Ligand Shaping in Induced Fit Docking of MraY Inhibitors. Polynomial Discriminant and Laplacian Operator as Biological Activity Descriptors


Abstract
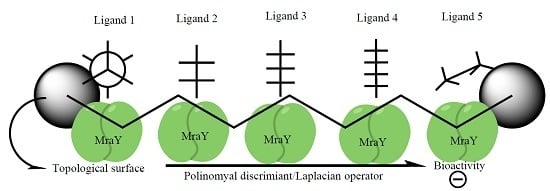
:
1. Introduction
2. Results
3. Discussion
4. Material and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chung, B.C.; Zhao, J.; Gillespie, R.A.; Kwon, D.-Y.; Guan, Z.; Hong, J.; Zhou, P.; Lee, S.-Y. Crystal Structure of MraY, an Essential Membrane Enzyme for Bacterial Cell Wall Synthesis. Science 2013, 341, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.; Lloyd, A.J.; Roper, D.I. Phospho-MurNAc-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect. Disord. Drug Targets 2006, 6, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Breukink, E. The Membrane Steps of Bacterial Cell Wall Synthesis as Antibiotic Targets. Antibiotics 2016, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Ganzha, V.G.; Mayr, E.W.; Vorozhtsov, E.V. (Eds.) Computer Algebra in Scientific Computing; CASC 2000; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Arfken, G. Mathematical Methods for Physicists, 3rd ed.; Academic Press: Orlando, FL, USA, 1985. [Google Scholar]
- Abrams, L.; Fishkind, D.E.; Priebe, C.E. A proof of the spherical homeomorphism conjecture for surfaces. IEEE Trans. Med. Imaging 2002, 21, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Boundary Conditions in Fluid Mechanics. Available online: http://web2.clarkson.edu/projects/subramanian/ch560/notes/Boundary%20Conditions%20in%20Fluid%20Mechanics.pdf (accessed on 23 June 2017).
- Rostami, M.; Michailovich, O.V.; Wang, Z. Surface reconstruction in gradient-field domain using compressed sensing. IEEE Trans. Image Process. 2015, 24, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Kuptsov, L.P. Gradient, Series: Encyclopedia of Mathematics; Hazewinkel, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Abrams, L.; Fishkind, D.E.; Priebe, C.E. The generalized spherical homeomorphism theorem for digital images. IEEE Trans. Med. Imaging 2004, 23, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Zarbafian, S.; Villar, E.; Mottarella, S.; Beglov, D.; Vajda, S.; Paschalidis, I.C.; Vakili, P.; Kozakov, D. Energy minimization on manifolds for docking flexible molecules. J. Chem. Theory Comput. 2015, 11, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Manin, Y.I.; Panchishkin, A.A. Introduction to Modern Number Theory, 2nd ed.; Series: Encyclopaedia of Mathematical Sciences; Springer: Berlin/Heidelberg, Germany, 2006; Volume 49, p. 130. [Google Scholar]
- Akritas, A.G. Elements of Computer Algebra with Applications; Wiley: New York, NY, USA, 1989. [Google Scholar]
- Kirby, R.C.; Siebenmann, L.C. Foundational Essays on Topological Manifolds. Smoothings, and Triangulations, a Detailed Study of the Category of Topological Manifolds; Princeton University Press: Princeton, NJ, USA, 1977. [Google Scholar]
- Hirano, S.; Ichikawa, S.; Matsuda, A. Synthesis of Caprazamycin Analogues and Their Structure-Activity Relationship for Antibacterial Activity. J. Org. Chem. 2008, 73, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, Y.; Hayashi, C.; Inoue, K.; Igarashi, M.; Takahashi, Y.; Pujari, V.; Crick, D.C.; Brennan, P.J.; Nomoto, A. Inhibition of the First Step in Synthesis of the Mycobacterial Cell Wall Core, Catalyzed by the GlcNAc-1-phosphate Transferase WecA, by the Novel Caprazamycin Derivative CPZEN-45. J. Biol. Chem. 2013, 88, 30309–30319. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.; Meier, P.S.; Heusser, R.; Berger-Bachi, B.; McCallum, N. Induction kinetics of the Staphylococcus aureus cell wall stress stimulon in response to different cell wall active antibiotics. BMC Microbiol. 2011, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Gualerzi, C.O.; Brandi, L.; Fabbretti, A.; Pon, C.L. (Eds.) Antibiotics Targets, Mechanism and Resistance; Wiley-Blackwell: New York, NY, USA, 2013; p. 137. [Google Scholar]
- Yu, Q.; Ding, X.; Xu, N.; Cheng, X.; Qian, K.; Zhang, B.; Xing, L.; Li, M. In vitro activity of verapamil alone and in combination with fluconazole or tunicamycin against Candida albicans biofilms. Int. J. Antimicrob. Agents 2013, 41, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C. Antibiotics: Challenges, Mechanisms, Opportunities, 1st ed.; American Society for Microbiology (ASM) Press: Washington, DC, USA, 2016. [Google Scholar]
- Takahashi, Y.; Igarashi, M.; Miyake, T.; Soutome, H.; Ishikawa, K.; Komatsuki, Y.; Koyama, Y.; Nakagawa, N.; Hattori, S.; Inoue, K.; et al. Novel semisynthetic antibiotics from caprazamycins A–G: Caprazene derivatives and their antibacterial activity. J. Antibiot. 2013, 66, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Mitachi, K.; Aleiwi, B.A.; Schnider, C.M.; Siricilla, S.; Kurosu, M. Stereocontrolled Total Synthesis of Muraymycin D1 Having a Dual Mode of Action against Mycobacterium tuberculosis. J. Am. Chem. Soc. 2016, 138, 12975–12980. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, B.; Henry, X.; El Ghachi, M.; Auger, G.; Blanot, D.; Parquet, C.; Mengin-Lecreulx, D.; Bouhss, A. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 2008, 47, 8919–8928. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yi, F.; Tian, Q.; Dang, G.; Si, W.; Liu, S.; Yu, S. Targeting the gram-negative bacteria peptidoglycan synthetase MarY as a new approach for monoclonal antibody anti-bacterial activity. Hum. Vaccin. Immunother. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Mashalidis, E.H.; Tanino, T.; Kim, M.; Matsuda, A.; Hong, J.; Ichikawa, S.; Lee, S.Y. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 2016, 533, 557–560. [Google Scholar] [CrossRef] [PubMed]
- AutoDock4.2 User Guide. Available online: http://autodock.scripps.edu/faqs-help/manual/autodock-4-2-user-guide (accessed on 23 June 2017).
