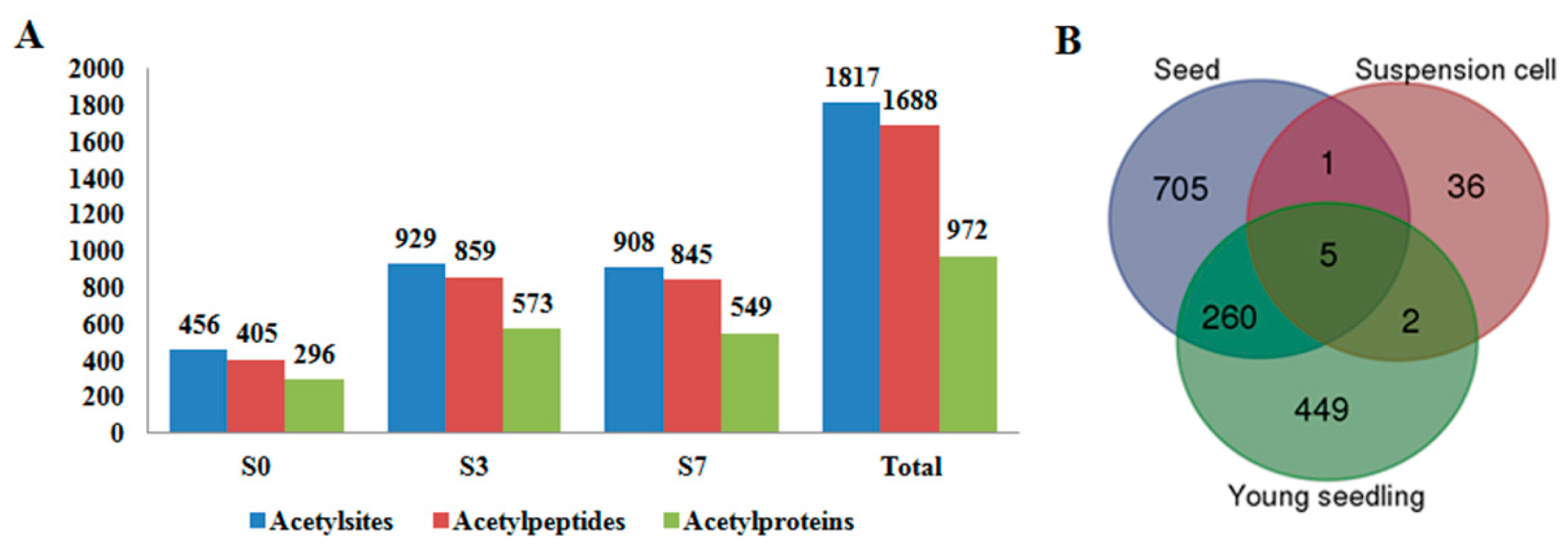
2.1. Global Profiling of PKA (Protein Lysine Acetylation) in Rice Pistil and Developing Seeds
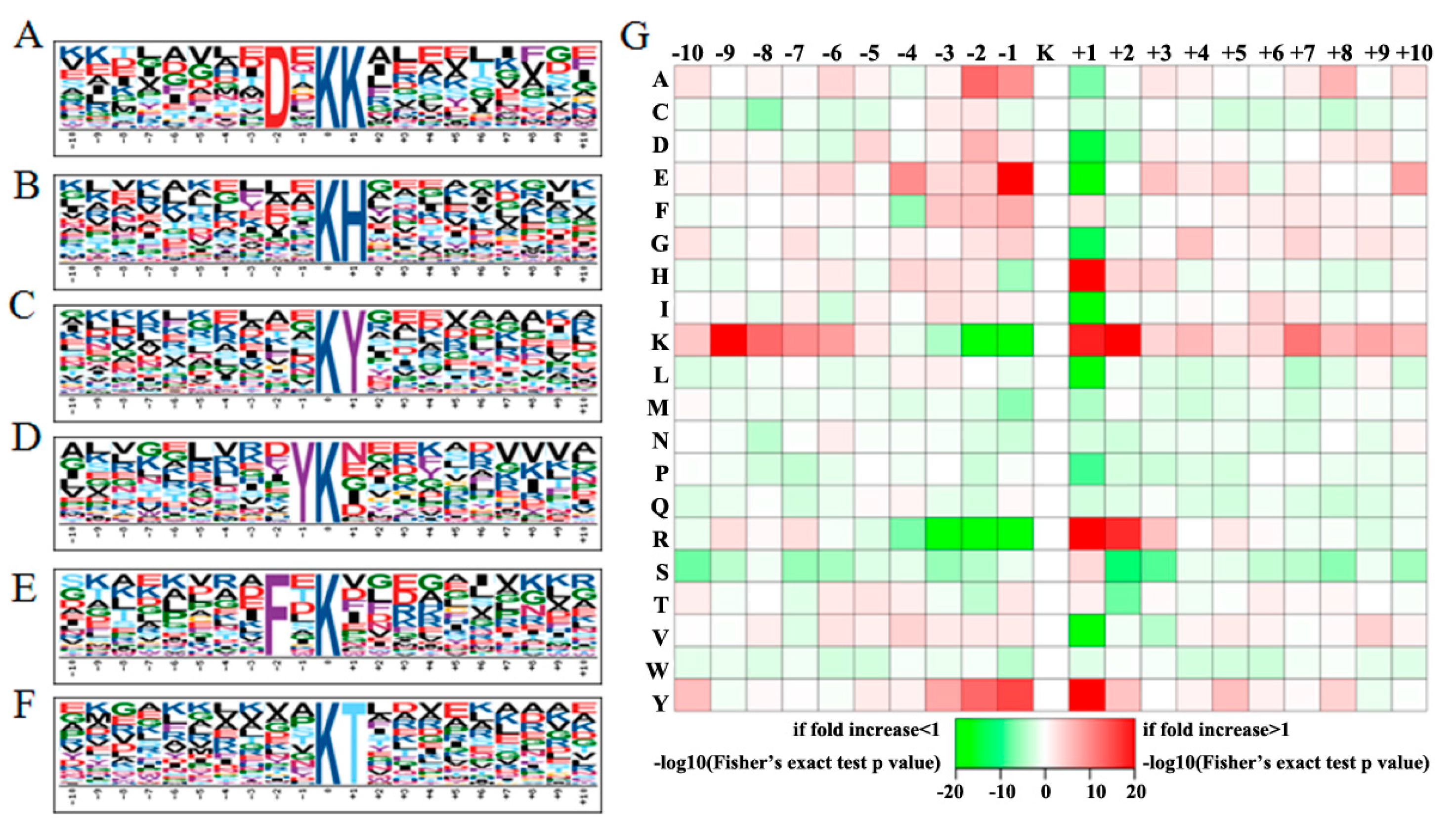
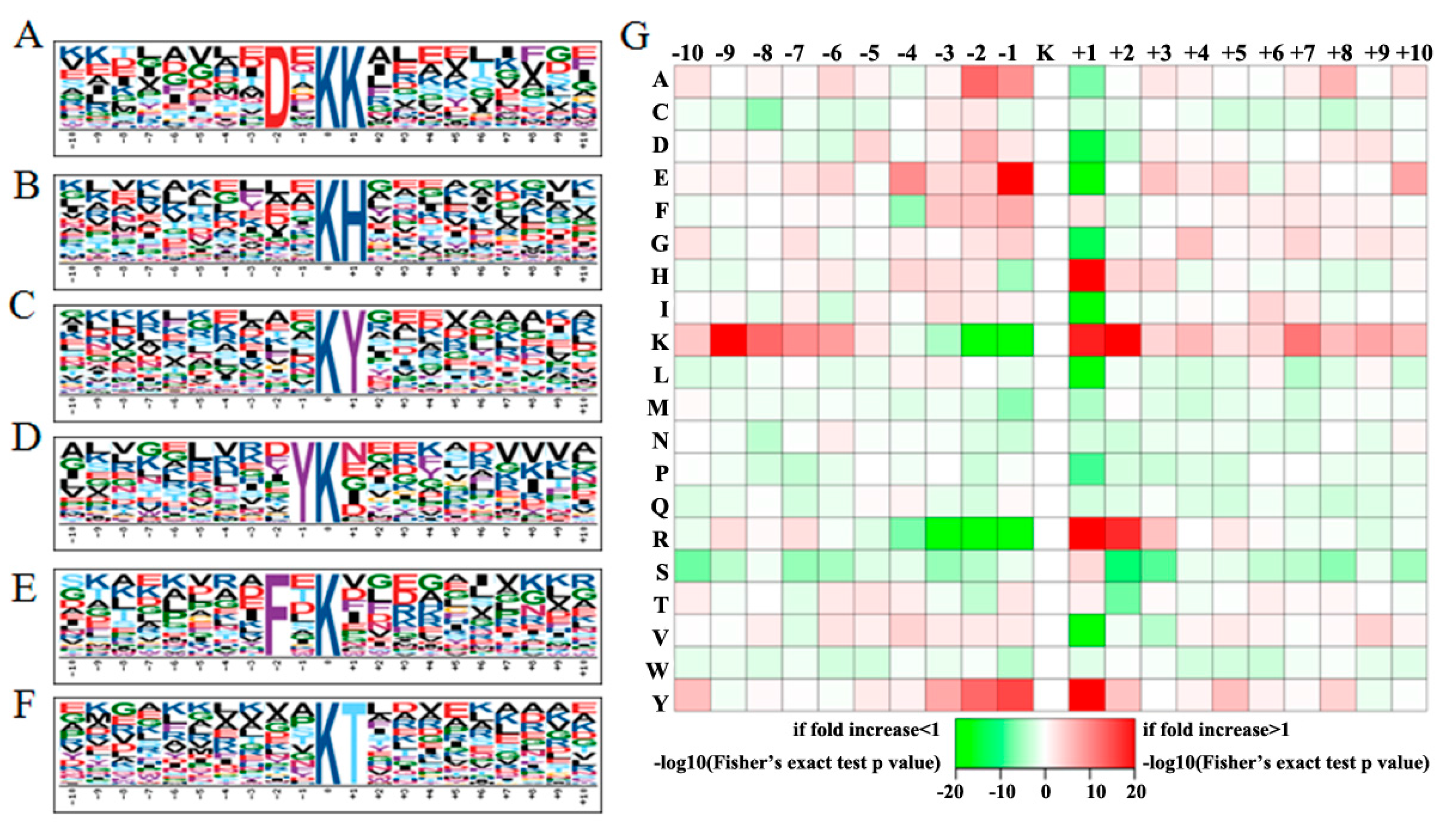
2.2. Conserved Motifs Flanking the Acetylsites
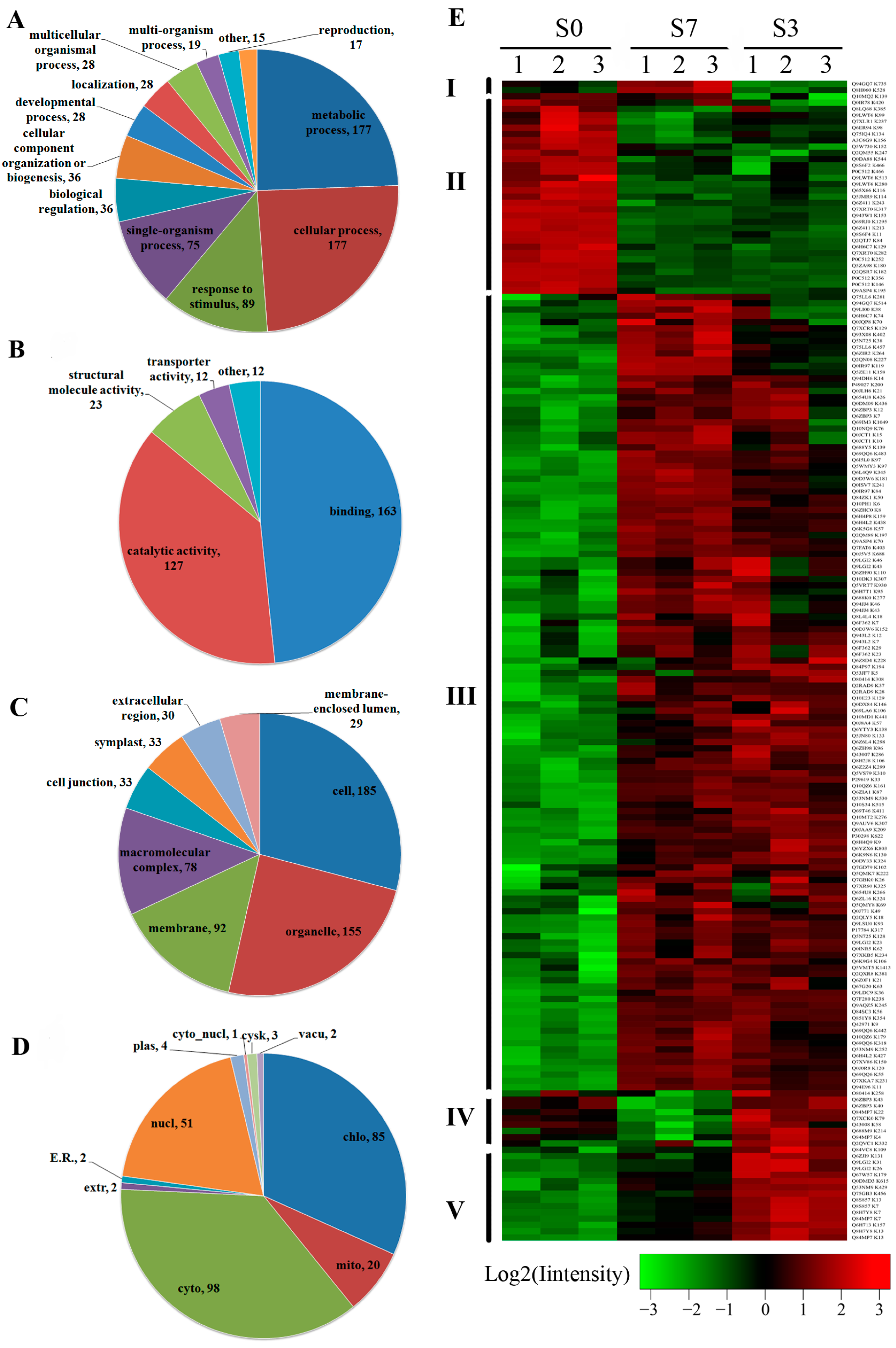
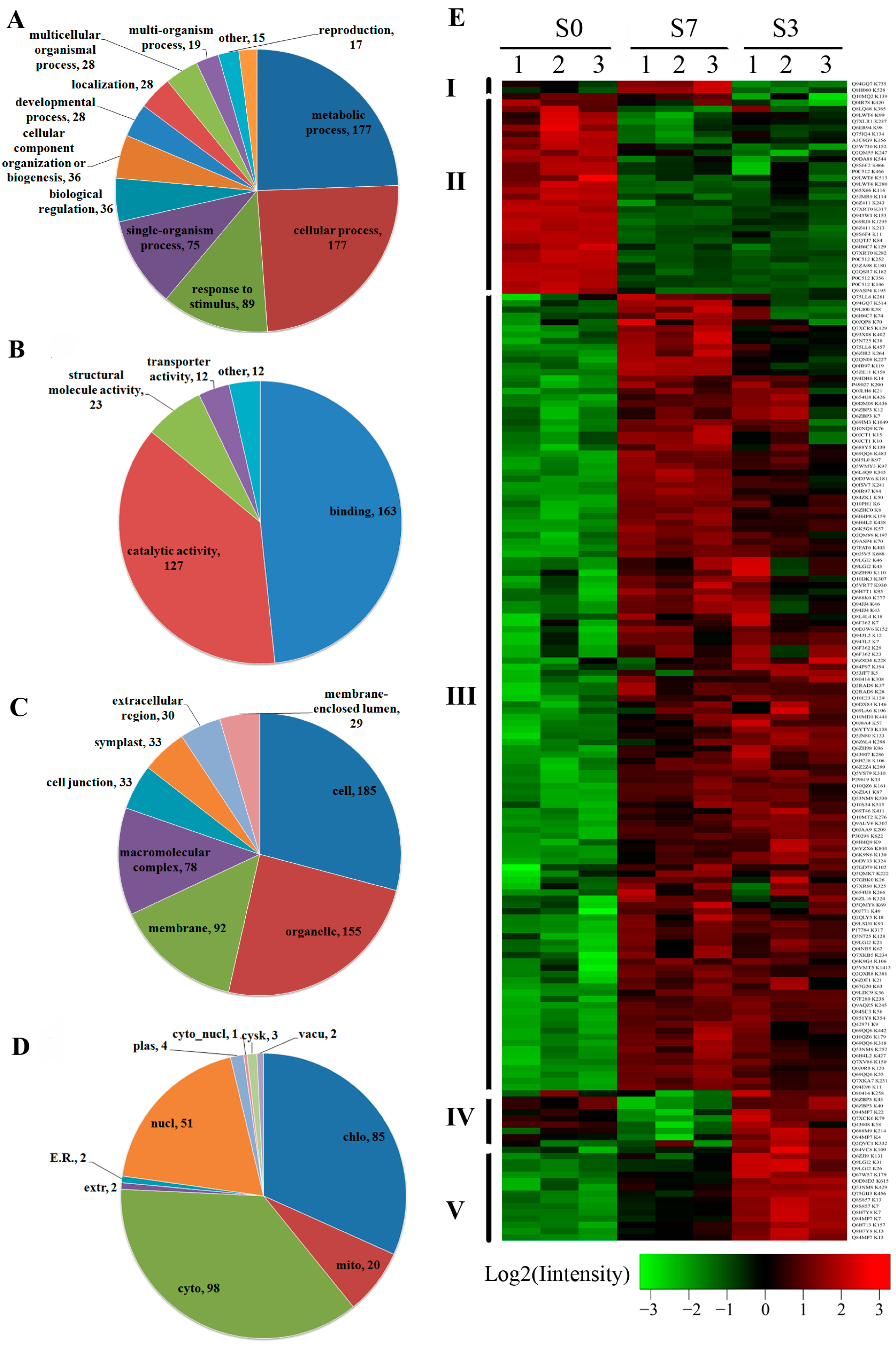
2.3. Differentially Acetylated (DA) Proteins in Rice Developing Seeds
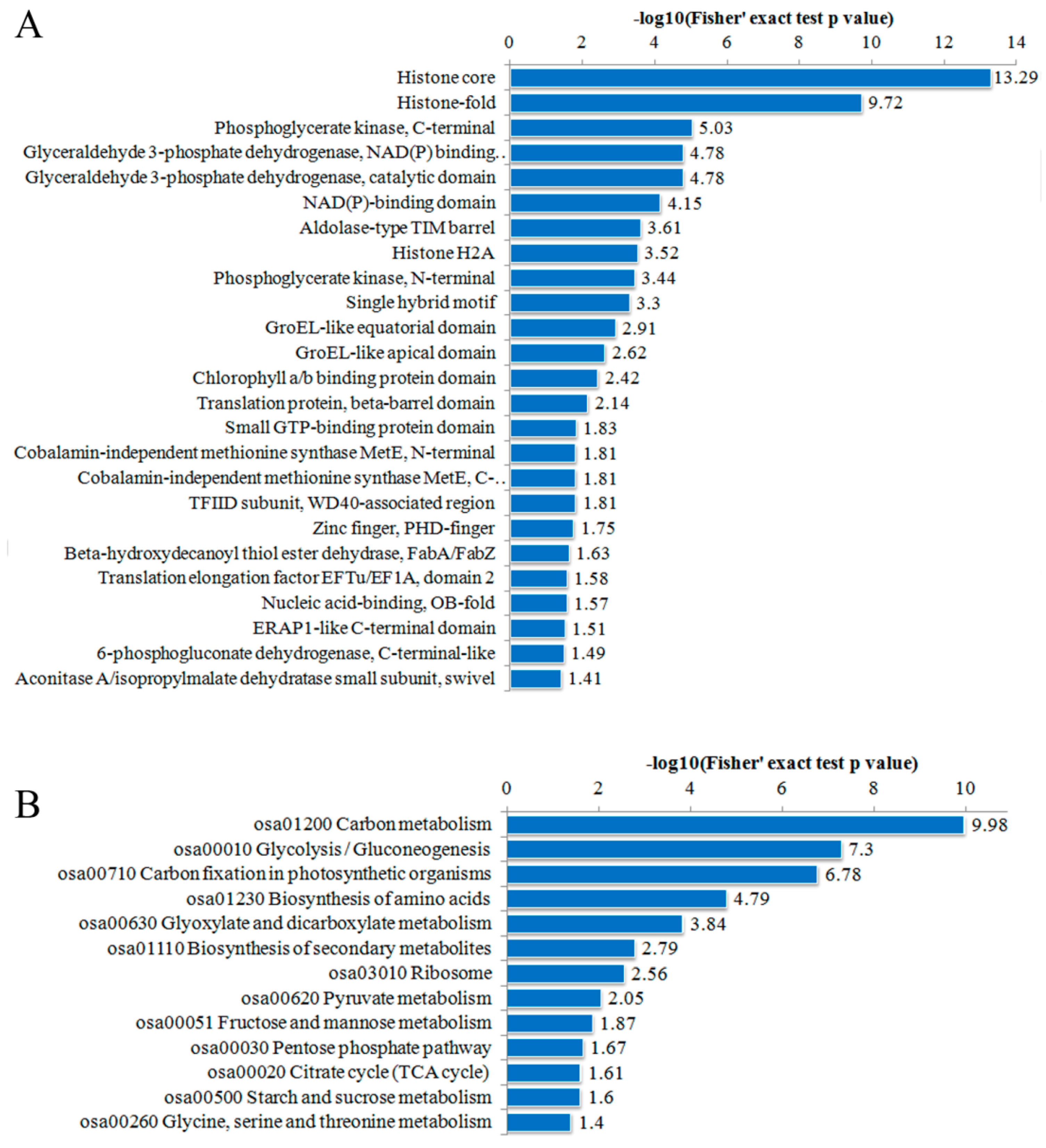
2.4. Functional Domain and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis of the DA Proteins
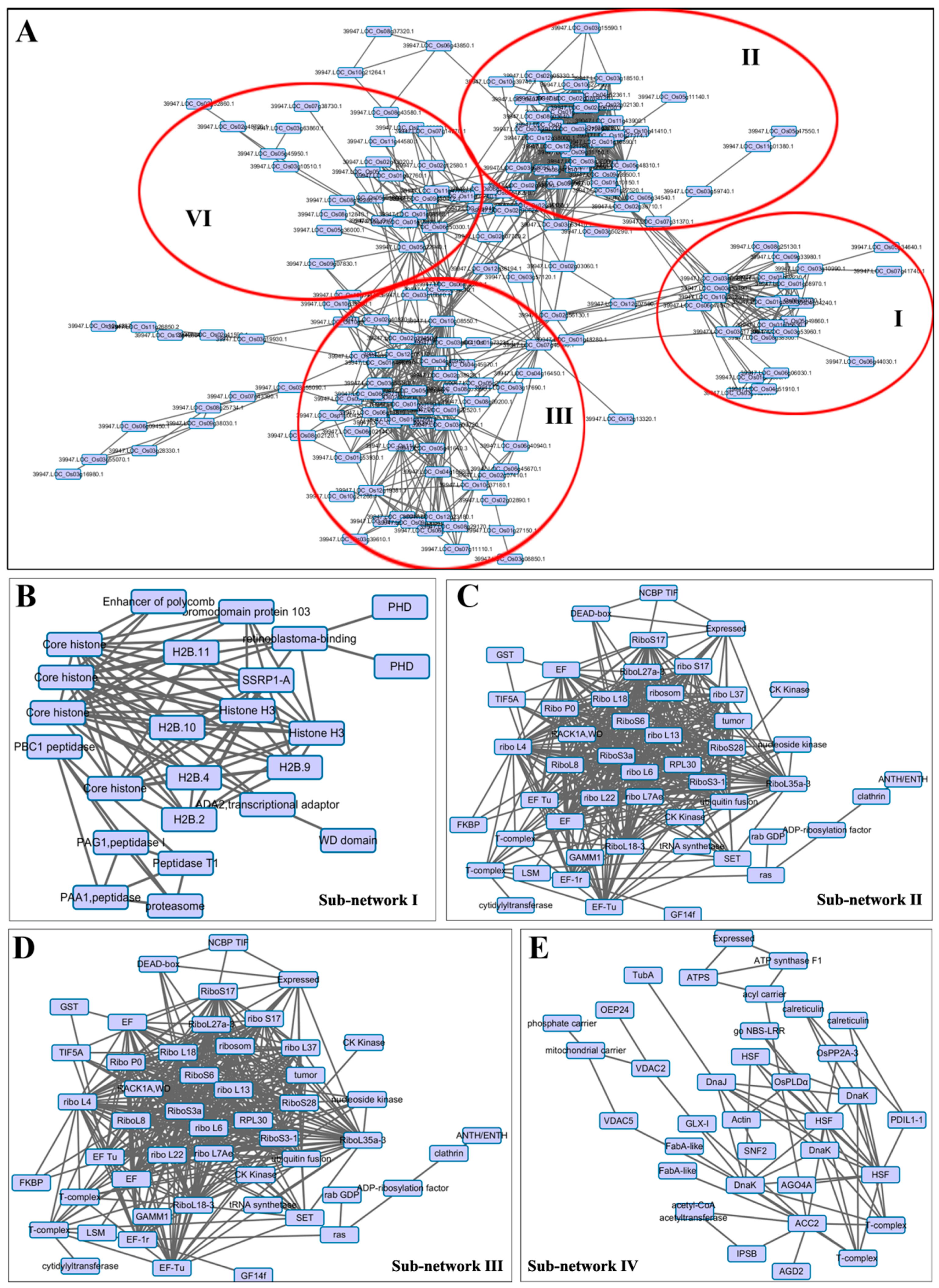
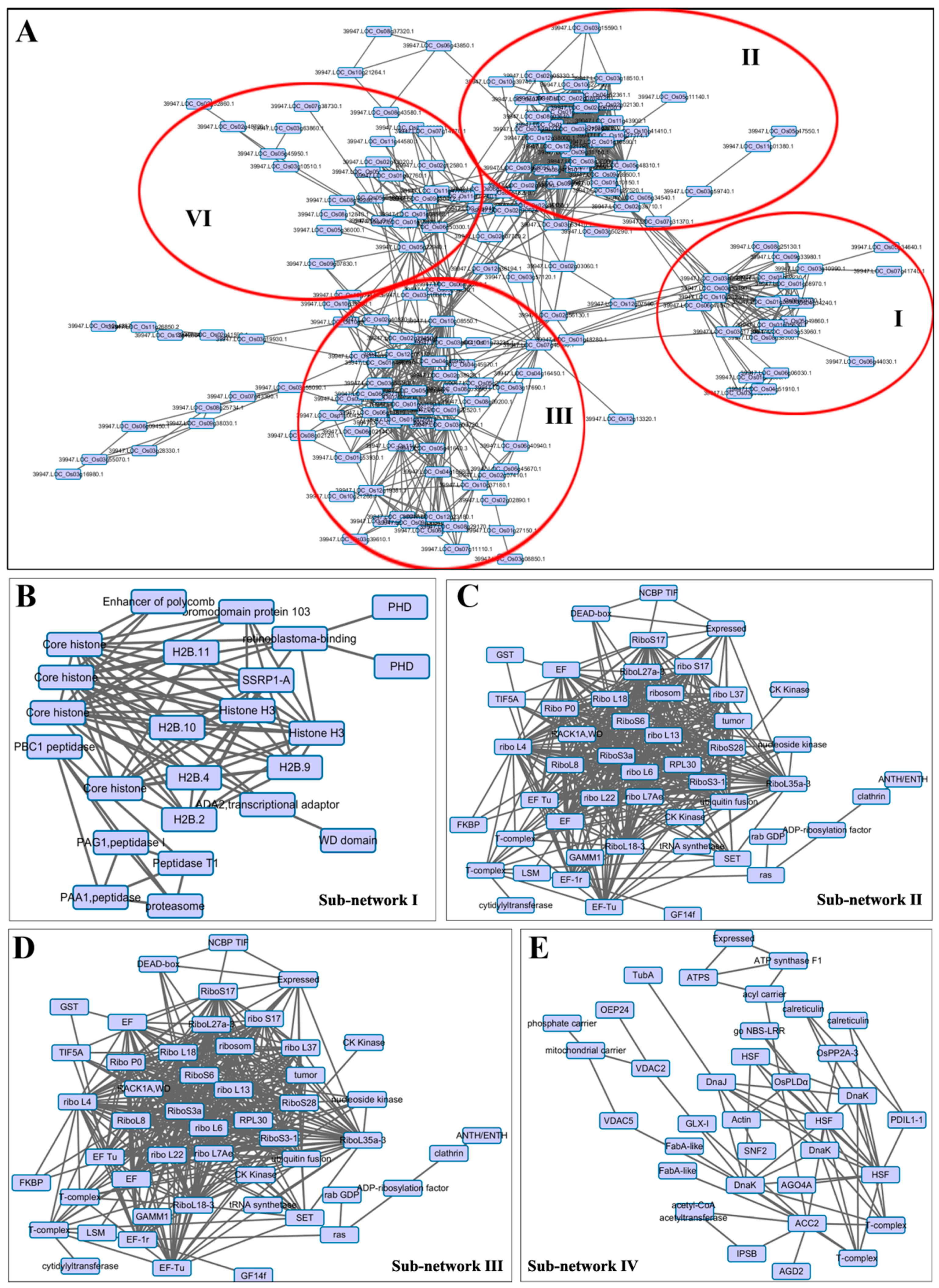
2.5. Protein–Protein Interaction (PPI) Analysis of DA Proteins
3. Discussion
4. Materials and Methods
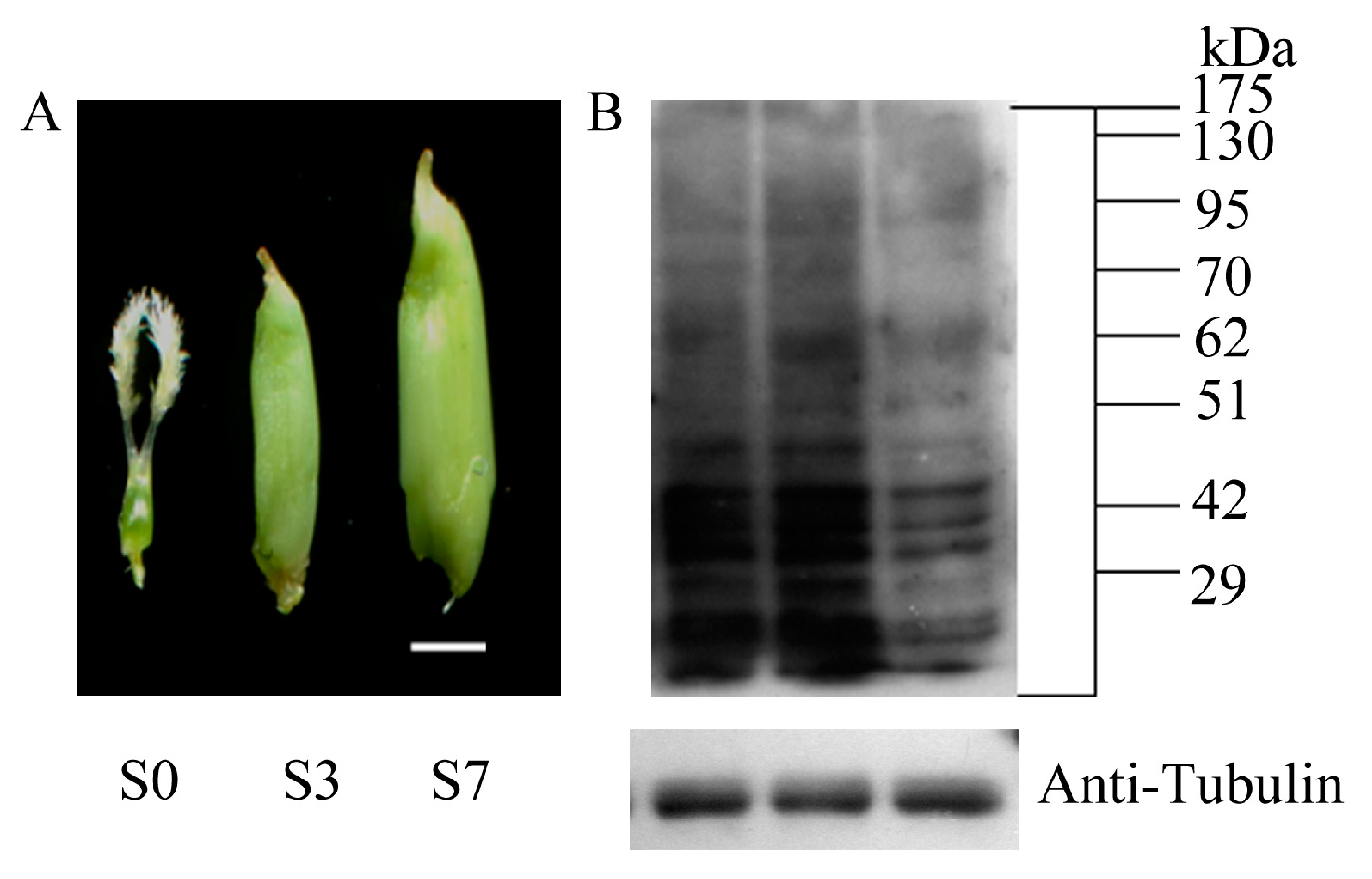
4.1. Collection and Preparation of Plant Materials
4.2. Protein Extraction
4.3. Western Blotting
4.4. Protein Digestion and Acetylpeptide Enrichment
4.5. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Analysis and Database Search
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, K.Y.; Su, M.G.; Kao, H.J.; Hsieh, Y.C.; Jhong, J.H.; Cheng, K.H.; Huang, H.D.; Lee, T.Y. dbPTM 2016: 10-Year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 2016, 44, D435–D446. