Inter-Strain Differences in LINE-1 DNA Methylation in the Mouse Hematopoietic System in Response to Exposure to Ionizing Radiation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
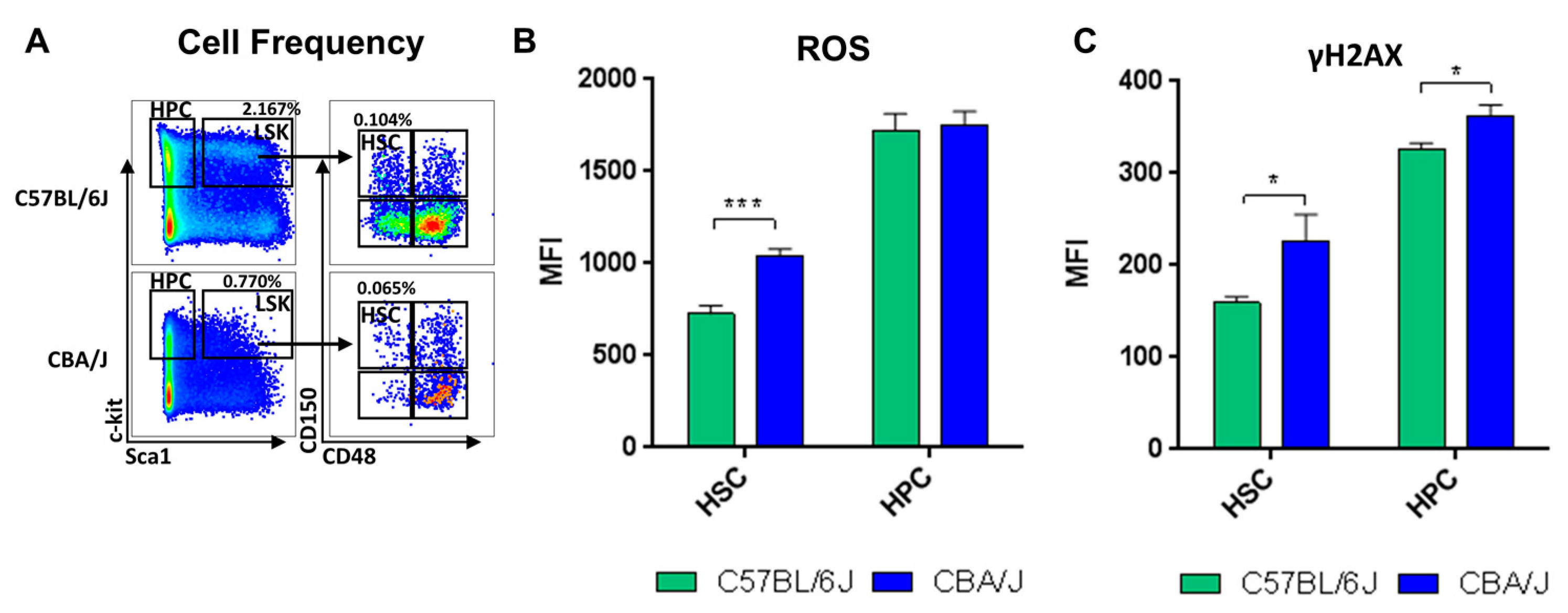
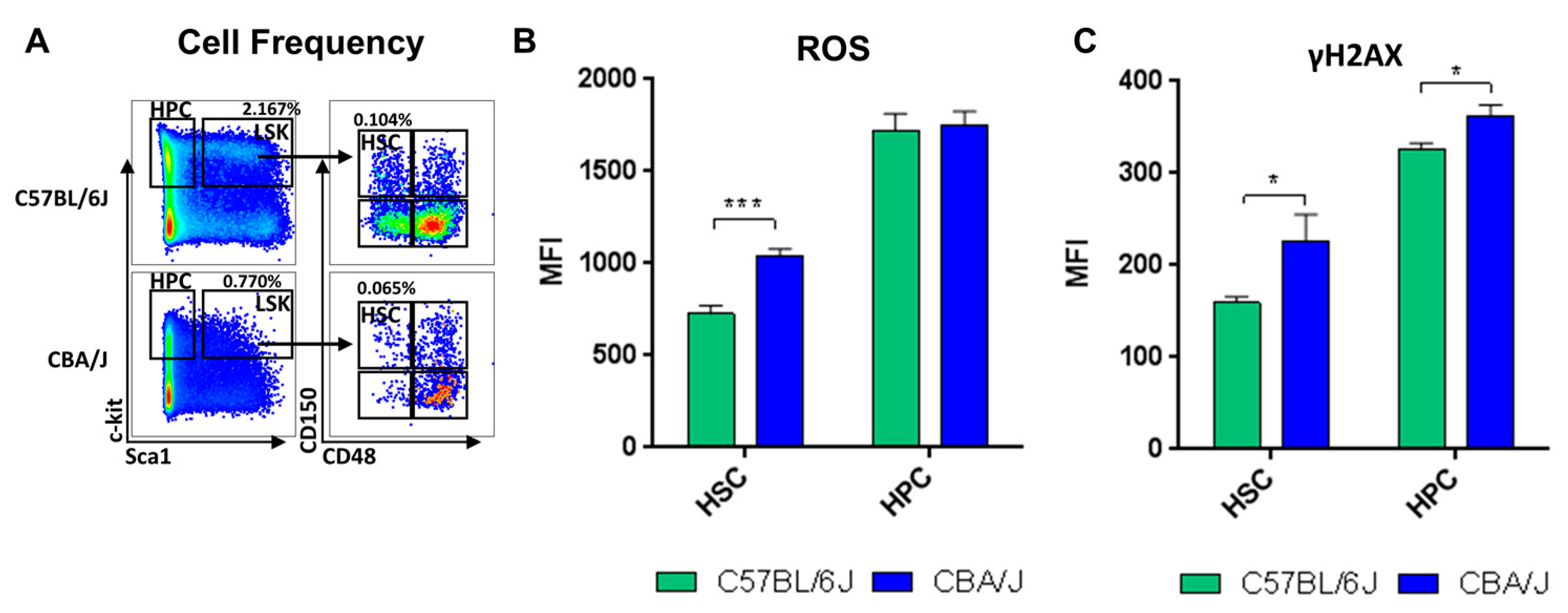
2.1. Comparative Analysis of Hematopoietic Profiles in Non-Irradiated Mice
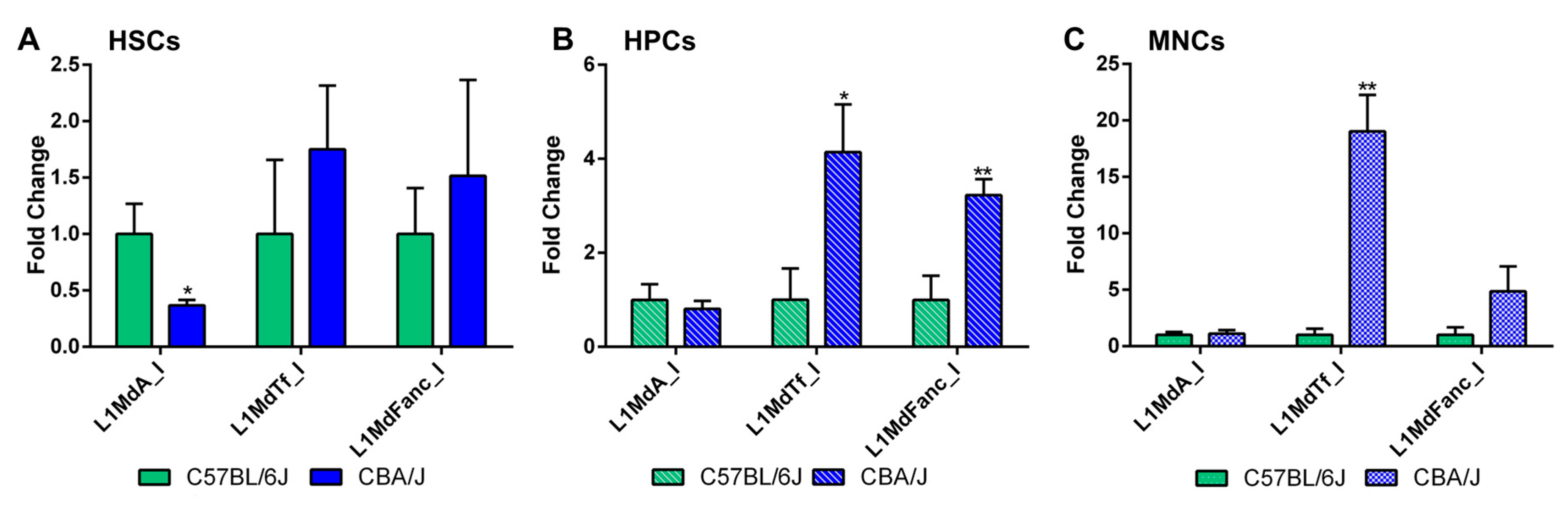
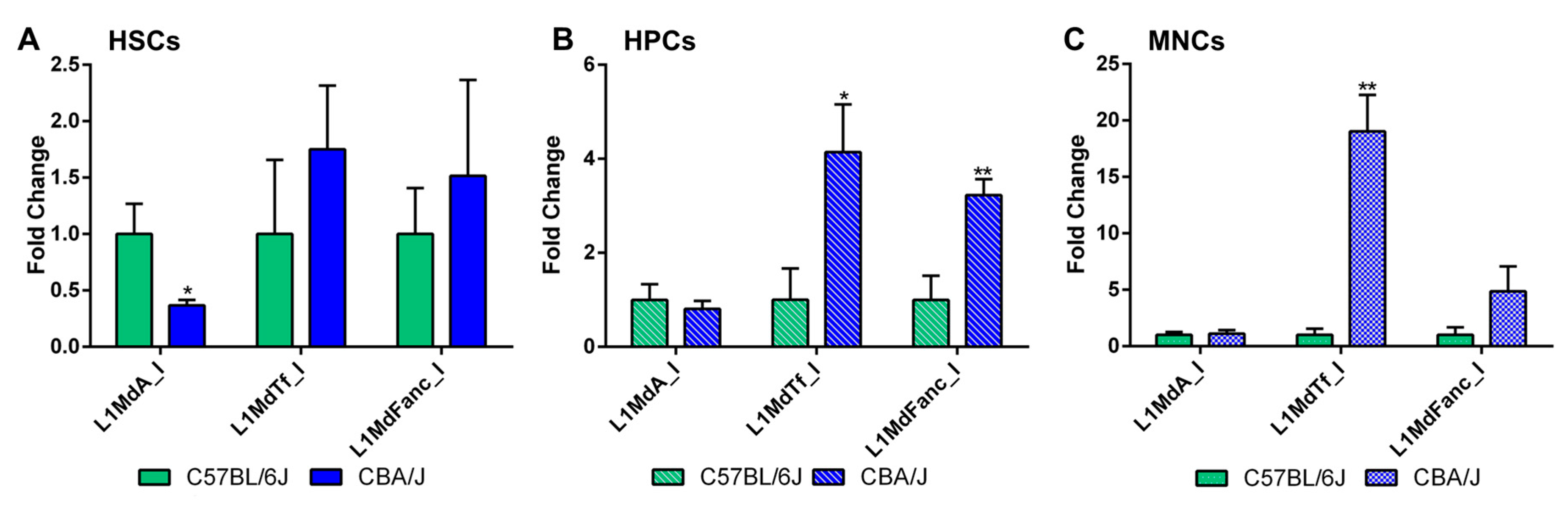
2.2. Comparative Analysis of LINE-1 Methylation in the Hematopoietic System in Non-Irradiated Mice
2.3. Comparative Analysis of LINE-1 Expression and DNA Methylation Machinery Expression in the Hematopoietic System in Non-Irradiated Mice
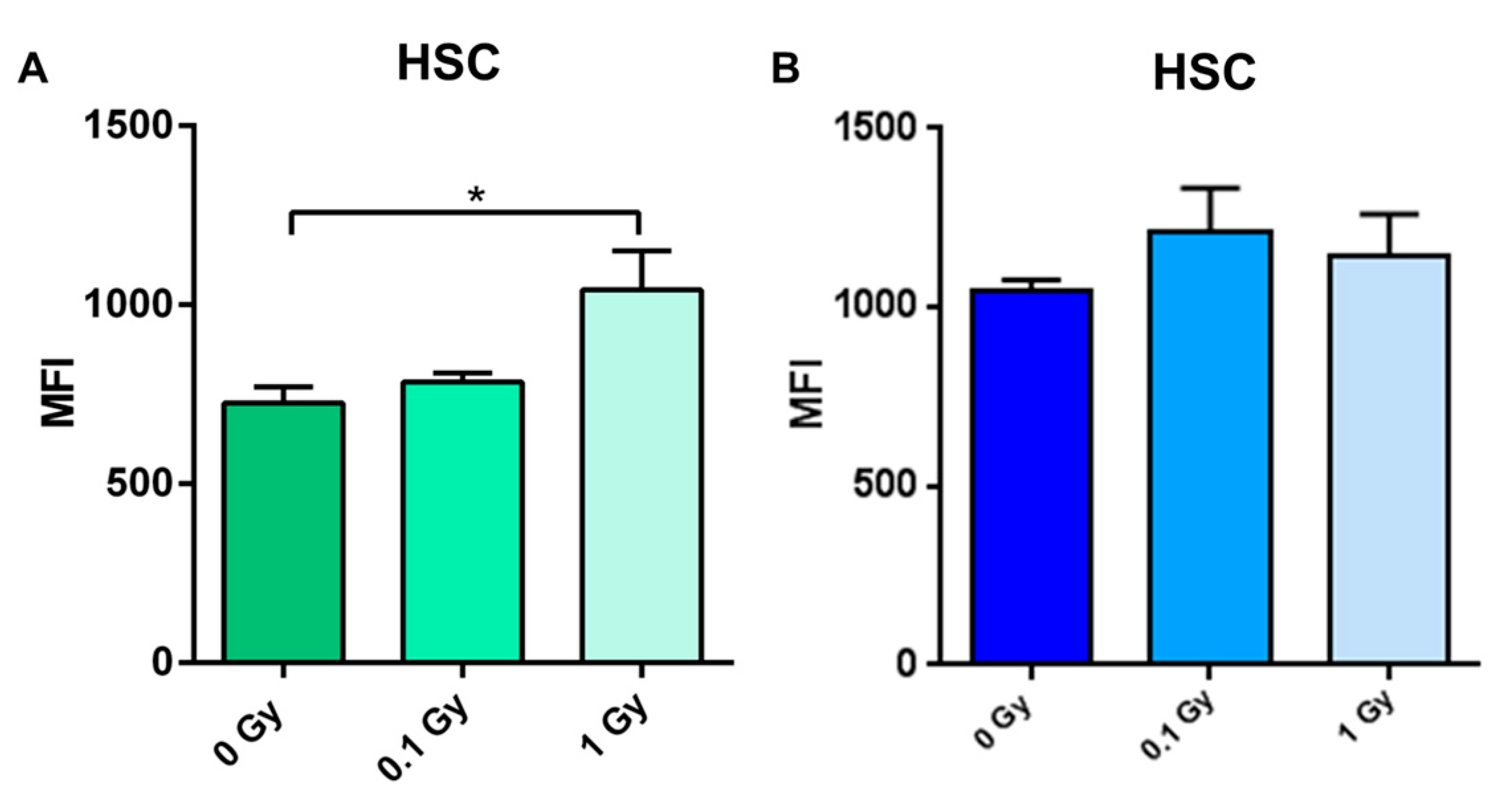
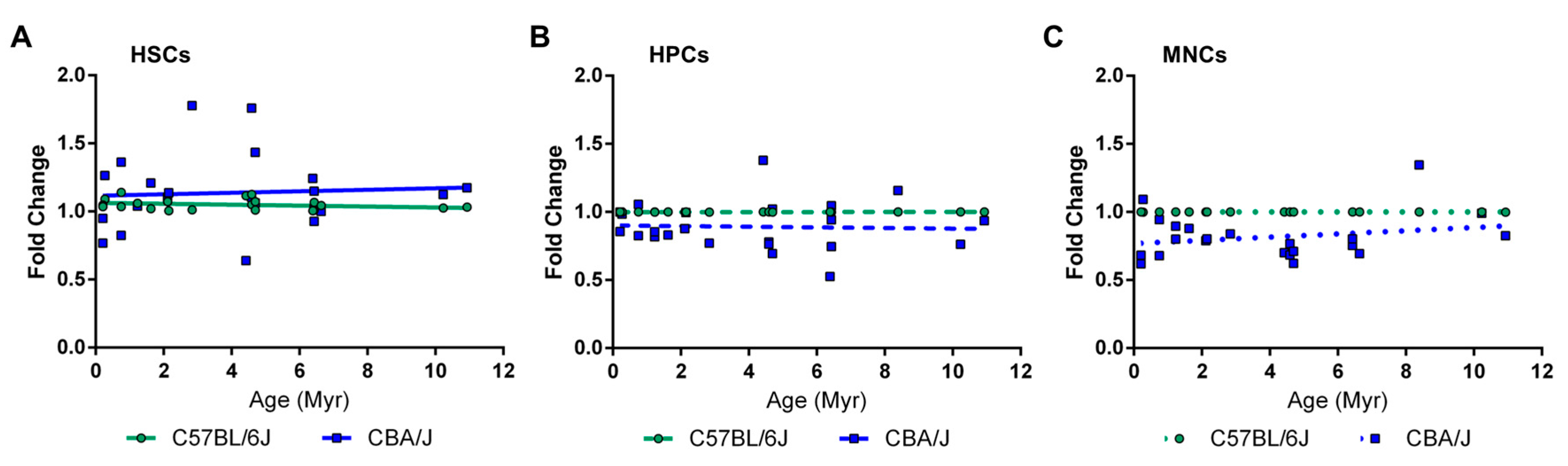
2.4. Long-Term Effects of Exposure to Ionizing Radiation on the Mouse Hematopoietic System
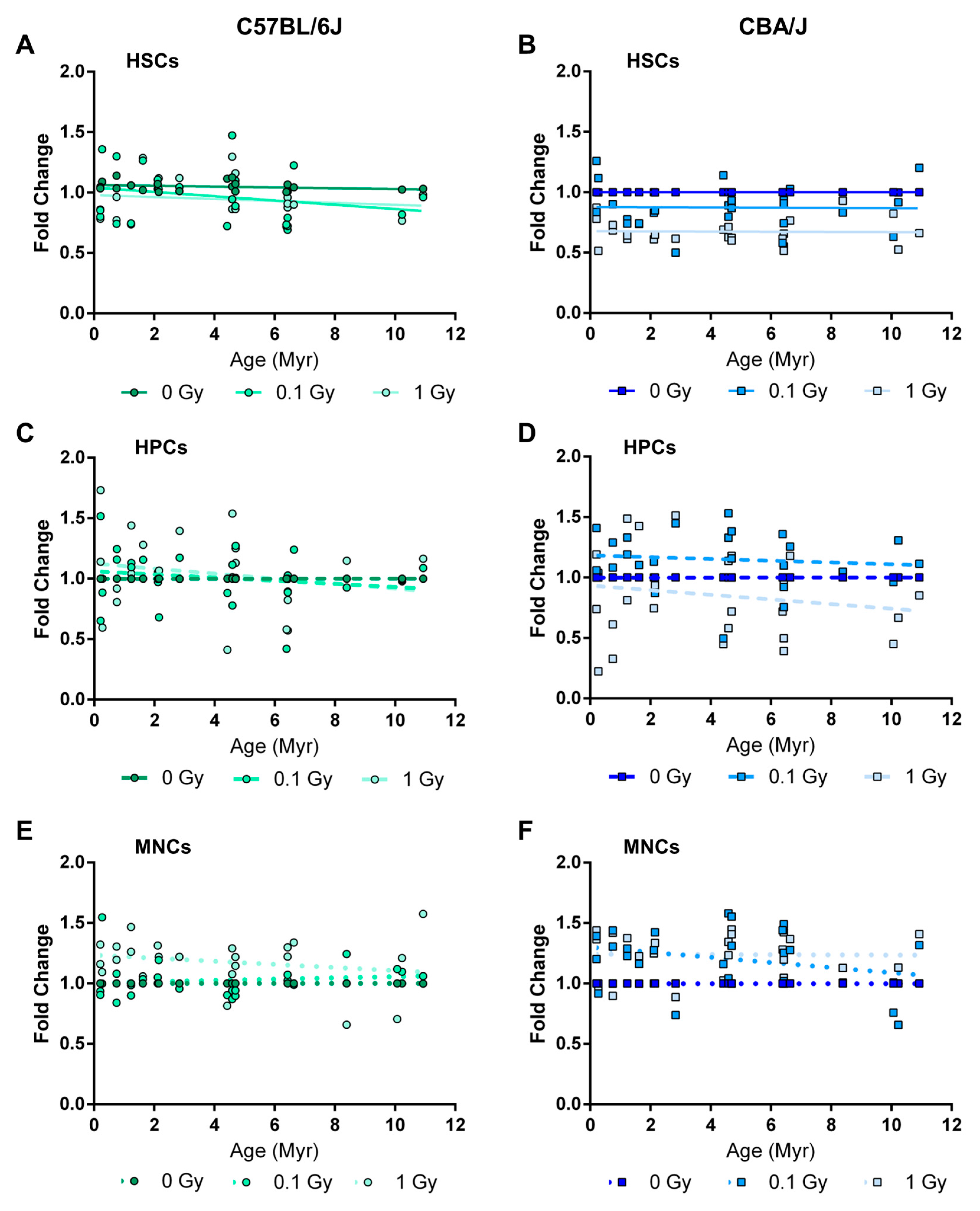
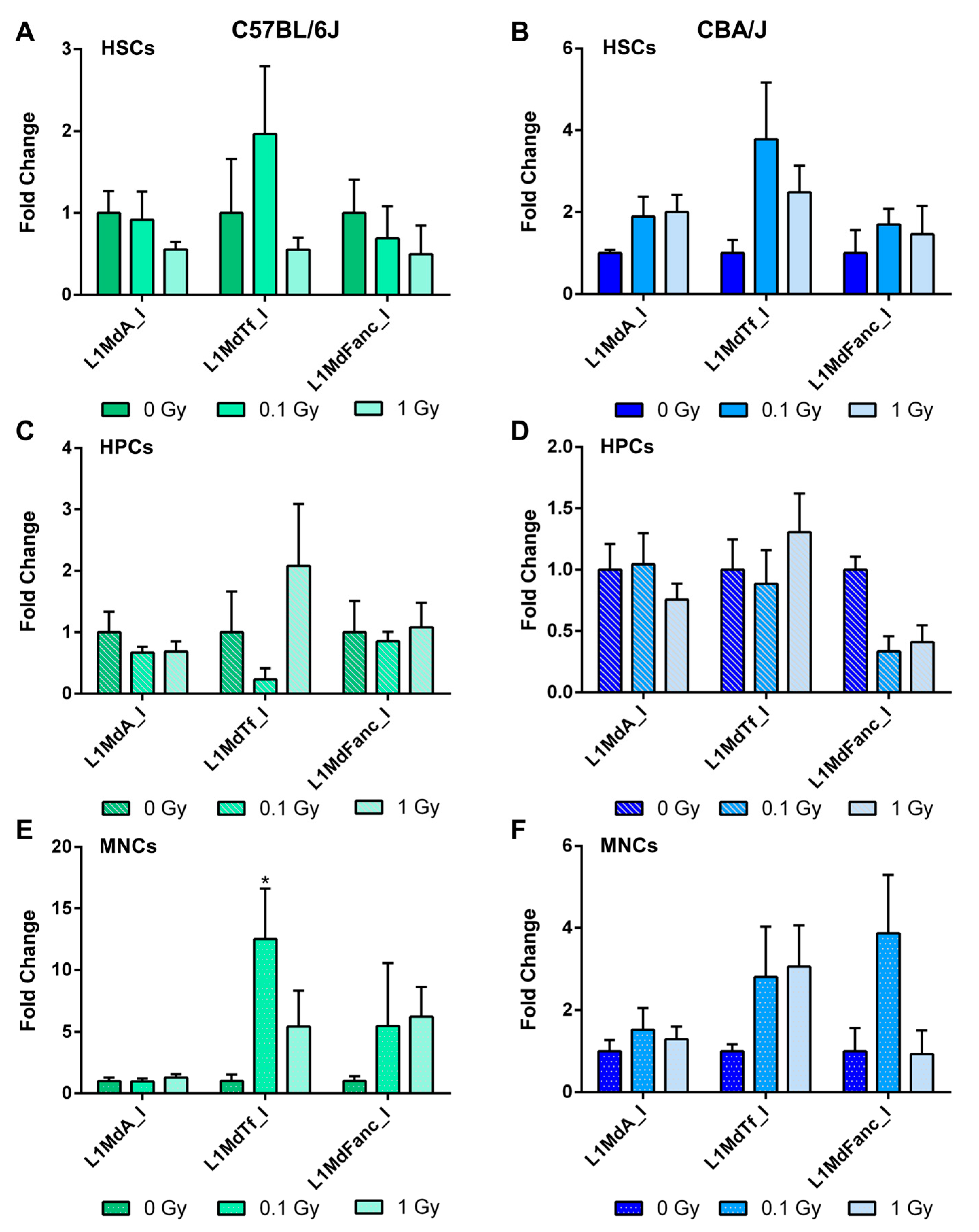
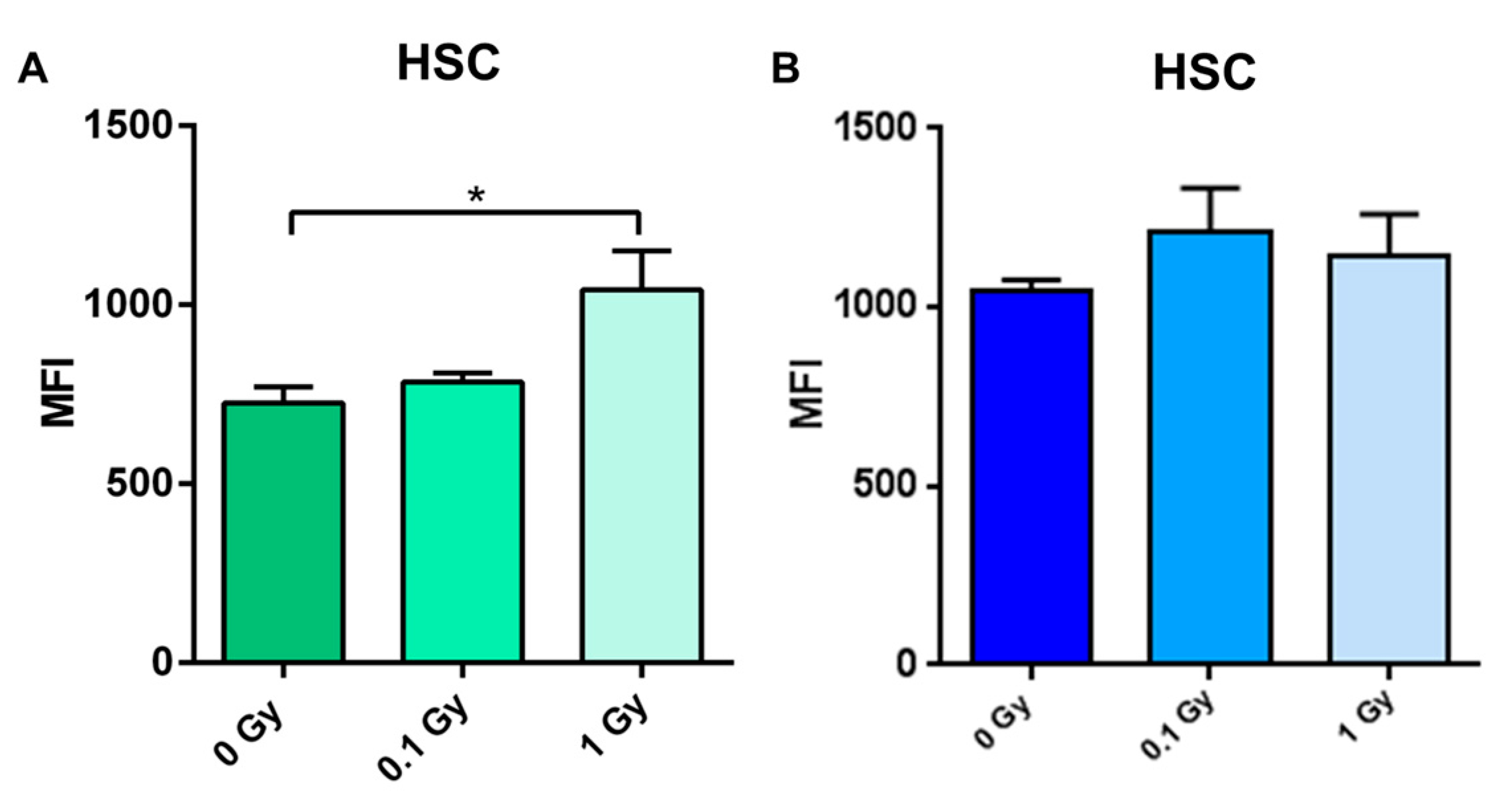
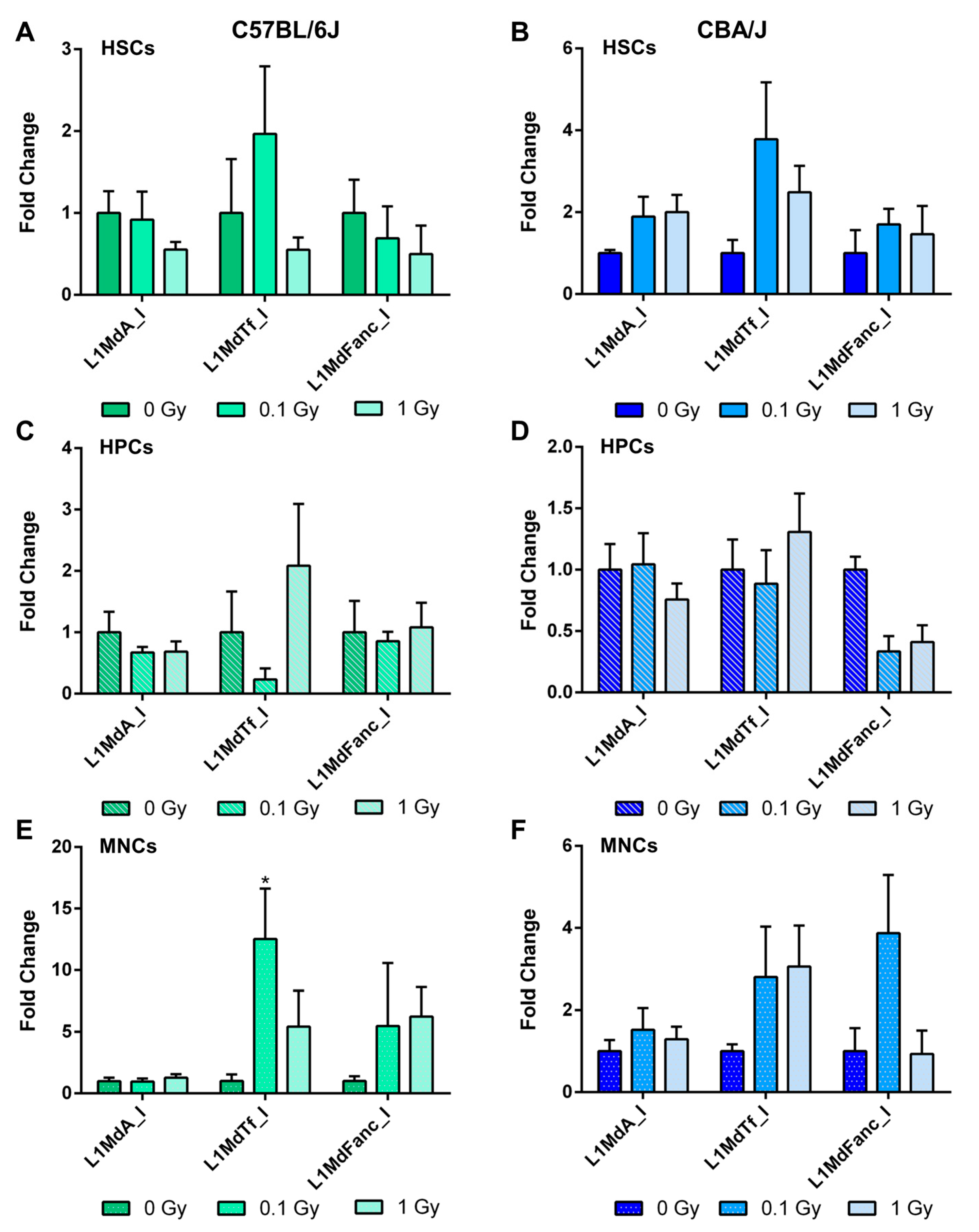
2.5. Long-Term Effects of Exposure to Ionizing Radiation on LINE-1 DNA Methylation in the Hematopoietic System
2.6. Long-Term Effects of Exposure to Ionizing Radiation on LINE-1 and DNA Methylation Machinery Expression in the Hematopoietic System
3. Discussion
4. Materials and Methods
4.1. Animals and Irradiation
4.2. Cell Collection
4.3. LINE-1 Families Consensus Sequences
4.4. Construction of LINE-1 Phylogenetic Tree
4.5. Nucleic Acid Isolation and Digestion
4.6. Analysis of DNA Methylation within the 5′-UTR of Selective LINE-1 Elements
4.7. Gene Expression Analysis
4.8. Analysis of the Frequencies and Numbers of Different Hematopoietic Cell Populations by Flow Cytometry