- Ramírez, D.; Caballero, J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.M. Handbook of Chemistry and Physics; CRC Press: Cleveland, OH, USA, 1974. [Google Scholar]
- Wiener, H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Schultz, H.P. Topological Organic Chemistry. 1. Graph Theory and Topological Indices of Alkanes. J. Chem. Inf. Comput. Sci. 1989, 29, 227–228. [Google Scholar] [CrossRef]
- Ahmad, I.; Aqil, F. New Strategies Combating Bacterial Infection; Wiley-Blackwell: New York, NY, USA, 2008; Volume 83. [Google Scholar]
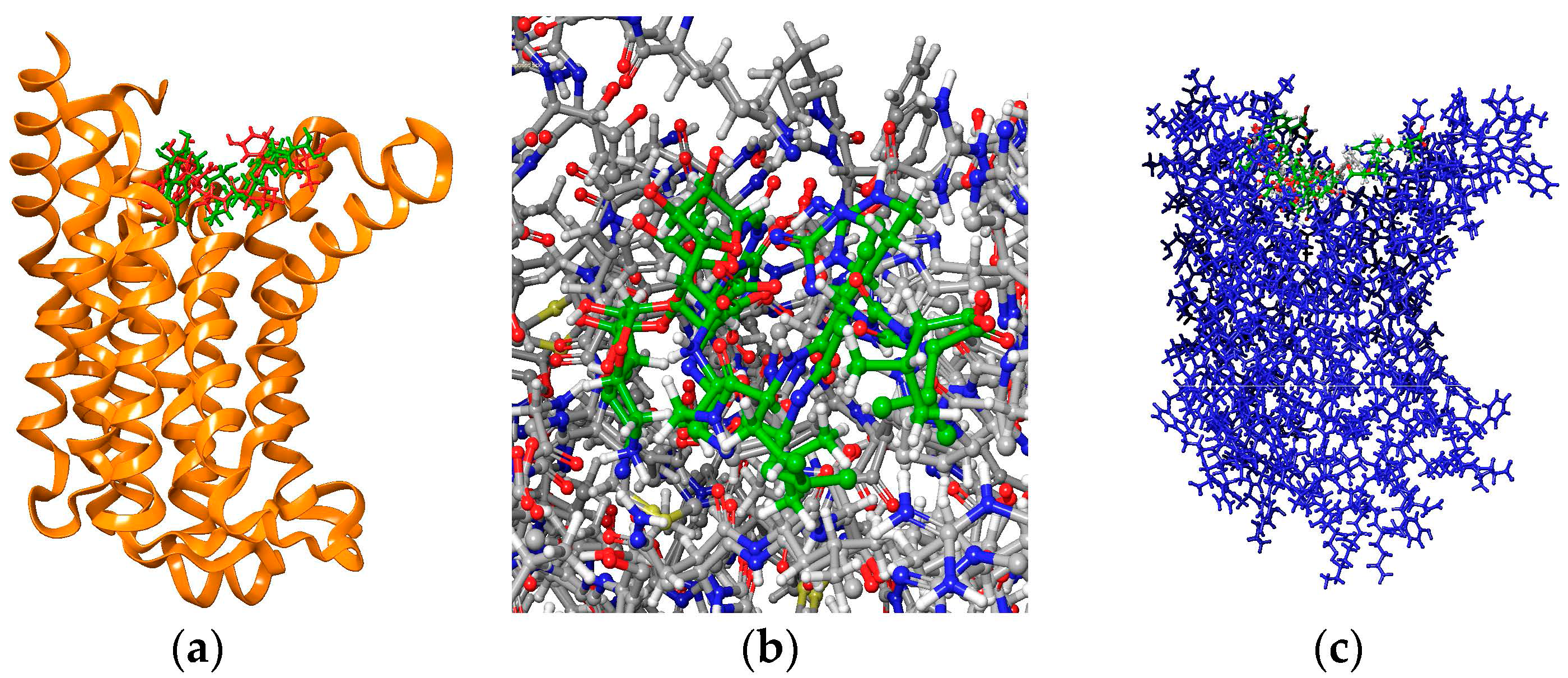
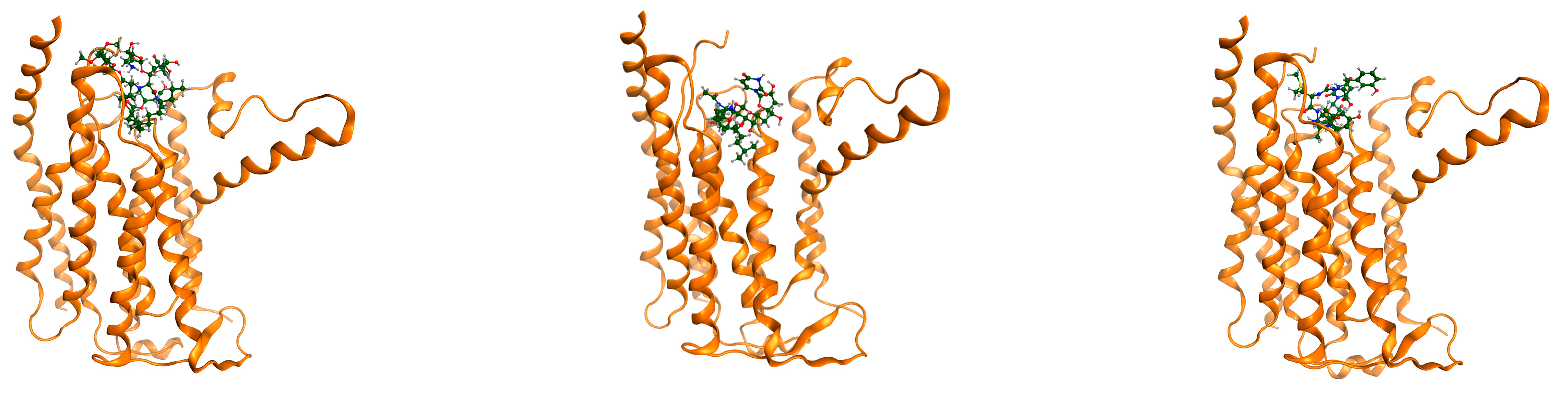
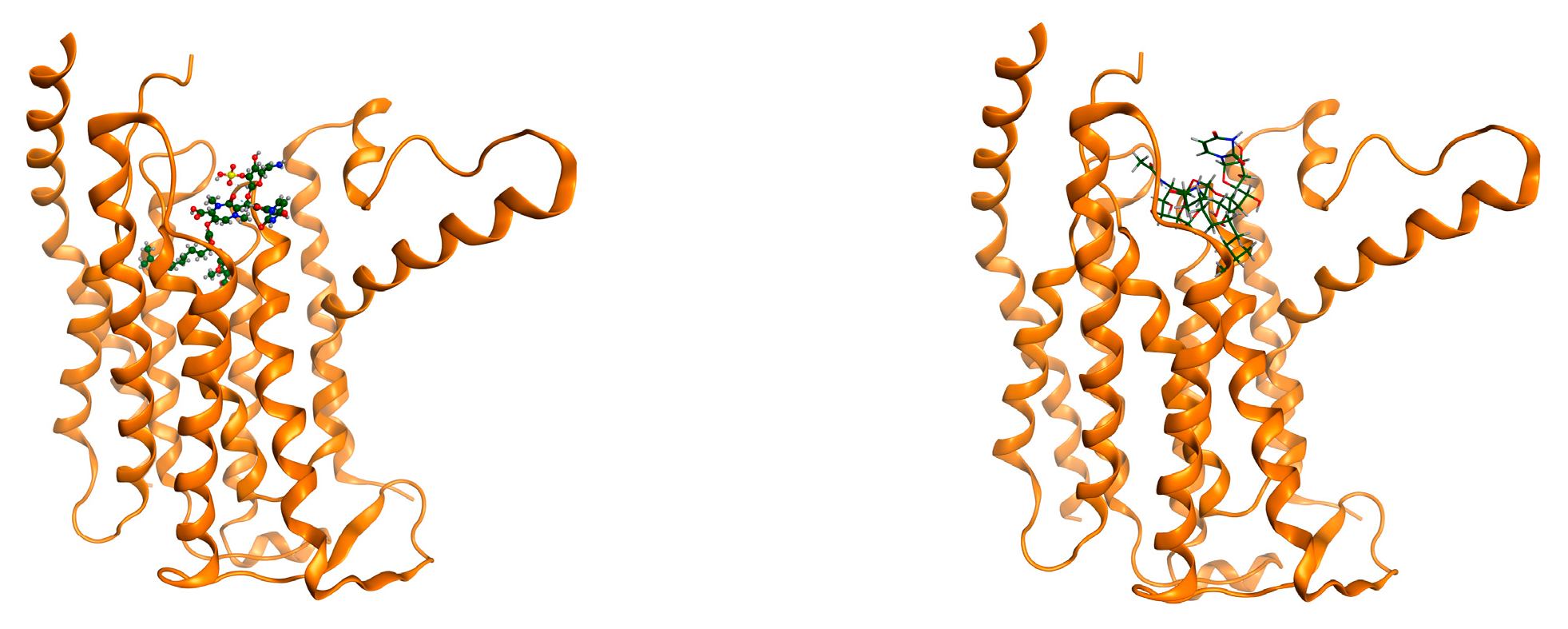
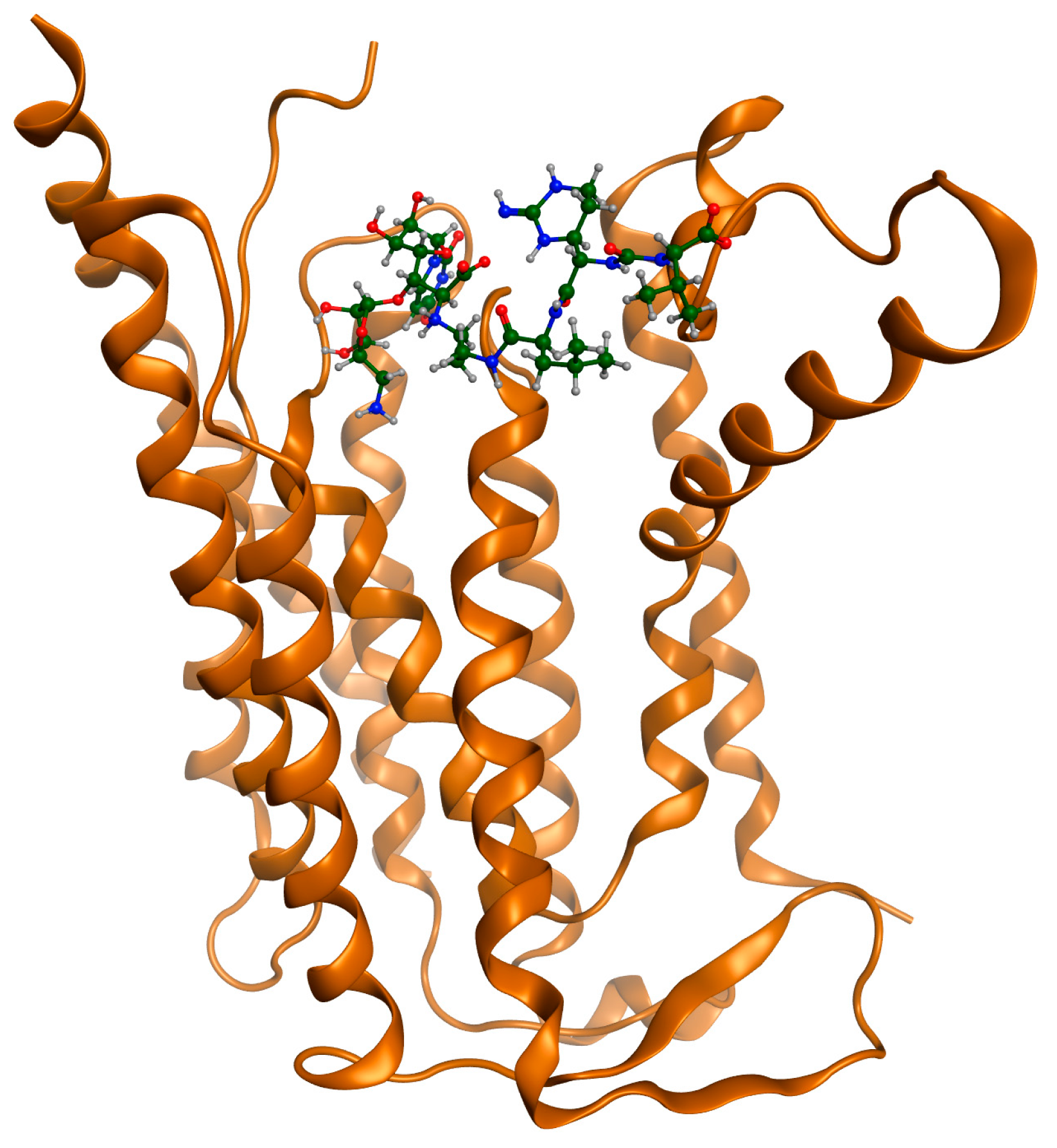

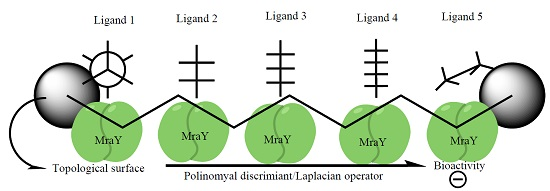{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
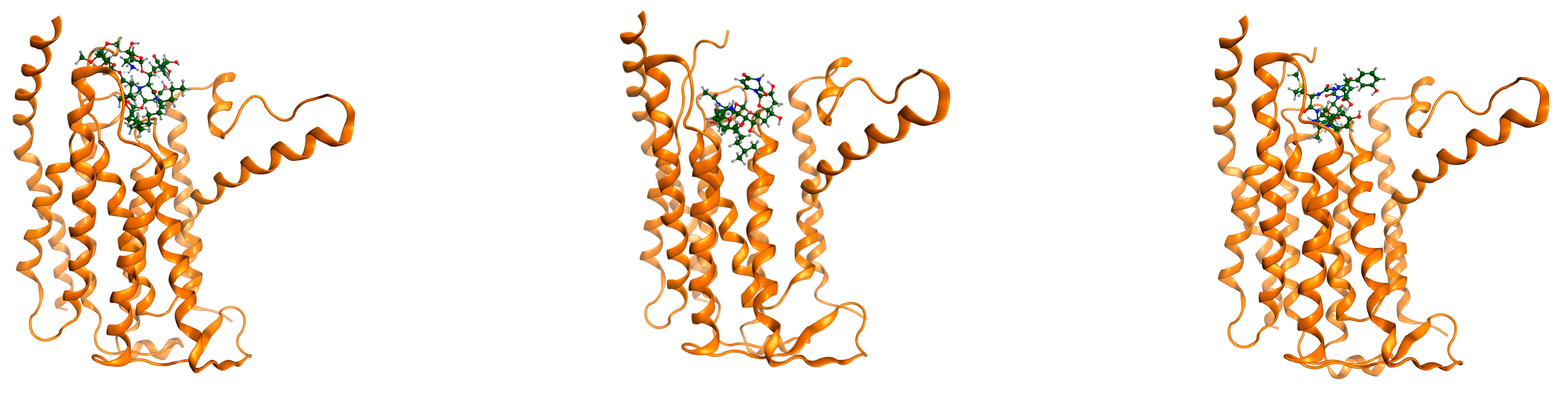{kind=link}
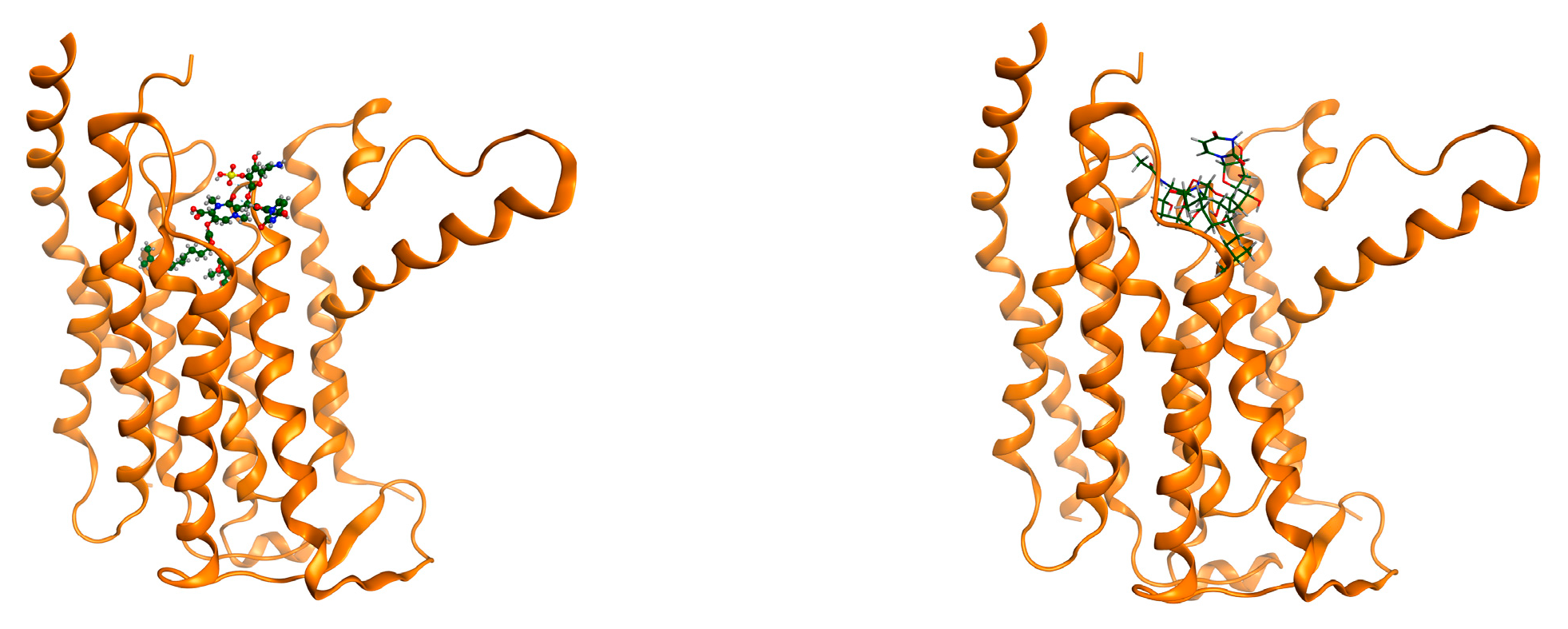{kind=link}
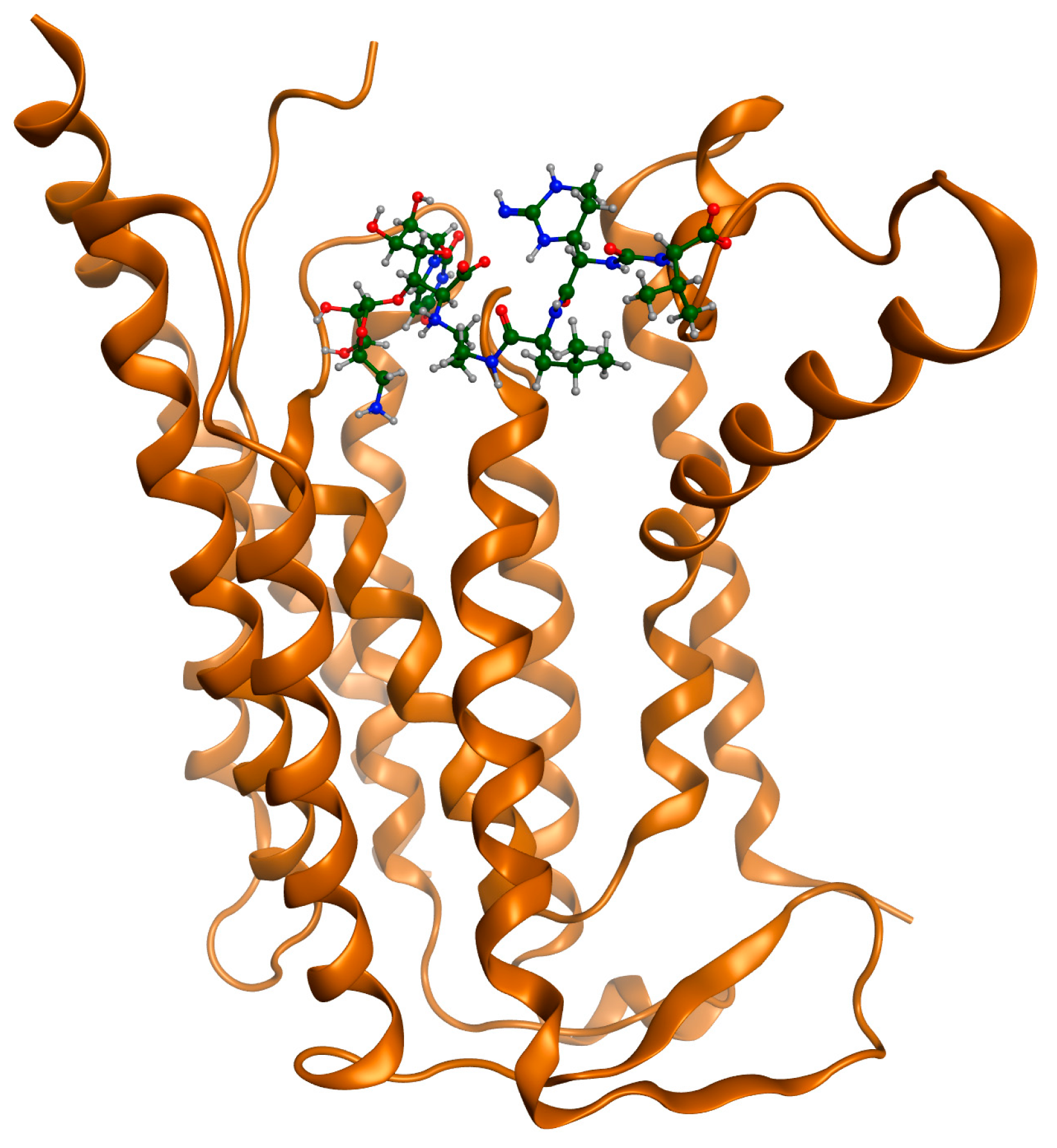{kind=link}