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.S.; Thelen, J.J.; Miernyk, J.A. Is Lys-Nε-acetylation the next big thing in post-translational modifications? Trends Plant Sci. 2014, 19, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wei, W.; Zhou, D.X. Histone Acetylation Enzymes Coordinate Metabolism and Gene Expression. Trends Plant Sci. 2015, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Poirier, Y. The protein acetylome and the regulation of metabolism. Trends Plant Sci. 2012, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Berr, A.; Shafiq, S.; Shen, W.H. Histone modifications in transcriptional activation during plant development. Biochim. Biophys. Acta 2011, 1809, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, D.; Chang, Y.; You, C.; Li, X.; Dai, X.; Weng, Q.; Zhang, J.; Chen, G.; Li, X.; et al. Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analyzing 13 804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant J. 2007, 49, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.Y.; Gong, C.Y.; Wang, T. Use of proteomics to understand seed development in rice. Proteomics 2013, 13, 1784. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.H.; Hou, Y.X.; Tong, X.H.; Wang, Y.F.; Lin, H.Y.; Liu, Q.; Zhang, W.; Li, Z.Y.; Nallamilli, B.R.; Zhang, J. Quantitative phosphoproteomic analysis of early seed development in rice (Oryza sativa L.). Plant Mol. Biol. 2016, 90, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, Y.; Zhao, Y.; Zhou, D.X. OsSRT1 