4.9. Analysis of the Levels of Intracellular ROS
4.10. DNA Damage Analysis
4.11. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FACS | Fluorescence activated cell sorting |
| HPC | Hematopoietic progenitor cell |
| HSC | Hematopoietic stem cell |
| HSPC | Hematopoietic stem and progenitor cell |
| IR | Ionizing radiation |
| LINE-1 | Long Interspersed Nucleotide Element 1 |
| MNC | Mononuclear cell |
| Myr | Million years |
| ORF | Open reading frame |
| ROS | Reactive oxygen species |
| SEM | Standard error of mean |
| TBI | Total body irradiation |
| UTR | Untranslated region |
References
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Koturbash, I. The Fine LINE: Methylation Drawing the Cancer Landscape. BioMed Res. Int. 2015, 2015, 131547. [Google Scholar] [CrossRef] [PubMed]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Belan, E. LINEs of evidence: Noncanonical DNA replication as an epigenetic determinant. Biol. Direct 2013, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.; Oakey, R.J. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet. 2013, 9, e1003234. [Google Scholar] [CrossRef] [PubMed]
- Cost, G.J.; Feng, Q.; Jacquier, A.; Boeke, J.D. Human L1 element target-primed reverse transcription in vitro. EMBO. J. 2002, 21, 5899–5910. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Narvaiza, I.; Kerman, B.E.; Pena, M.; Benner, C.; Marchetto, M.C.N.; Diedrich, J.K.; Aslanian, A.; Ma, J.; Moresco, J.J.; et al. Primate-specific ORF0 contributes to retrotransposon-mediated diversity. Cell 2015, 163, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Smit, A.; Boissinot, S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006, 16, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Sookdeo, A.; Hepp, C.M.; McClure, M.A.; Boissinot, S. Revisiting the evolution of mouse LINE-1 in the genomic era. Mob. DNA 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Kutanzi, K.R.; Koturbash, I. Effects of ionizing radiation on DNA methylation: From experimental biology to clinical applications. Int. J. Radiat. Biol. 2017, 93, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Boyko, A.; Rodriguez-Juarez, R.; McDonald, R.J.; Tryndyak, V.P.; Kovalchuk, I.; Pogribny, I.P.; Kovalchuk, O. Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis 2007, 28, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Chalbot, M.-C.G.; Lumen, A.; Ferguson, A.; Kavouras, I.G.; Koturbash, I. Response of transposable elements to environmental stressors. Mutat. Res. Rev. Mutat. Res. 2015, 765, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.R.; Sykes, P.J.; Blyth, B.J.; Bezak, E.; Lawrence, M.D.; Morel, K.L.; Ormsby, R.J. The methylation of DNA repeat elements is sex-dependent and temporally different in response to X radiation in radiosensitive and radioresistant mouse strains. Radiat. Res. 2014, 181, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.R.; Sykes, P.J.; Blyth, B.J.; Bezak, E.; Lawrence, M.D.; Morel, K.L.; Ormsby, R.J. A single whole-body low dose X-irradiation does not affect L1, B1 and IAP repeat element DNA methylation longitudinally. PLoS ONE 2014, 9, e93016. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Luo, Y.; Zhou, D. Hematopoietic stem cell injury induced by ionizing radiation. Antioxid. Redox Signal. 