| State | Undocked | Docked | ||||
|---|---|---|---|---|---|---|
| Ligand | PM | W | MTI | PM | W | MTI |
| Caprazamycin A | 10,307.536 | 34,977 | 236,428 | 17,708.229 | 34,977 | 236,428 |
| Liposidomycin B | 8447.889 | 23,994 | 161,237 | 10,855.062 | 23,994 | 161,237 |
| Muraymycin Cl | 8196.615 | 21,822 | 143,843 | 9364.920 | 21,822 | 143,843 |
| Mureidomycin A | 7980.791 | 16,639 | 113,829 | 9722.197 | 16,639 | 113,829 |
| Tunicamycin I | 5628.636 | 14,696 | 99,741 | 11,318.951 | 14,696 | 99,741 |
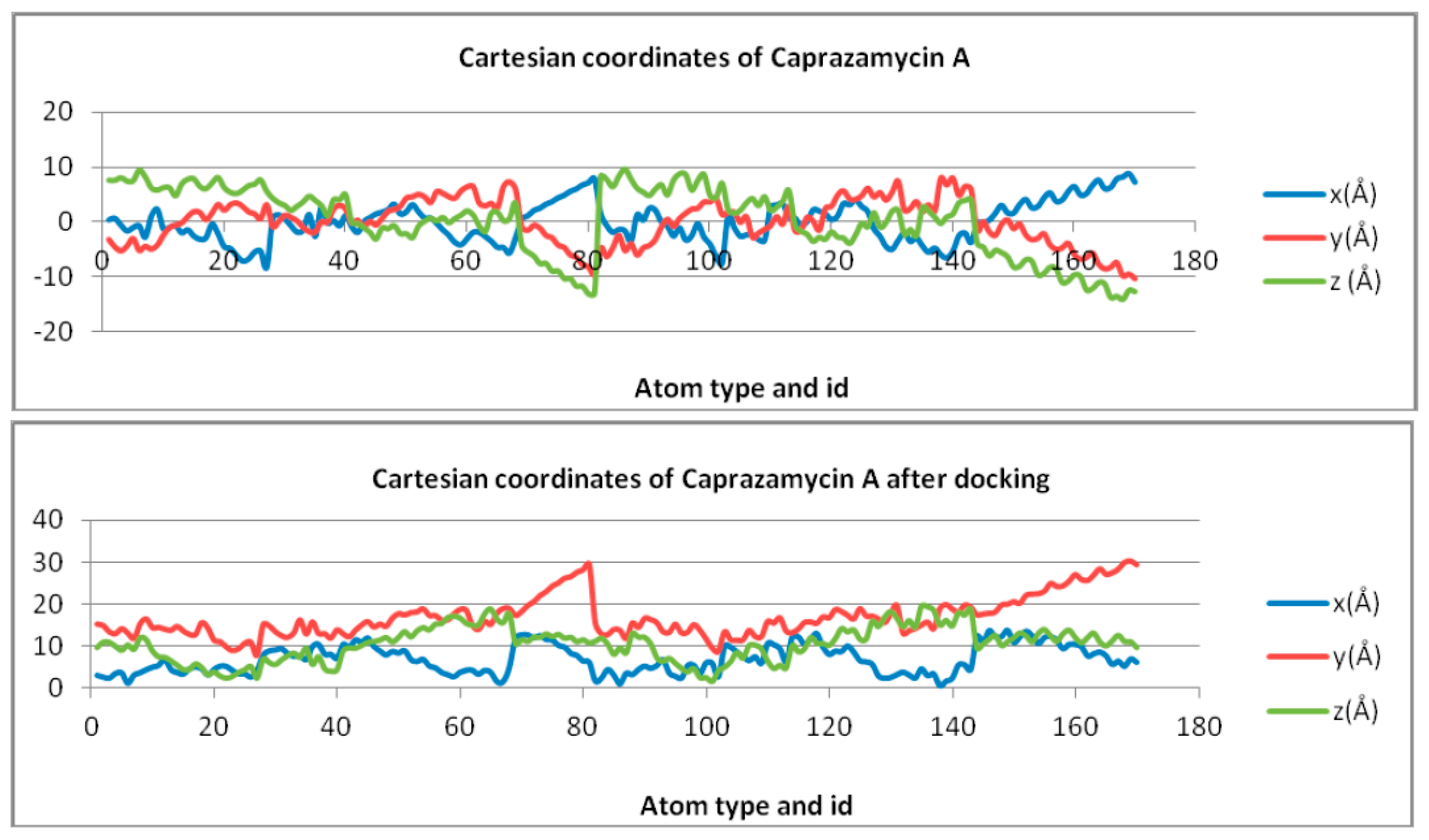
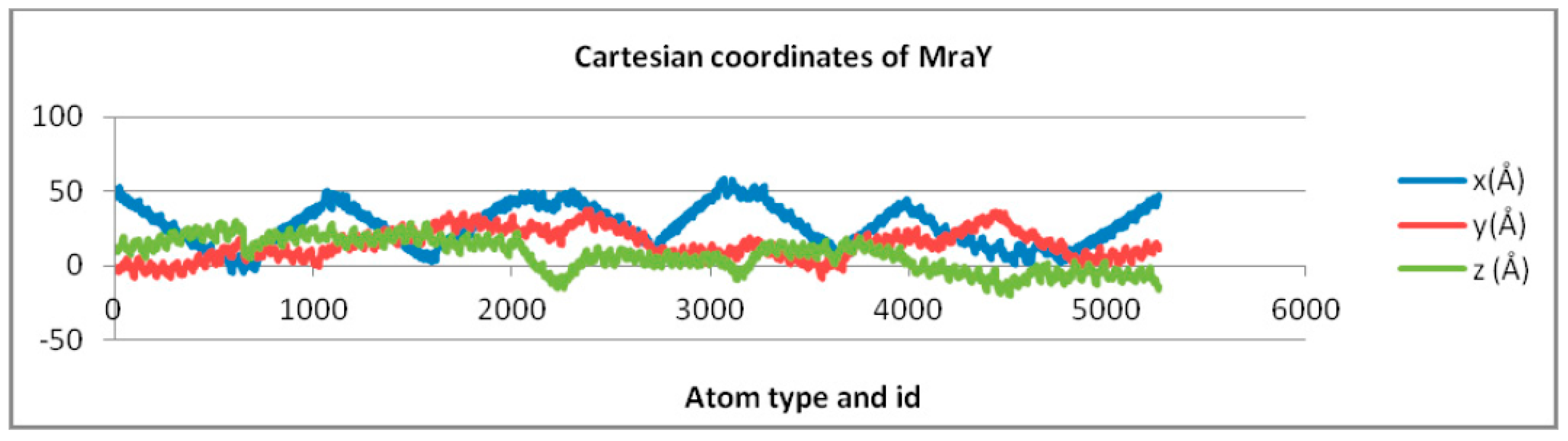
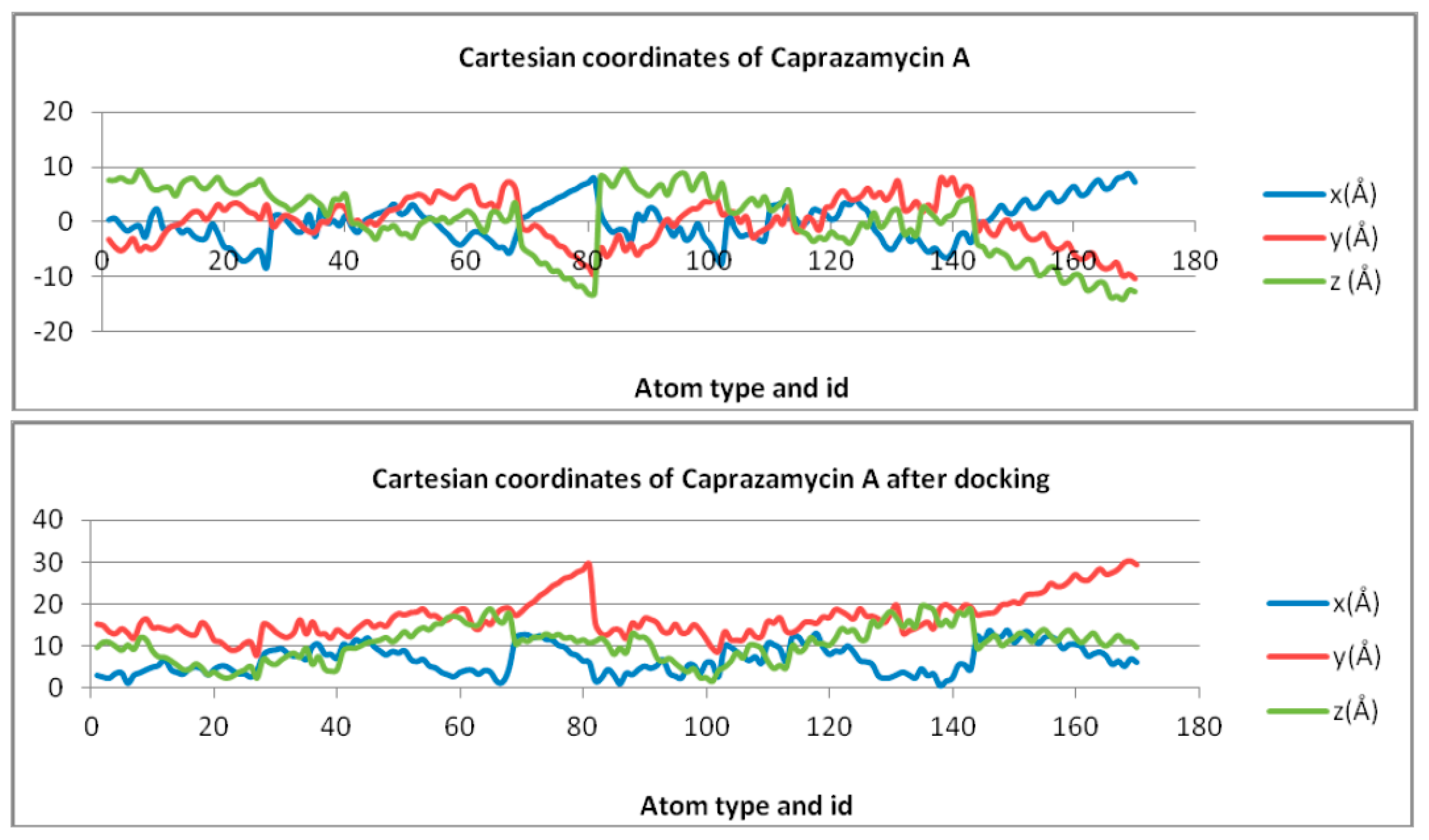
| Free Molecule | X Coordinate | Z Coordinate | Y Coordinate |
| Caprazamycin A | Y = 0.9741ln(x) − 4.0486 | Y = 0.1524ln(x) − 0.6333 | Y = −3.588ln(x) + 14.913 |
| Liposidomycin B | Y = 0.2486ln(x) − 4.9995 | Y = −2.49ln(x) + 8.0632 | y = −2.371ln(x) + 6.8077 |
| Muraymycin Cl | Y = 0.3637ln(x) + 8.2632 | Y = − 2.35ln(x) + 9.3795 | Y = −0.105ln(x) + 0.4055 |