is involved in rice seed development through regulation of starch metabolism gene expression. Plant Sci. 2016, 248, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, L.; Liang, W.; Mu, P.; Wang, S.; Lin, Q. Comprehensive profiling of lysine acetylproteome analysis reveals diverse functions of lysine acetylation in common wheat. Sci. Rep. 2016, 6, 21069. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Deng, X.; Wang, J.; Zhu, G.; Cao, H.; Yuan, L.; Yan, Y. First Comprehensive Proteome Analyses of Lysine Acetylation and Succinylation in Seedling Leaves of Brachypodium distachyon L. Sci. Rep. 2016, 6, 31576. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Chen, W.; Zhao, Y.; Ruan, S.; Zhang, H.; Yan, C.; Jin, L.; Cao, L.; Zhu, J.; Ma, H.; et al. Global analysis of lysine acetylation in strawberry leaves. Front. Plant Sci. 2015, 6, 739. [Google Scholar] [CrossRef] [PubMed]
- Finkemeier, I.; Laxa, M.; Miguet, L.; Howden, A.J.; Sweetlove, L.J. Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant physiol. 2011, 155, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Konig, A.C.; Hartl, M.; Boersema, P.J.; Mann, M.; Finkemeier, I. The mitochondrial lysine acetylome of Arabidopsis. Mitochondrion 2014, 19, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Melo-Braga, M.N.; Verano-Braga, T.; Leon, I.R.; Antonacci, D.; Nogueira, F.C.; Thelen, J.J.; Larsen, M.R.; Palmisano, G. Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Mol. Cell. Proteom. 2012, 11, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Nallamilli, B.R.; Edelmann, M.J.; Zhong, X.; Tan, F.; Mujahid, H.; Zhang, J.; Nanduri, B.; Peng, Z. Global analysis of lysine acetylation suggests the involvement of protein acetylation in diverse biological processes in rice (Oryza sativa). PLoS ONE 2014, 9, e89283. [Google Scholar] [CrossRef] [PubMed]
- Smith-Hammond, C.L.; Hoyos, E.; Miernyk, J.A. The pea seedling mitochondrial Nε-lysine acetylome. Mitochondrion 2014, 19, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Smith-Hammond, C.L.; Swatek, K.N.; Johnston, M.L.; Thelen, J.J.; Miernyk, J.A. Initial description of the developing soybean seed protein Lys-Nε-acetylome. J. Proteom. 2014, 96, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Peng, X.; Cheng, Z.; Liu, W.; Wang, G.L. A comprehensive catalog of the lysine-acetylation targets in rice (Oryza sativa) based on proteomic analyses. J. Proteom. 2016, 138, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Salvato, F.; Havelund, J.F.; Chen, M.; Rao, R.S.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Gang, D.R.; Thelen, J.J.; Moller, I.M. The potato tuber mitochondrial proteome. Plant Physiol. 