2014, 20, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. DNMT3A and DNMT3B have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2015, 263, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Pogribny, I.; Kovalchuk, O. Stable loss of global DNA methylation in the radiation-target tissue—A possible mechanism contributing to radiation carcinogenesis? Biochem. Biophys. Res. Commun. 2005, 337, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Shao, L.; Chang, J.; Feng, W.; Wang, Y.; Allen, A.R.; Turner, J.; Stewart, B.; Raber, J.; Zhou, D.; et al. Exposure to low-dose 56Fe-ion radiation induces long-term epigenetic alterations in mouse bone marrow hematopoietic progenitor and stem cells. Radiat. Res. 2014, 182, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Scholz, B.; Marschalek, R. Epigenetics and blood disorders. Br. J. Haematol. 2012, 158, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017, 168, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Giotopoulos, G.; McCormick, C.; Cole, C.; Zanker, A.; Jawad, M.; Brown, R.; Plumb, M. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp. Hematol. 2006, 34, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, J.; Koturbash, I.; Baker, M.; Kutanzi, K.; Kathiria, P.; Pogribny, I.P.; Sutherland, R.J.; Kovalchuk, O. Paternal cranial irradiation induces distant bystander DNA damage in the germline and leads to epigenetic alterations in the offspring. Cell Cycle Georget. Tex 2008, 7, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ehrlich, L.I.R.