| Mureidomycin A | Y = 2.3747ln(x) − 7.7925 | Y = 1.7282ln(x) − 5.4429 | Y = −0.13ln(x) + 0.4852 |
| Tunicamycin I | Y = −1.382ln(x) − 1.382 | Y = 1.219ln(x) + 3.022 | Y = −1.325ln(x) + 1.95 |
| MraY | Y = −0.991ln(x) + 34.353 | Y = 3.639ln(x) − 12.969 | Y = −6.486ln(x) + 56.661 |
| Docked Molecule | X Coordinate | Z Coordinate | Y Coordinate |
| Caprazamycin A | Y = 1.1474ln(x) + 2.1222 | Y = 2.2825ln(x) + 7.4555 | y = 1.4935ln(x) + 4.3625 |
| Liposidomycin B | Y = 0.6125ln(x) + 8.986 | Y = 1.8278ln(x) + 9.8712 | Y = 0.1306ln(x) + 9.6344 |
| Muraymycin Cl | Y = 1.9153ln(x) + 2.4093 | Y = −0.492ln(x) + 22.764 | Y = 0.1996ln(x) + 7.3225 |
| Mureidomycin A | Y = 0.9779ln(x) + 6.4303 | Y = −0.93ln(x) + 19.552 | Y = 0.6179ln(x) + 7.1176 |
| Tunicamycin I | Y = 0.6101ln(x) + 10.26 | Y = 1.5282ln(x) + 14.268 | Y = 0.851ln(x) + 6.0062 |
| MraY | Y = −0.991ln(x) + 34.353 | Y = 3.639ln(x) − 12.969 | Y = −6.486ln(x) + 56.661 |