2014, 164, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.F.; Schwartz, D. Biological sequence motif discovery using motif-X. Curr. Protoc. Bioinform. 2011, 13, 15–24. [Google Scholar]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Ohdan, T.; Francisco, P.B.; Sawada, J.T.; Hirose, T.; Terao, T.; Satoh, H.; Nakamura, Y. Expression profiling of genes involved in starch synthesis in sink and source organs of rice. J. Exp. Bot. 2005, 56, 3229–3244. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Hwang, S.K.; Han, M.; Eom, J.S.; Kang, H.G.; Han, Y.; Choi, S.B.; Cho, M.H.; Bhoo, S.H.; An, G.; et al. Identification of the ADP-glucose pyrophosphorylase isoforms essential for starch synthesis in the leaf and seed endosperm of rice (Oryza sativa L.). Plant Mol. Biol. 2007, 65, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, H.; Sugimoto, T.; Tanaka, K.; Kasai, Z. Biosynthesis of storage proteins in developing rice seeds. Plant Physiol. 1982, 70, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Cruz, M.; Crofts, A.J.; Takemoto-Kuno, Y.; Sugino, A.; Washida, H.; Crofts, N.; Okita, T.W.; Ogawa, M.; Satoh, H.; Kumamaru, T. Protein disulfide isomerase like 1–1 participates in the maturation of proglutelin within the endoplasmic reticulum in rice endosperm. Plant Cell Physiol. 