Effects of an Inhibitor of Monocyte Recruitment on Recovery from Traumatic Brain Injury in Mice Treated with Granulocyte Colony-Stimulating Factor

Abstract
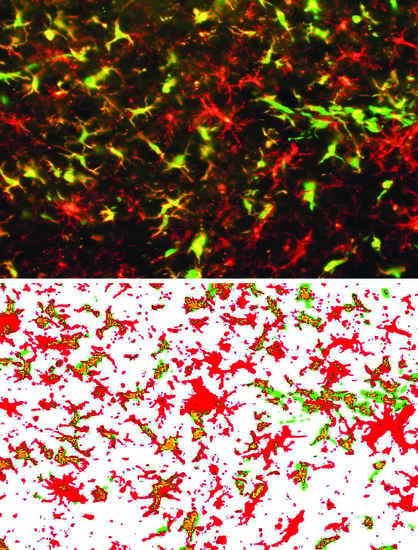
:
1. Introduction
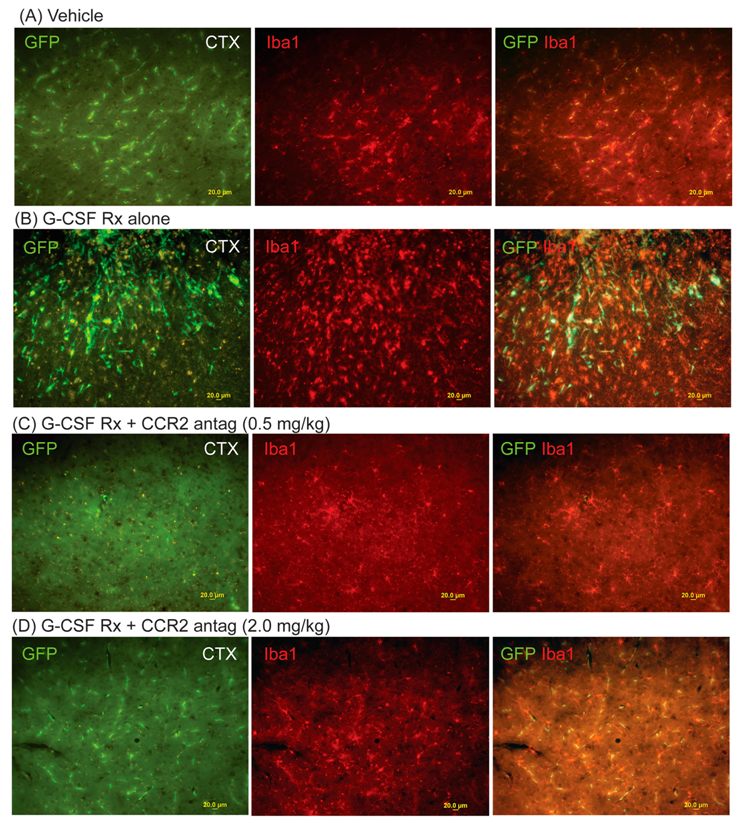
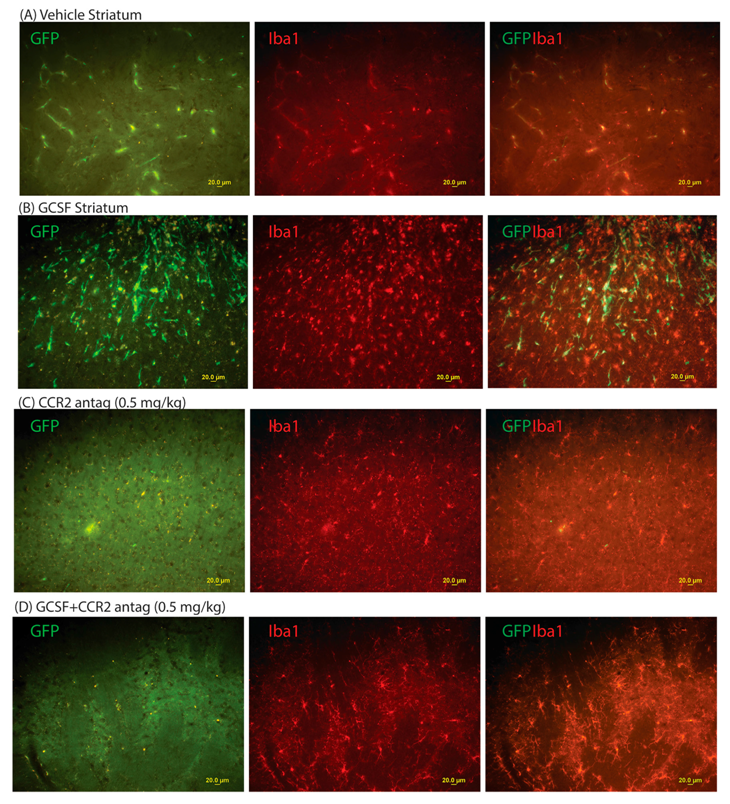
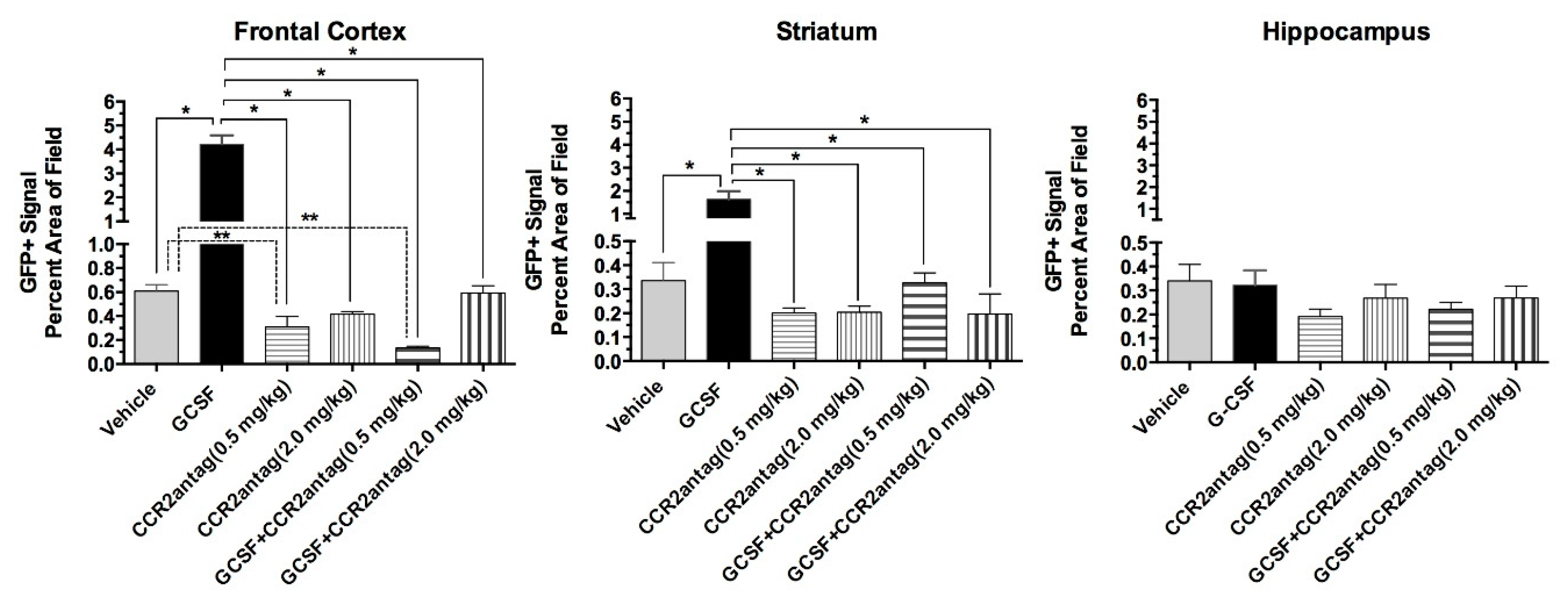
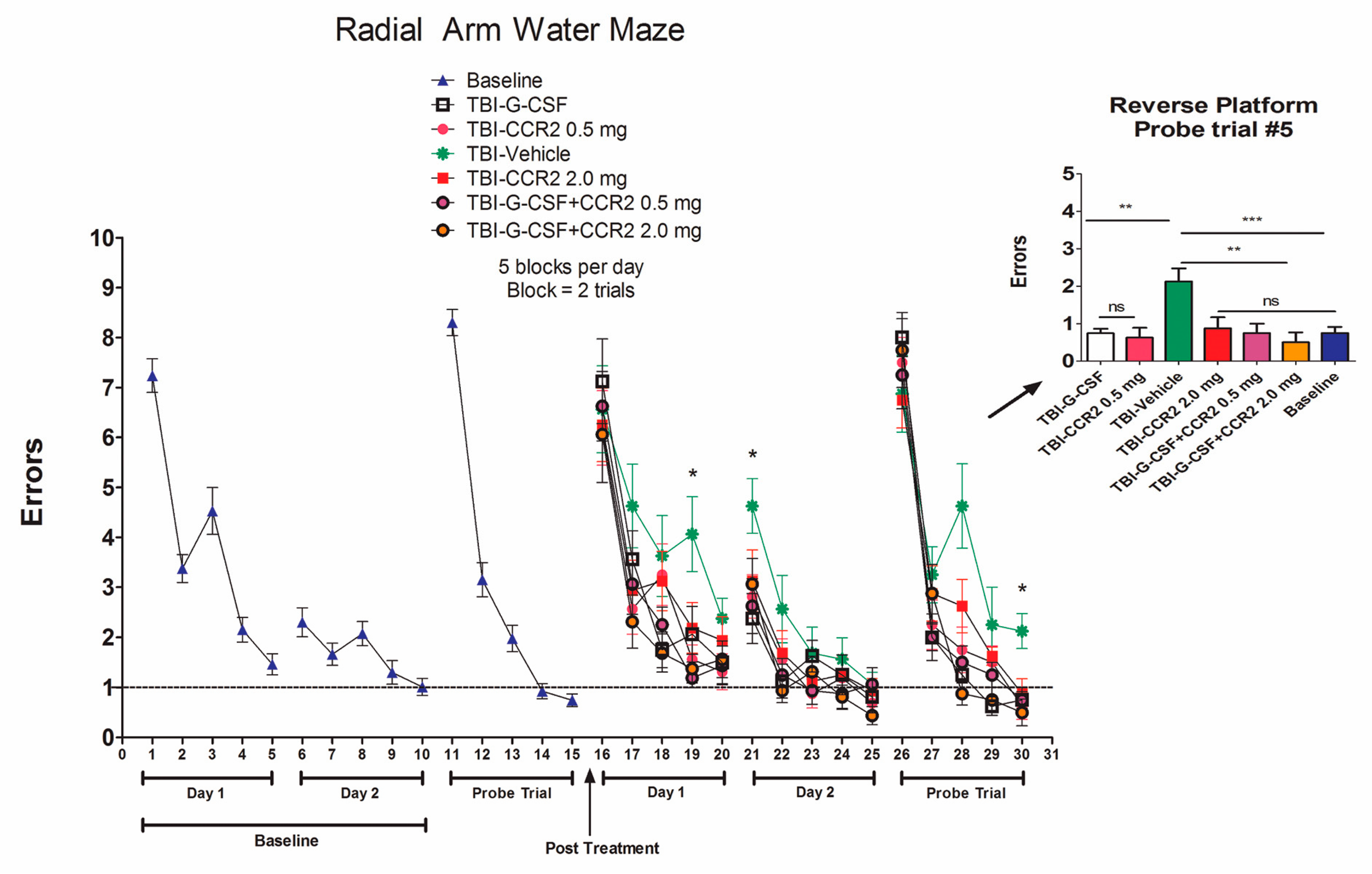
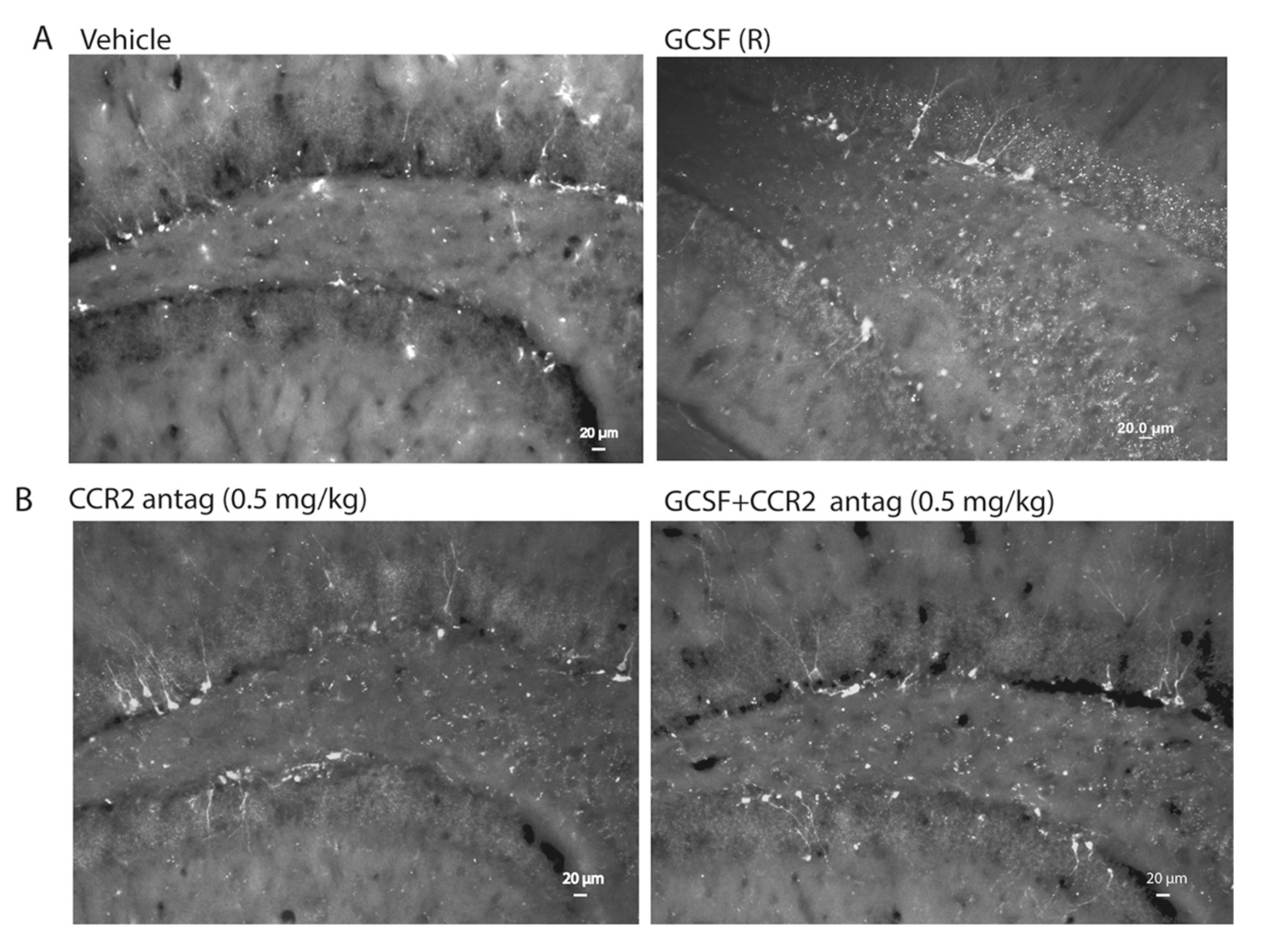
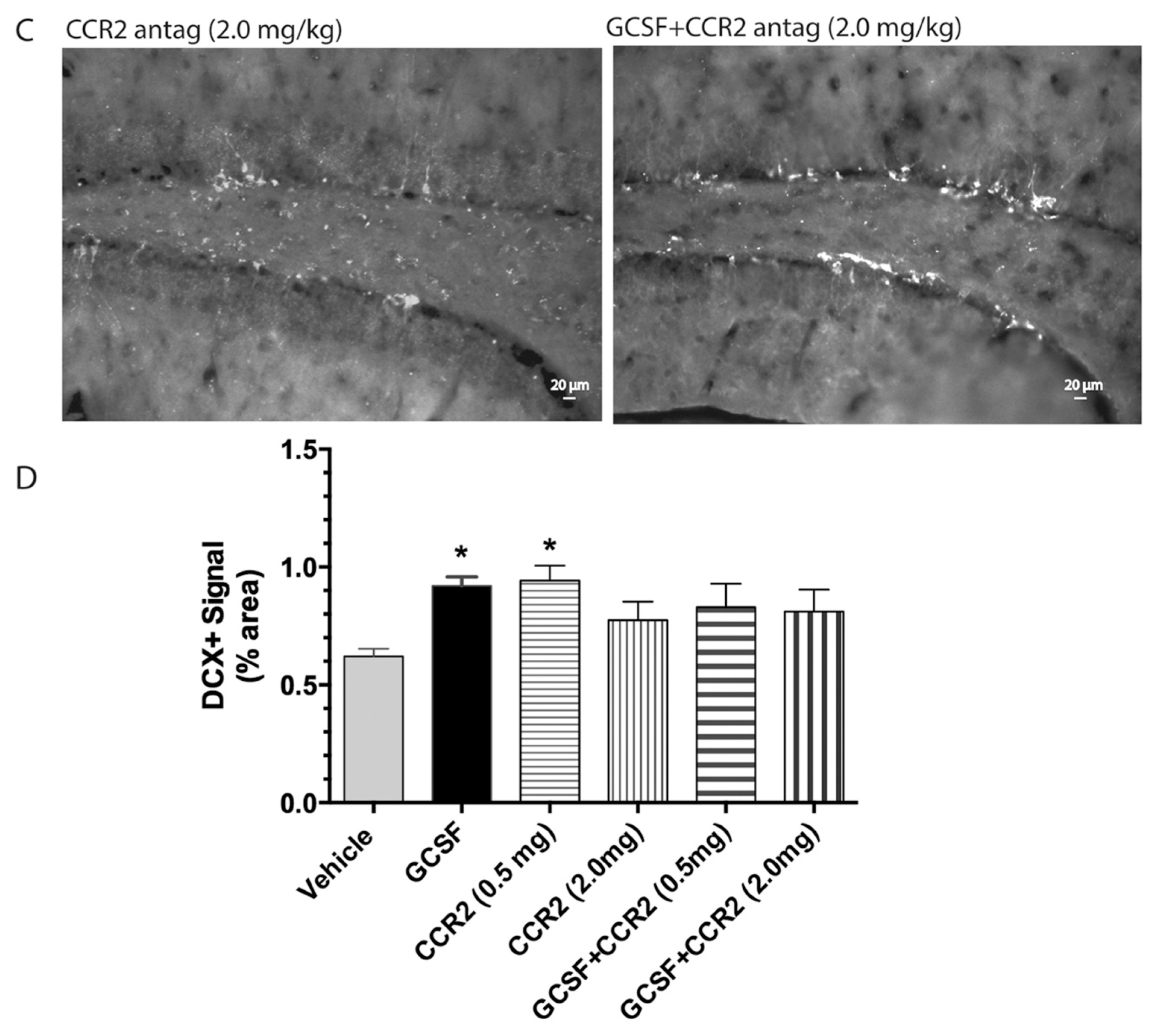
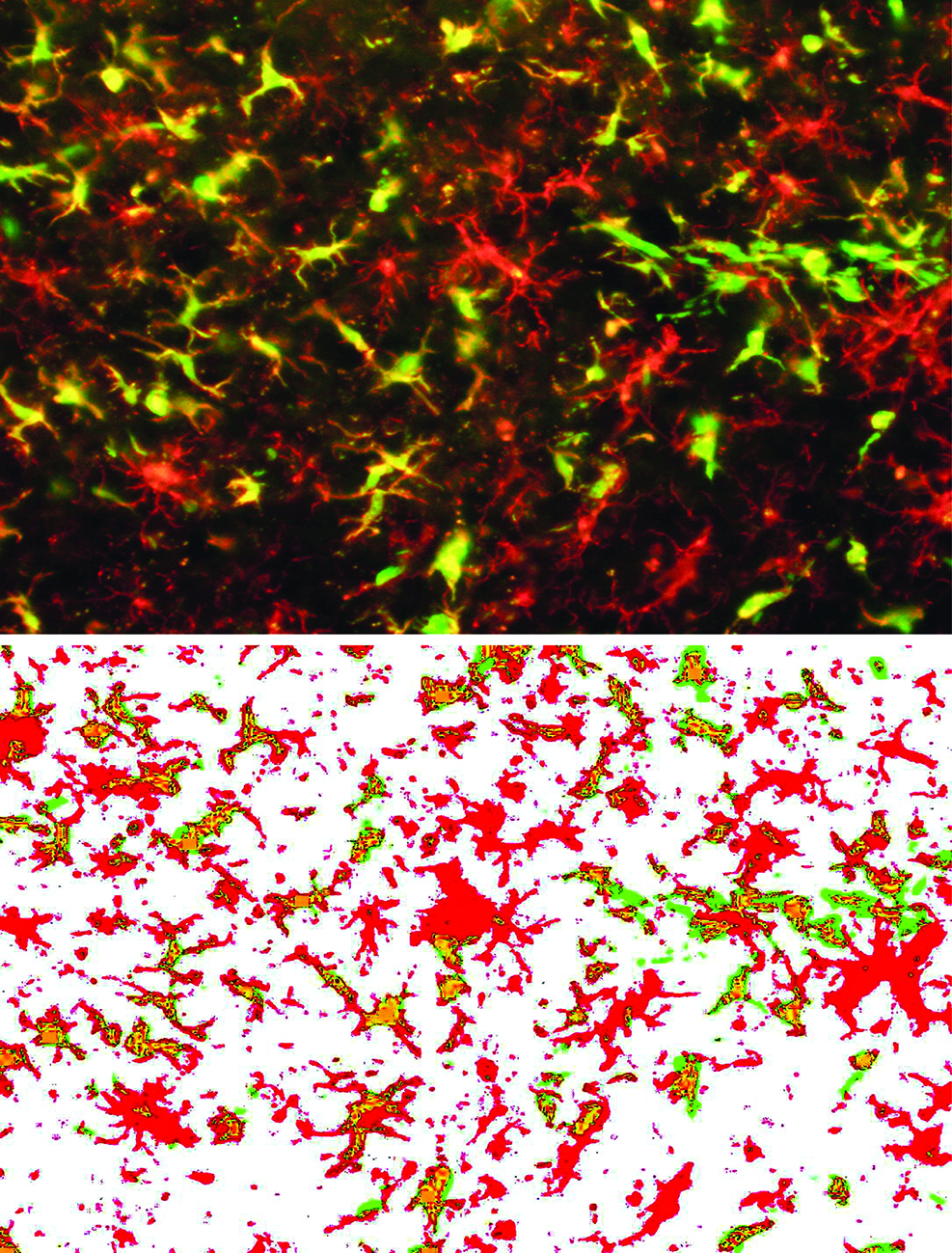
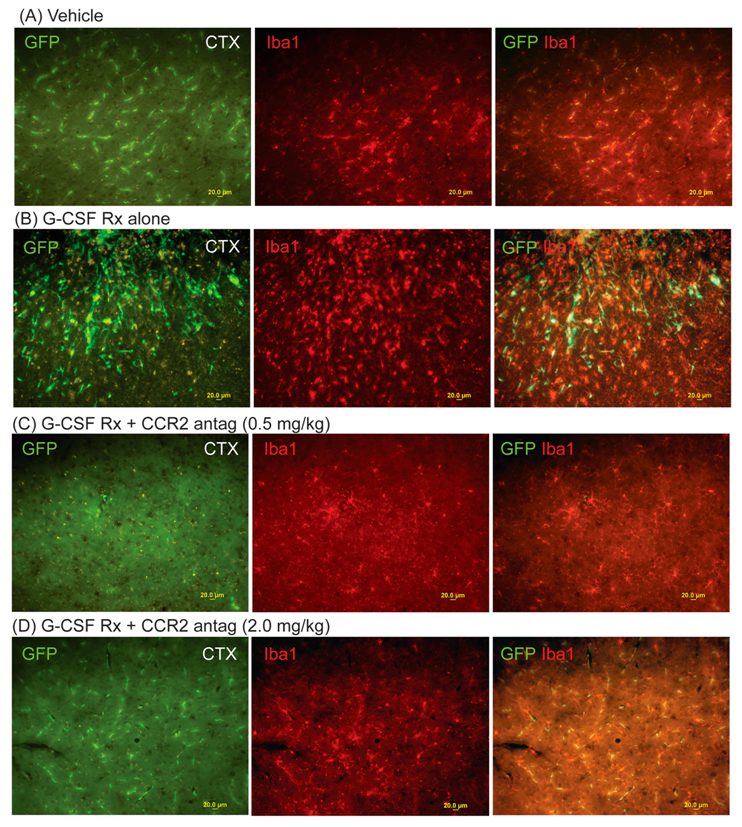
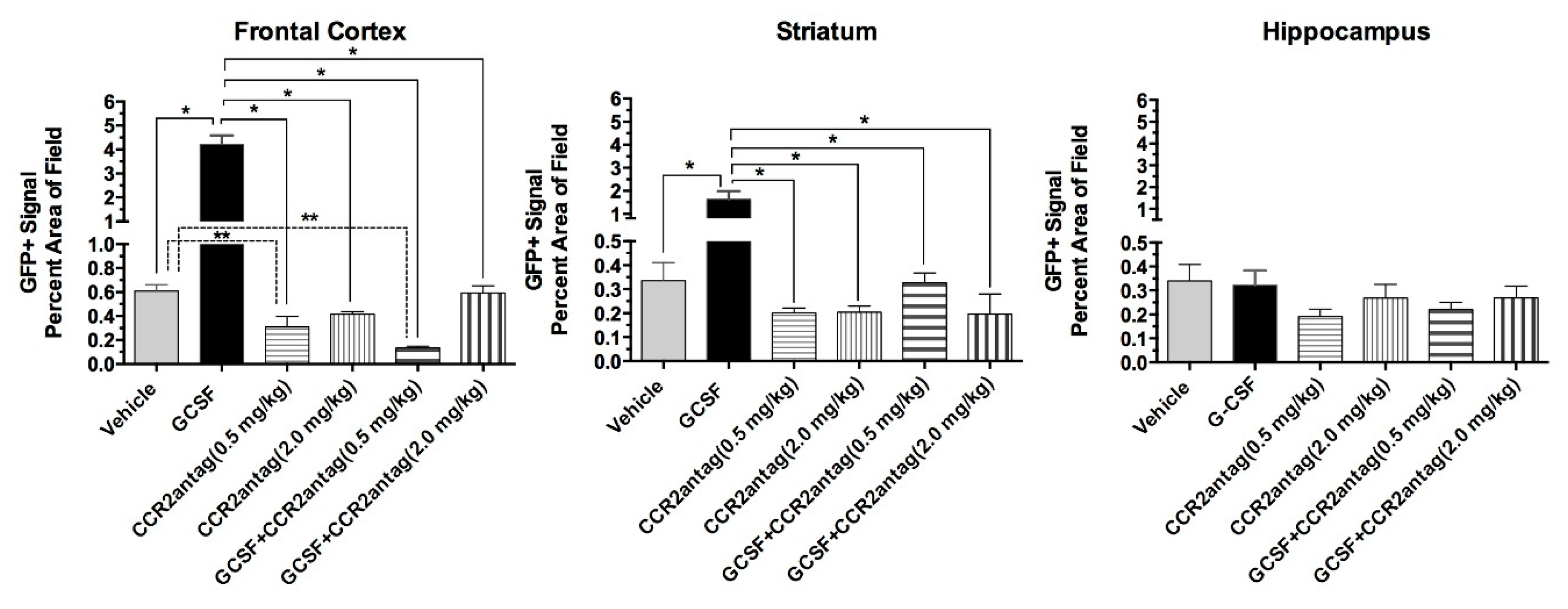
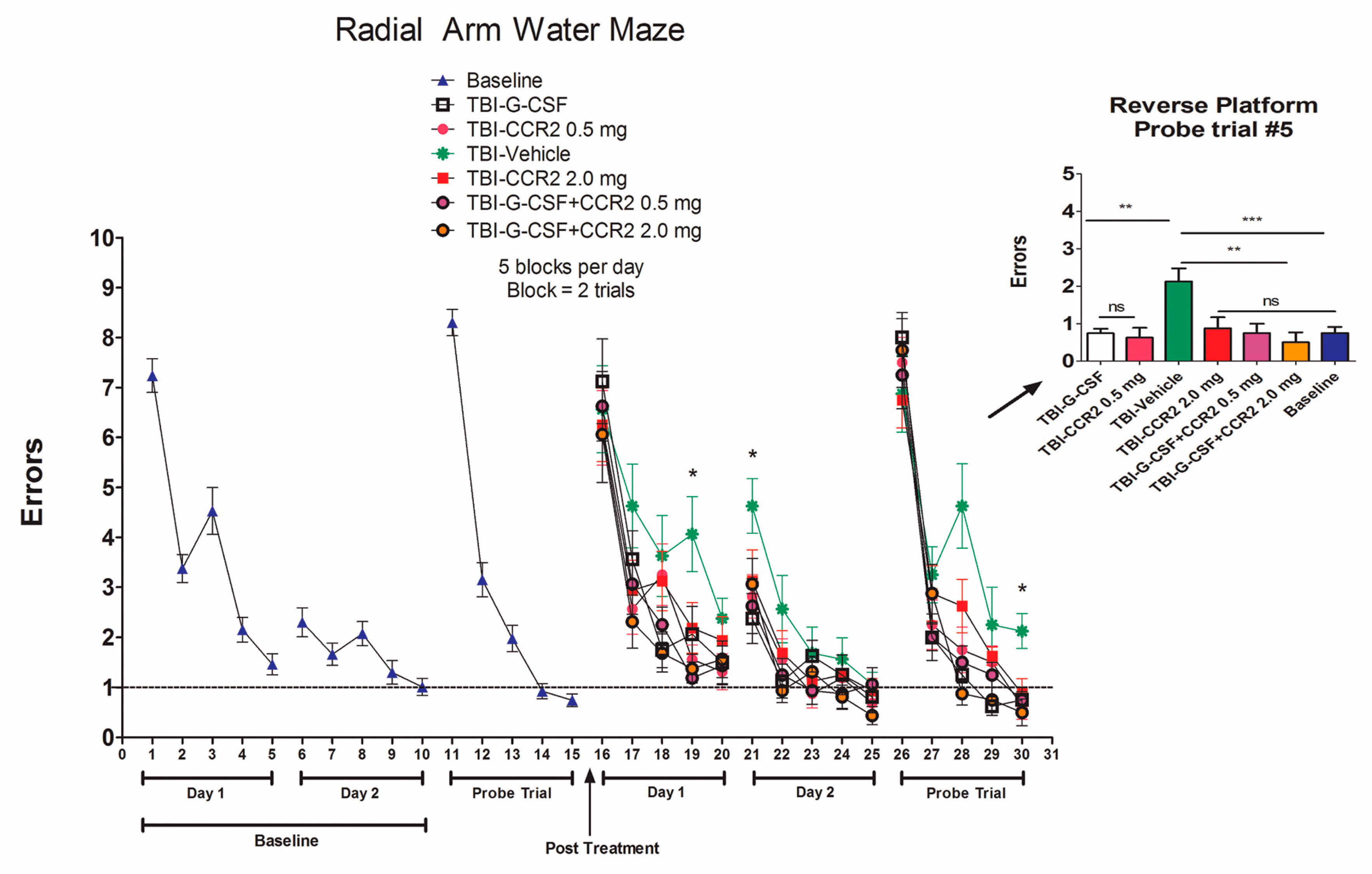
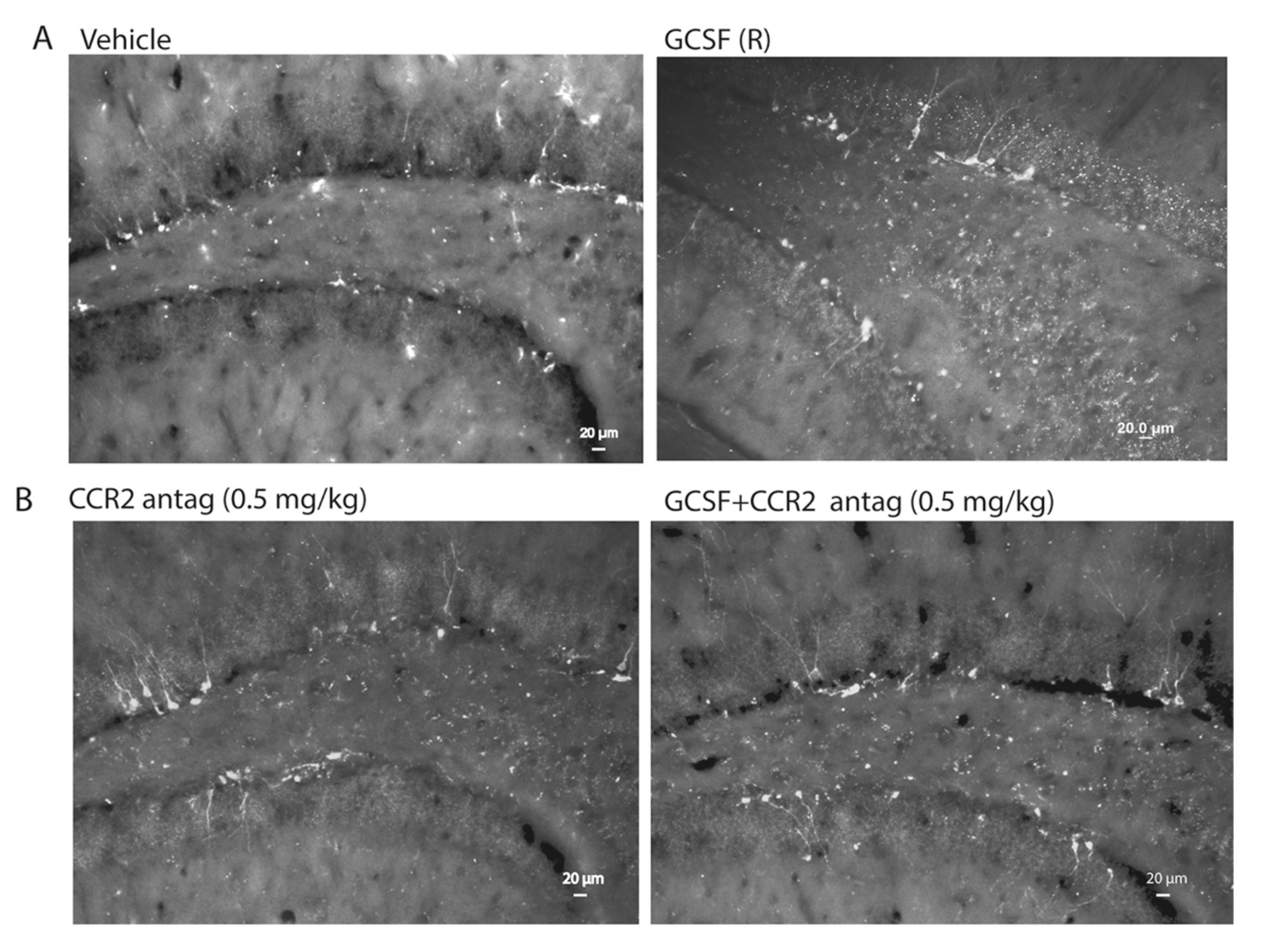
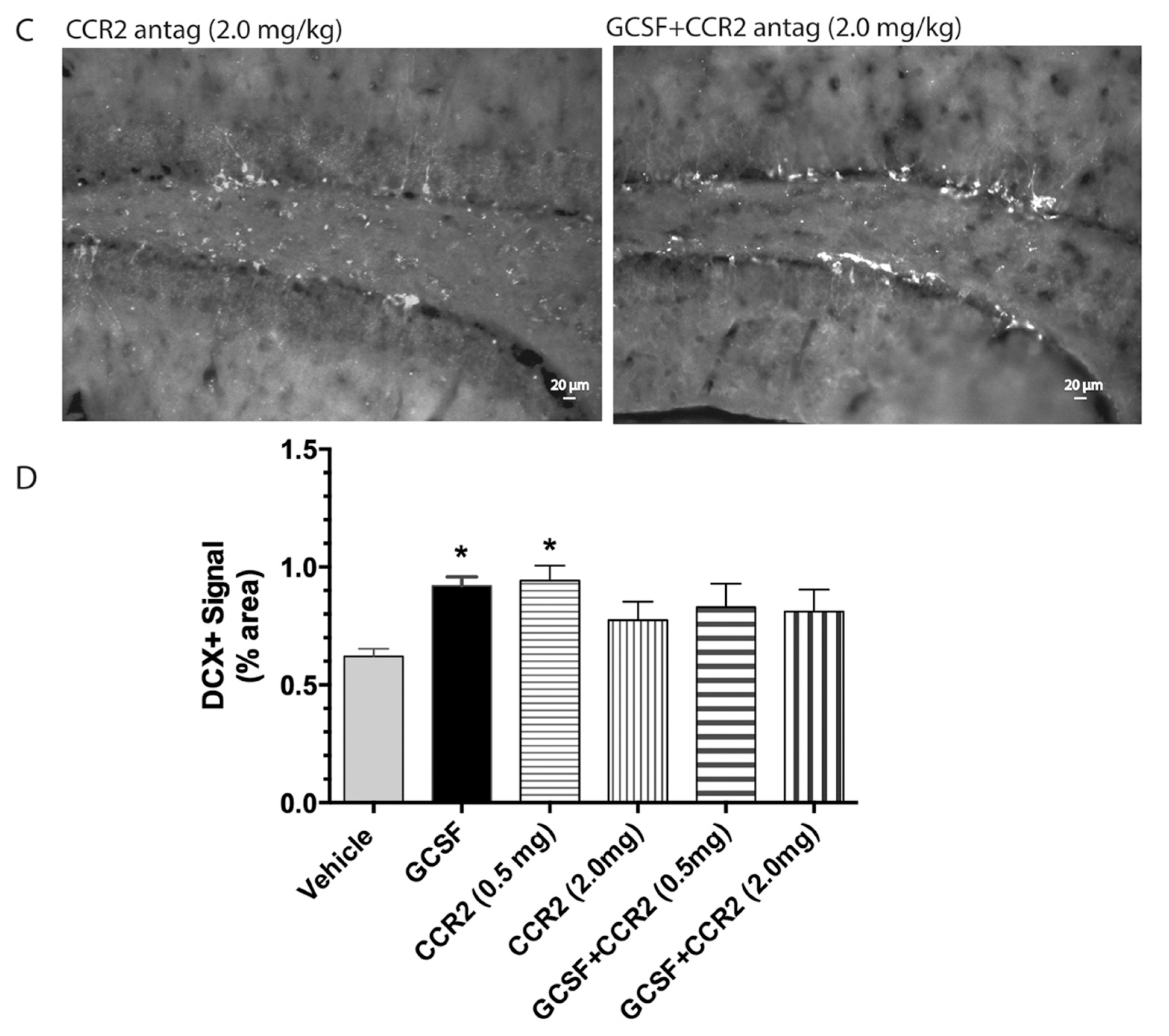
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Surgery and Controlled Cortical Impact
4.3. Drugs
4.4. Radial Arm Water Maze (RAWM)
4.5. Immunohistology
4.6. Quantitative Assessment of Bone Marrow-Derived Cells (GFP+ cells), Microglial Cells (Iba1+) and Hippocampal Neurogenesis (DCX+ Cells)
4.7. Data Analysis and Statistics
5. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
Abbreviations