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Miousse, I.R.; Nzabarushimana, E.; Pathak, R.; Skinner, C.; Kutanzi, K.R.; Allen, A.R.; Raber, J.; Tackett, A.J.; Hauer-Jensen, M.; et al. Densely ionizing radiation affects DNA methylation of selective LINE-1 elements. Environ. Res. 2016, 150, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Miousse, I.R.; Sridharan, V.; Nzabarushimana, E.; Skinner, C.M.; Melnyk, S.B.; Pavliv, O.; Hauer-Jensen, M.; Nelson, G.A.; Boerma, M. Radiation-induced changes in DNA methylation of repetitive elements in the mouse heart. Mutat. Res. 2016, 787, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Rübe, C.E.; Grudzenski, S.; Kühne, M.; Dong, X.; Rief, N.; Löbrich, M.; Rübe, C. DNA double-strand break repair of blood lymphocytes and normal tissues analysed in a preclinical mouse model: Implications for radiosensitivity testing. Clin. Cancer Res. 2008, 14, 6546–6555. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Nakamura, A.; Kovalchuk, O.; Koturbash, I.; Mitchell, S.A.; Marino, S.A.; Brenner, D.J.; Bonner, W.M. DNA double-strand breaks form in bystander cells after microbeam irradiation of three-dimensional human tissue models. Cancer Res. 2007, 67, 4295–4302. [Google Scholar] [CrossRef] [PubMed]
- Rithidech, K.N.; Honikel, L.M.; Reungpathanaphong, P.; Tungjai, M.; Jangiam, W.; Whorton, E.B. Late-occurring chromosome aberrations and global DNA methylation in hematopoietic stem/progenitor cells of CBA/CaJ mice exposed to silicon (28Si) ions. Mutat. Res. 2015, 781, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Hájková, H.; Marková, J.; Haškovec, C.; Sárová, I.; Fuchs, O.; Kostečka, A.; Cetkovský, P.; Michalová, K.; Schwarz, J. Decreased