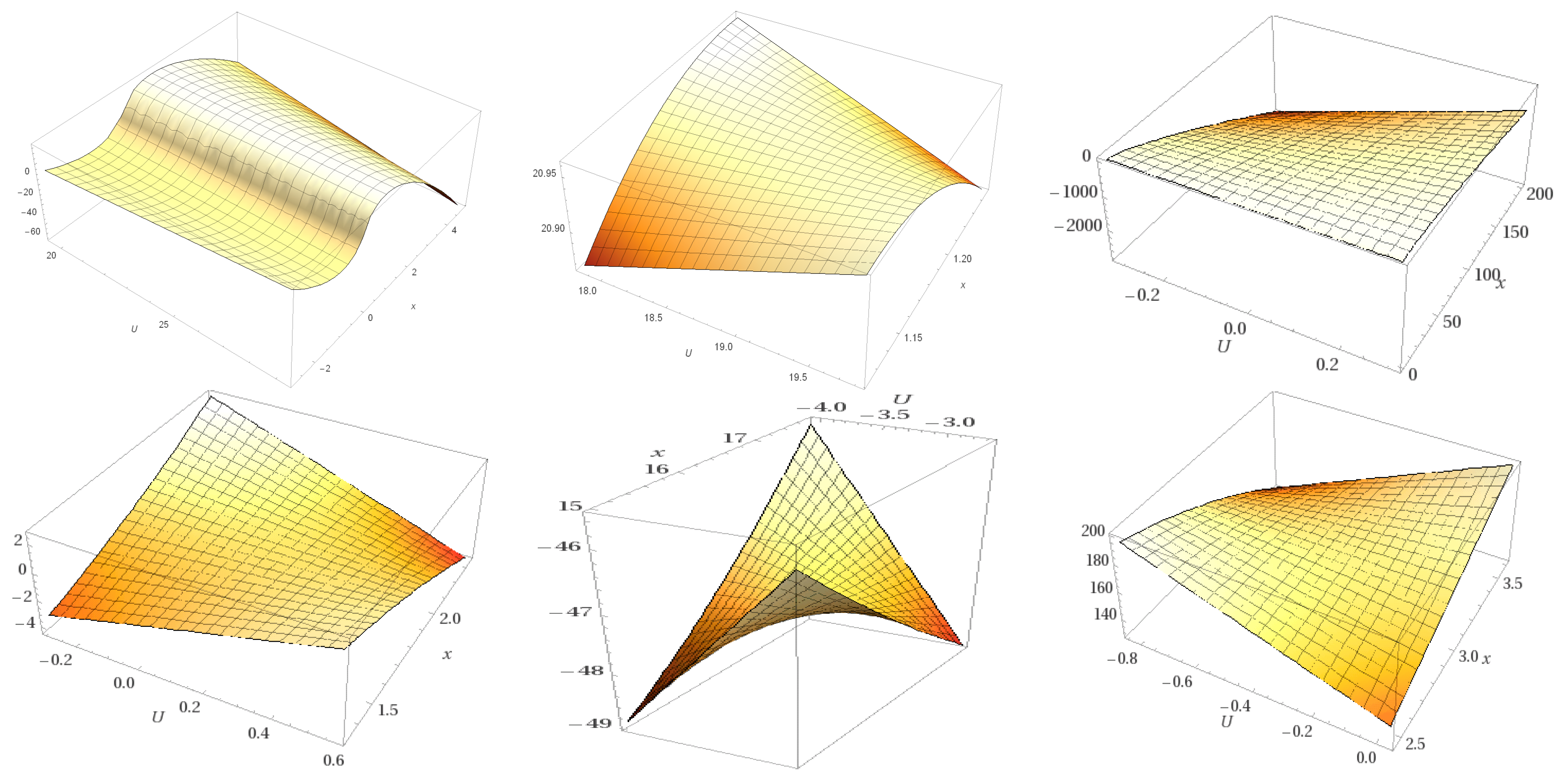
| Molecule | Cartesian Equation before Docking | Cartesian Equation after Docking |
| Caprazamycyn A | Y = −0.0453x2 − 0.7281x + 1.5735 | Y = −0.0143x2 + 0.670x + 13.167 |
| Liposidomycin B | Y = 0.1597x2 + 0.9449x − 1.1911 | Y = −0.0736x2 + 2.5211x − 1.7814 |
| Muraymycin Cl | Y = 0.0151x2 + 0.4133x + 4.8176 | Y = −0.0378x2 + 0.6574x + 18.339 |
| Mureidomycin A | Y = 0.0179x2 + 0.6583x − 0.0096 | Y = 0.0833x2 − 1.6429x + 23.528 |
| Tunicamycin I | Y = −0.0871x2 + 0.7366x + 2.0081 | Y = 0.0546x2 − 0.6779x + 19.579 |
| MraY | Y = 0.0003x2 − 0.0085x + 14.496 | Y = 0.0003x2 − 0.0085x + 14.496 |
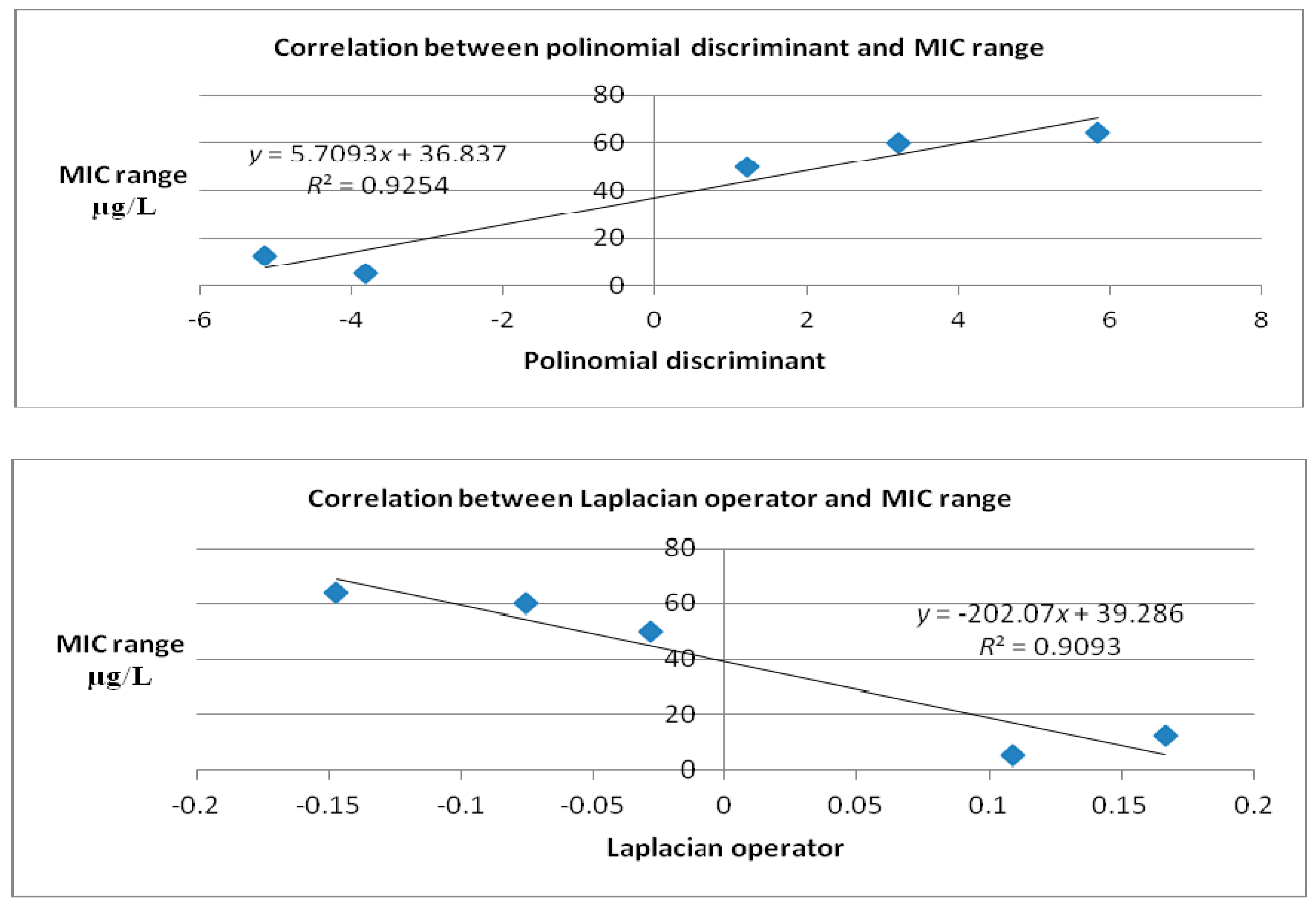
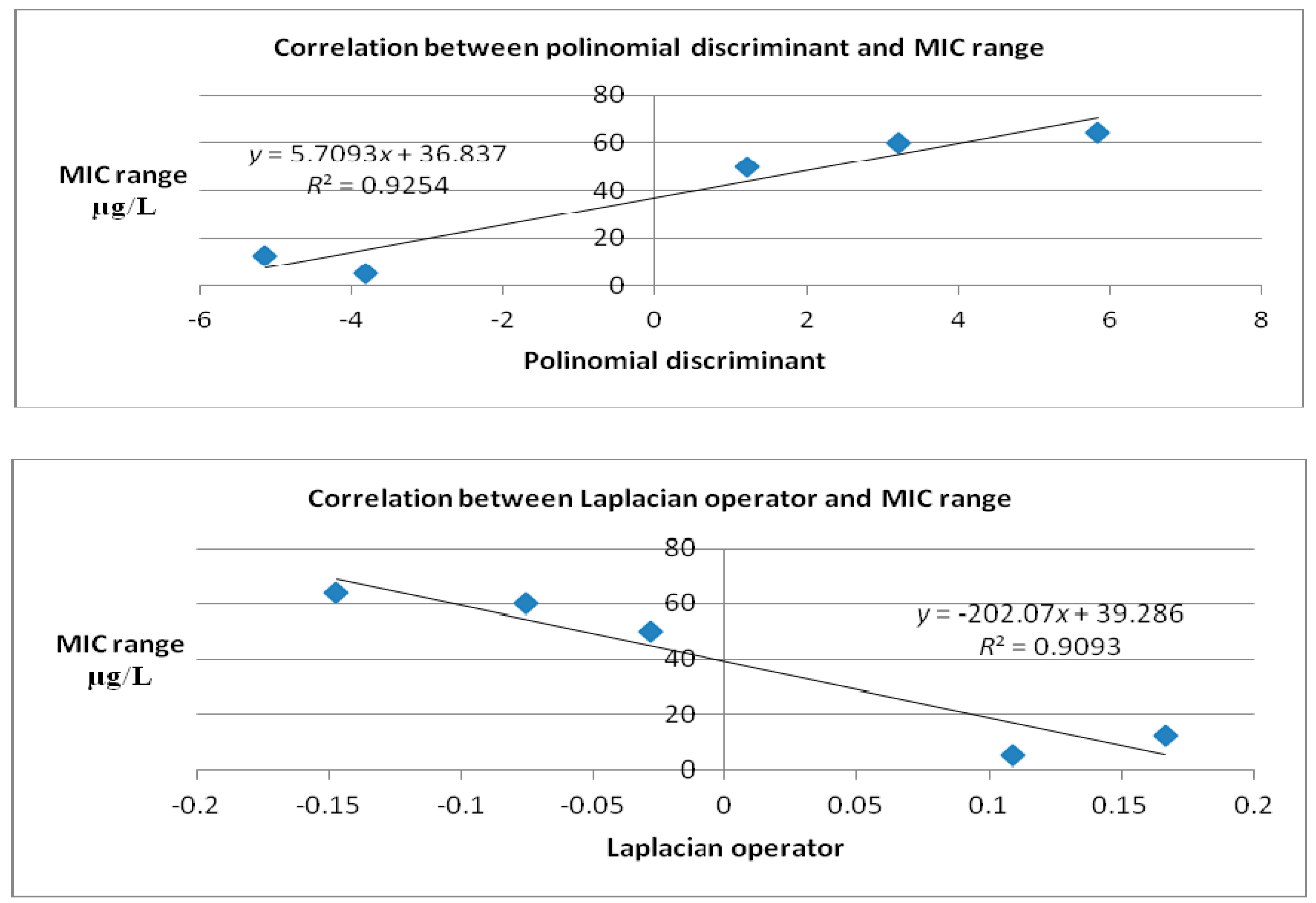
| Polynomial Discriminant | ||
| Molecule | Cartesian equation before docking | Cartesian equation after docking |
| Caprazamycin A | 0.815248 | 1.20205 |
| Liposidomicyn B | 1.65371 | 5.8315 |
| Muraymycin Cl | −0.120166 | 3.20503 |