| TBI | Traumatic brain injury |
| CCI | Controlled cortical impact |
| CCR2 | C-C motif receptor 2 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| RAWM | Radial arm water maze |
| DCX | Doublecortin |
| CCI | Controlled cortical impact |
| GFP | Green fluorescent protein |
References
- Schabitz, W.R.; Kollmar, R.; Schwaninger, M.; Juettler, E.; Bardutzky, J.; Scholzke, M.N.; Sommer, C.; Schwab, S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke 2003, 34, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Lin, S.Z.; Lee, C.C.; Liu, D.D.; Li, H. Granulocyte colony-stimulating factor for acute ischemic stroke: A randomized controlled trial. Can. Med. Assoc. J. 2006, 174, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Gasan, G.; Mura, E.; Bordet, R. Beneficial effect of pharmacological mobilization of bone marrow in experimental cerebral ischemia. Eur. J. Pharmacol. 2003, 458, 327–338. [Google Scholar] [CrossRef]
- Solaroglu, I.; Tsubokawa, T.; Cahill, J.; Zhang, J.H. Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience 2006, 143, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.Y.; Chen, Y.J.; Wang, M.F.; Pan, H.C.; Chen, S.Y.; Cheng, F.C. Granulocyte colony-stimulating factor enhances cellular proliferation and motor function recovery on rats subjected to traumatic brain injury. Neurol. Res. 2010, 32, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Sanberg, P.R.; Sanchez-Ramos, J.; Song, S.; Kaneko, Y.; Borlongan, C.V. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS ONE 2014, 9, e90953. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Sava, V.; Kong, X.; Acosta, S.; Borlongan, C.; Sanchez-Ramos, J. Granulocyte-colony stimulating factor promotes brain repair following traumatic brain injury by recruitment of microglia and increasing neurotrophic factor expression. Restor. Neurol Neurosci. 2016, 34, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, S.J.; Sinn, D.I.; Kim, S.U.; Kim, M.; Roh, J.K. Granulocyte colony-stimulating factor stimulates neurogenesis via vascular endothelial growth factor with STAT activation. Brain Res. 2006, 1073, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ramos, J.; Song, S.; Sava, V.; Catlow, B.; Lin, X.; Mori, T.; Cao, C.; Arendash, G.W. Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer’s mice. Neuroscience 2009, 163, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.R.; Navalitloha, Y.; Singhal, S.; Mehta, J.; Piao, C.S.; Guo, W.P.; Kessler, J.A.; Groothuis, D.R. Hematopoietic growth factors pass through the blood-brain barrier in intact rats. Exp. Neurol. 2007, 204, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Kruger, C.; Steigleder, T.; Weber, D.; Pitzer, C.; Laage, R.; Aronowski, J.; Maurer, M.H.; Gassler, N.; Mier, W.; Hasselblatt, M. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Investig. 2005, 115, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Kuhn, H.G.; Schabitz, W.R. A role for G-CSF (granulocyte-colony stimulating factor) in the central nervous system. Cell Cycle 2005, 4, 1753–1757. [Google Scholar] [PubMed]
- Song, S.; Kong, X.; Acosta, S.; Sava, V.; Borlongan, C.; Sanchez-Ramos, J. Granulocyte colony-stimulating factor promotes behavioral recovery in a mouse model of traumatic brain injury. J. Neurosci. Res. 2016, 94, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiary, M.; Marzban, M.; Mehdizadeh, M.; Joghataei, M.T.; Khoei, S.; Mahabadi, V.P.; Laribi, B.; Tondar, M.; Moshkforoush, A. Comparison of transplantation of bone marrow stromal cells (BMSC) and stem cell mobilization by granulocyte colony stimulating factor after traumatic brain injury in rat. Iran. Biomed. J. 2010, 14, 142–149. [Google Scholar] [PubMed]
- Pennington, A.; Sava, V.; Song, S.; Patel, N.A.; Sanchez-Ramos, J. Direct actions of granulocyte-colony stimulating factor on human neuronal and monocytic cell lines. J. Alzheimer’s Dis. Parkinsonism. 2013, 3, 121. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ransohoff, R.M. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol. 2003, 15, 23–32. [Google Scholar] [CrossRef]
- Berman, J.W.; Guida, M.P.; Warren, J.; Amat, J.; Brosnan, C.F. Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J. Iummunol. 1996, 156, 3017–3023. [Google Scholar]
- Umehara, F.; Izumo, S.; Takeya, M.; Takahashi, K.; Sato, E.; Osame, M. Expression of adhesion molecules and monocyte chemoattractant protein-1 (MCP-1) in the spinal cord lesions in HTLV-I-associated myelopathy. Acta Neuropathol. 