DNA methylation in acute myeloid leukemia patients with DNMT3A mutations and prognostic implications of DNA methylation. Leuk. Res. 2012, 36, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Holz-Schietinger, C.; Matje, D.M.; Reich, N.O. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef] [PubMed]
- Nzabarushimana, E.; Prior, S.; Miousse, I.R.; Pathak, R.; Allen, A.R.; Latendresse, J.; Olsen, R.H.J.; Raber, J.; Hauer-Jensen, M.; Nelson, G.A.; et al. Combined exposure to protons and 56Fe leads to overexpression of Il13 and reactivation of repetitive elements in the mouse lung. Life Sci. Space Res. 2015, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aypar, U.; Morgan, W.F.; Baulch, J.E. Radiation-induced epigenetic alterations after low and high LET irradiations. Mutat. Res. 2011, 707, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Goetz, W.; Morgan, M.N.M.; Baulch, J.E. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat. Res. 2011, 175, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Nzabarushimana, E.; Miousse, I.R.; Shao, L.; Chang, J.; Allen, A.R.; Turner, J.; Stewart, B.; Raber, J.; Koturbash, I. Long-term epigenetic effects of exposure to low doses of 56Fe in the mouse lung. J. Radiat. Res. 2014, 55, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.; Ding, D.; Goetz, W.; Yang, A.J.; Baulch, J.E. High LET 56Fe ion irradiation induces tissue-specific changes in DNA methylation in the mouse. Environ. Mol. Mutagen. 2014, 55, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gu, L.; Adey, A.; Radlwimmer, B.; Wang, W.; Hovestadt, V.; Bähr, M.; Wolf, S.; Shendure, J.; Eils, R.; et al. Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 2013, 8, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Perez, J.L.; Moran, J.V. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Wang, Y.; Chang, J.; Luo, Y.; Meng, A.; Zhou, D. Hematopoietic stem cell senescence and cancer therapy-induced long-term bone marrow injury. Transl. Cancer Res. 2013, 2, 397–411. [Google Scholar] [PubMed]
- Laznicka, M.; Libansky, J.; Svoboda, M. Post-Irradiation Leukaemia in CBA Mice. Neoplasma 1964, 11, 385–388. [Google Scholar] [PubMed]
- Koturbash, I.; Beland, F.A.; Pogribny, I.P. Role of epigenetic events in chemical carcinogenesis--a justification for incorporating epigenetic evaluations in cancer risk assessment. Toxicol. Mech. Methods 2011, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Lambert, M.P.; van, V.K.; Demetriou, C.; Vineis, P.; Smith, M.T.; Straif, K.; Wild, C.P. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis 2013, 34, 1955–1967. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H.; Moran, J.V. The impact of L1 retrotransposons on the human genome. Nat. Genet. 1998, 19, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.M.; Duffield, A.S.; Symer, D.E.; Burns, K.H. Roles of retrotransposons in benign and malignant hematologic disease. Cellscience 2009, 6, 121–145. [Google Scholar] [PubMed]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Wolff, E.M.; Byun, H.