| Mureidomycin A | 0.434046 | −5.14041 |
| Tunicamycin I | 1.2422 | −3.81651 |
| MraY | −0.017323 | −0.017323 |
| Laplacian | ||
| Molecule | Cartesian equation before docking | Cartesian equation after docking |
| Caprazamycin A | −0.0906 | −0.0286 |
| Liposidomicyn B | 0.3194 | −0.1472 |
| Muraymycin Cl | 0.0302 | −0.0756 |
| Mureidomycin A | 0.0358 | 0.1666 |
| Tunicamycin I | −0.1742 | 0.1092 |
| MraY | 0.0006 | 0.0006 |
| Molecule | Cartesian Equation before Docking | Cartesian Equation after Docking |
| Caprazamycin A | Y = −0.001x4 − 0.0074x + 0.0146x2 − 0.4585x + 1.3125 | Y = 0.0052x4 − 0.1596x3 + 1.6554x2 − 6.0837x + 21.636 |
| Liposidomycin B | Y = −0.0163x4 − 0.3471x3 − 2.3041x2 − 5.5628x − 6.0698 | Y = −0.0107x4 + 0.3649x3 − 4.2848x2 + 20.373x − 18.692 |
| Muraymycin Cl | Y = 0.0008x4 − 0.0256x3 + 0.1962x2 + 0.4938x + 3.5622 | Y = 0.0038x4 − 0.1135x3 + 1.017x2 − 2.1836x + 16.887 |
| Mureidomycin A | Y = −0.0014x4 − 0.0139x3 + 0.1119x2 + 1.1734x − 0.854 | Y = −0.0039x4 + 0.1384x3 − 1.6016x2 + 6.5958x + 10.301 |