2010, 51, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Onda, Y.; Nagamine, A.; Sakurai, M.; Kumamaru, T.; Ogawa, M.; Kawagoe, Y. Distinct Roles of Protein Disulfide Isomerase and P5 Sulfhydryl Oxidoreductases in Multiple Pathways for Oxidation of Structurally Diverse Storage Proteins in Rice. Plant Cell 2011, 23, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, J.; Li, M.; Fragner, L.; Weckwerth, W.; Yang, P. Metabolomic and Proteomic Profiles Reveal the Dynamics of Primary Metabolism during Seed Development of Lotus (Nelumbo nucifera). Front. Plant Sci. 2016, 7, 750. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Liu, S.J.; Song, S.Q.; Moller, I.M. Proteomics of seed development, desiccation tolerance, germination and vigor. Plant Physiol. Biochem. 2015, 86, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-I.; Ryoo, N.; Eom, J.-S.; Lee, D.-W.; Kim, H.-B.; Jeong, S.-W.; Lee, Y.-H.; Kwon, Y.-K.; Cho, M.-H.; Bhoo, S.H. Role of the Rice Hexokinases OsHXK5 and OsHXK6 as Glucose Sensors. Plant Physiol. 2009, 149, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Huq, E.; Grover, A.; Dennis, E.S.; Peacock, W.J.; Hodges, T.K. Characterization of pyruvate decarboxylase genes from rice. Plant Mol. Biol. 1996, 31, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Geigenberger, P. Response of plant metabolism to too little oxygen. Curr. Opin. Plant Biol. 2003, 6, 247–256. [Google Scholar] [CrossRef]