1996, 91, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.K.; Sharkey, J.; Andrews, P.J. The temporal expression, cellular localization, and inhibition of the chemokines MIP-2 and MCP-1 after traumatic brain injury in the rat. J. Neurotrauma 2009, 26, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Galasso, J.M.; Miller, M.J.; Cowell, R.M.; Harrison, J.K.; Warren, J.S.; Silverstein, F.S. Acute excitotoxic injury induces expression of monocyte chemoattractant protein-1 and its receptor, CCR2, in neonatal rat brain. Exp. Neurol. 2000, 165, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Van Der Voorn, P.; Tekstra, J.; Beelen, R.H.; Tensen, C.P.; van Der Valk, P.; de Groot, C.J. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am. J. Pathol. 1999, 154, 45–51. [Google Scholar] [CrossRef]
- Che, X.; Ye, W.; Panga, L.; Wu, D.C.; Yang, G.Y. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001, 902, 171–177. [Google Scholar] [CrossRef]
- Sozzani, S.; Zhou, D.; Locati, M.; Rieppi, M.; Proost, P.; Magazin, M.; Vita, N.; van Damme, J.; Mantovani, A. Receptors and transduction pathways for monocyte chemotactic protein-2 and monocyte chemotactic protein-3. Similarities and differences with MCP-1. J. Immunol. 1994, 152, 3615–3622. [Google Scholar] [PubMed]
- Liu, S.; Zhang, L.; Wu, Q.; Wu, Q.; Wang, T. Chemokine CCL2 induces apoptosis in cortex following traumatic brain injury. J. Mol. Neurosci. 2013, 51, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Kaneko, Y.; Bae, E.; Stahl, C.E.; Wang, Y.; van Loveren, H.; Sanberg, P.R.; Borlongan, C.V. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009, 1287, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Boyd, T.D.; Bennett, S.P.; Mori, T.; Governatori, N.; Runfeldt, M.; Norden, M.; Padmanabhan, J.; Neame, P.; Wefes, I.; Sanchez-Ramos, J.; Arendash, G.W. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J. Alzheimer’s Dis. Arkinsonism. 2010, 21, 507–518. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Mice | Treatments | Schedule |
|---|---|---|---|
| A | 8 | Saline + Saline | Daily × 3 days |
| B | 8 | Saline + G-CSF | Daily × 3 days |
| C | 16 | CCR-2 antag (0.5 or 2 mg/kg)+ Saline | Daily × 3 days |