| Tunicamycin I | Y = 0.0013x4 − 0.0044x3 − 0.1304x2 + 0.8329x + 2.1574 | Y = 0.0052x4 − 0.2714x3 + 5.2418x2 − 43.377x + 146.8 |
| MraY | Y = −0.0001x4 + 0.0032x3 − 0.1071x2 + 1.1968x + 11.556 | Y = −0.001x4 + 0.0032x3 − 0.1071x2 + 1.1968x + 11.556 |
| Polynomial Discriminant | ||
| Molecule | Cartesian equation before docking | Cartesian equation after docking |
| Caprazamycin A | −2.80872 × 10−6 | 0.181815 |
| Liposidomycin B | −0.0183835 | −1.11938 |
| Muraymycin Cl | 0.0000417636 | 0.0936006 |
| Mureidomycin A | −0.0000169638 | −0.0829902 |
| Tunicamycin I | −0.0000339967 | 0.3223 |
| MraY | −2.97825 × 10−6 | −2.97825 × 10−6 |
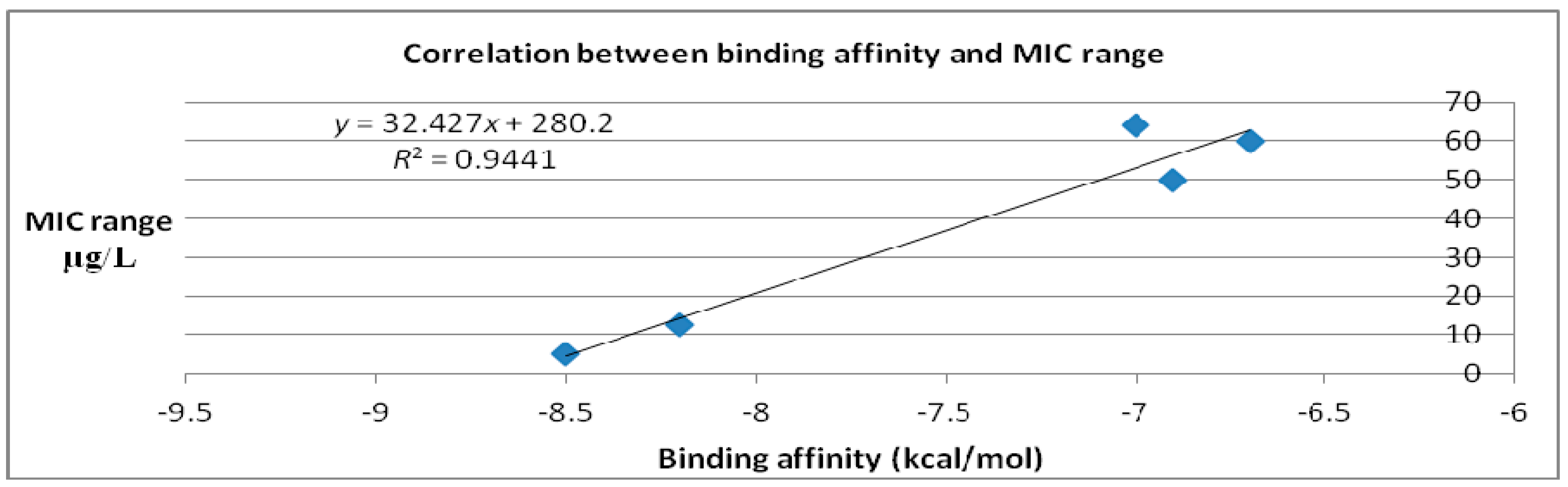
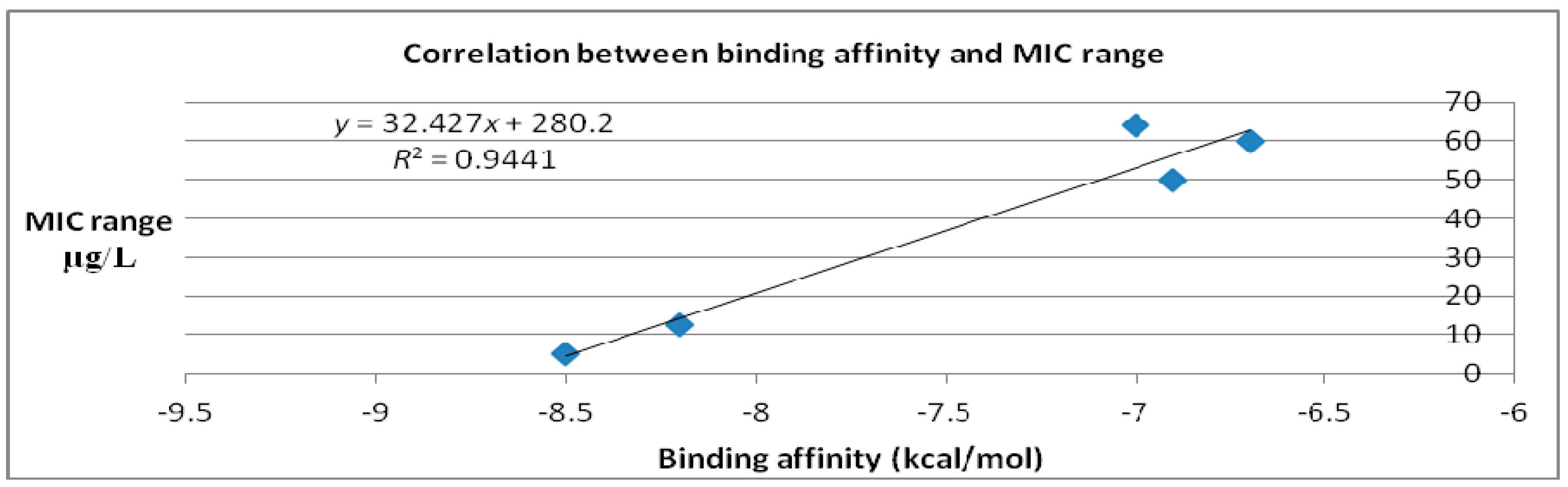
| Molecules | Binding Affinity (kcal/mol) | MIC Range (μg/mL) |
|---|---|---|
| Caprazamycin A | −6.9 | 50 [15,16] |
| Liposidomycin B | −7 | 64 [17] |
| Muraymycin Cl | −6.7 | 60 [18] |
| Mureidomycin A | −8.2 | 12.5 [19] |
| Tunicamycin I | −8.5 | 5 [19] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lungu, C.N.; Diudea, M.V.; Putz, M.V. Ligand Shaping in Induced Fit Docking of MraY Inhibitors. Polynomial Discriminant and Laplacian Operator as Biological Activity Descriptors. Int. J. Mol. Sci. 2017, 18, 1377. https://doi.org/10.3390/ijms18071377
Lungu CN, Diudea MV, Putz MV. Ligand Shaping in Induced Fit Docking of MraY Inhibitors. Polynomial Discriminant and Laplacian Operator as Biological Activity Descriptors. International Journal of Molecular Sciences. 2017; 18(7):1377. https://doi.org/10.3390/ijms18071377
Chicago/Turabian StyleLungu, Claudiu N., Mircea V. Diudea, and Mihai V. Putz. 2017. "Ligand Shaping in Induced Fit Docking of MraY Inhibitors. Polynomial Discriminant and Laplacian Operator as Biological Activity Descriptors" International Journal of Molecular Sciences 18, no. 7: 1377. https://doi.org/10.3390/ijms18071377