- Xu, S.B.; Li, T.; Deng, Z.Y.; Chong, K.; Xue, Y.; Wang, T. Dynamic proteomic analysis reveals a switch between central carbon metabolism and alcoholic fermentation in rice filling grains. Plant Physiol. 2008, 148, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Lv, D.; Yan, X.; Subburaj, S.; Ge, P.; Li, X.; Hu, Y.; Yan, Y. Proteome characterization of developing grains in bread wheat cultivars (Triticum aestivum L.). BMC Plant Biol. 2012, 12, 147. [Google Scholar] [CrossRef] [PubMed]
- Rolletschek, H.; Weschke, W.; Weber, H.; Wobus, U.; Borisjuk, L. Energy state and its control on seed development: Starch accumulation is associated with high ATP and steep oxygen gradients within barley grains. J. Exp. Bot. 2004, 55, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Govindjee, A. Role for a Light-Harvesting Antenna Complex of Photosystem II in Photoprotection. Plant Cell 2002, 14, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ohkubo, M.; Hatakeyama, H.; Ohashi, K.; Yoshizawa, R.; Kojima, S.; Hayakawa, T.; Yamaya, T.; Mae, T.; Makino, A. Increased Rubisco content in transgenic rice transformed with the “sense” rbcS gene. Plant Cell Physiol. 2007, 48, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Tissue | Acetylsites | Acetylproteins | Reference |
|---|---|---|---|---|
| Triticum aestivum | leaf | 416 | 277 | [14] |