| D | 16 | CCR-2 antag (0.5 or 2 mg/kg)+ G-CSF | Daily × 3 days |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Kong, X.; Acosta, S.; Sava, V.; Borlongan, C.V.; Sanchez-Ramos, J. Effects of an Inhibitor of Monocyte Recruitment on Recovery from Traumatic Brain Injury in Mice Treated with Granulocyte Colony-Stimulating Factor. Int. J. Mol. Sci. 2017, 18, 1418. https://doi.org/10.3390/ijms18071418
Song S, Kong X, Acosta S, Sava V, Borlongan CV, Sanchez-Ramos J. Effects of an Inhibitor of Monocyte Recruitment on Recovery from Traumatic Brain Injury in Mice Treated with Granulocyte Colony-Stimulating Factor. International Journal of Molecular Sciences. 2017; 18(7):1418. https://doi.org/10.3390/ijms18071418
Chicago/Turabian StyleSong, Shijie, Xiaoyuan Kong, Sandra Acosta, Vasyl Sava, Cesar V. Borlongan, and Juan Sanchez-Ramos. 2017. "Effects of an Inhibitor of Monocyte Recruitment on Recovery from Traumatic Brain Injury in Mice Treated with Granulocyte Colony-Stimulating Factor" International Journal of Molecular Sciences 18, no. 7: 1418. https://doi.org/10.3390/ijms18071418