| Oryza sativa | Suspension cell, young seedling | 1403 | 760 | [20,23] |
| Arabidopsis thaliana | Suspension cell, young seedling | 398 | 251 | [17,18] |
| Glycine max | developing seeds | 190 | 121 | [22] |
| Pisum sativum | seedling | 664 | 358 | [21] |
| Solanum tuberosum | tuber | 35 | 31 | [24] |
| Fragaria X ananassa | leaf | 1392 | 684 | [16] |
| Vitis vinifera | mesocarp and exocarp | 138 | 97 | [19] |
| Brachypodium distachyon | leaf | 636 | 353 | [15] |
| Oryza sativa | pistil and developing seeds | 1817 | 972 | this study |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Hou, Y.; Qiu, J.; Li, Z.; Zhao, J.; Tong, X.; Zhang, J. A Quantitative Acetylomic Analysis of Early Seed Development in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2017, 18, 1376. https://doi.org/10.3390/ijms18071376
Wang Y, Hou Y, Qiu J, Li Z, Zhao J, Tong X, Zhang J. A Quantitative Acetylomic Analysis of Early Seed Development in Rice (Oryza sativa L.). International Journal of Molecular Sciences. 2017; 18(7):1376. https://doi.org/10.3390/ijms18071376
Chicago/Turabian StyleWang, Yifeng, Yuxuan Hou, Jiehua Qiu, Zhiyong Li, Juan Zhao, Xiaohong Tong, and Jian Zhang. 2017. "A Quantitative Acetylomic Analysis of Early Seed Development in Rice (Oryza sativa L.)" International Journal of Molecular Sciences 18, no. 7: 1376. https://doi.org/10.3390/ijms18071376