-M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Tobacyk, J.; Melnyk, S.; James, S.J.; Cheema, A.K.; Boerma, M.; Hauer-Jensen, M.; Koturbash, I. One-carbon metabolism and ionizing radiation: A multifaceted interaction. Biomol. Concepts 2017, 8, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Batra, V.; Verma, P. Dietary l-methionine supplementation mitigates gamma-radiation induced global DNA hypomethylation: Enhanced metabolic flux towards S-adenosyl-l-methionine (SAM) biosynthesis increases genomic methylation potential. Food Chem. Toxicol. 2014, 69, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evol. Int. J. Org. Evol. 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miousse, I.R.; Chang, J.; Shao, L.; Pathak, R.; Nzabarushimana, É.; Kutanzi, K.R.; Landes, R.D.; Tackett, A.J.; Hauer-Jensen, M.; Zhou, D.; et al. Inter-Strain Differences in LINE-1 DNA Methylation in the Mouse Hematopoietic System in Response to Exposure to Ionizing Radiation. Int. J. Mol. Sci. 2017, 18, 1430. https://doi.org/10.3390/ijms18071430
Miousse IR, Chang J, Shao L, Pathak R, Nzabarushimana É, Kutanzi KR, Landes RD, Tackett AJ, Hauer-Jensen M, Zhou D, et al. Inter-Strain Differences in LINE-1 DNA Methylation in the Mouse Hematopoietic System in Response to Exposure to Ionizing Radiation. International Journal of Molecular Sciences. 2017; 18(7):1430. https://doi.org/10.3390/ijms18071430
Chicago/Turabian StyleMiousse, Isabelle R., Jianhui Chang, Lijian Shao, Rupak Pathak, Étienne Nzabarushimana, Kristy R. Kutanzi, Reid D. Landes, Alan J. Tackett, Martin Hauer-Jensen, Daohong Zhou, and et al. 2017. "Inter-Strain Differences in LINE-1 DNA Methylation in the Mouse Hematopoietic System in Response to Exposure to Ionizing Radiation" International Journal of Molecular Sciences 18, no. 7: 1430. https://doi.org/10.3390/ijms18071430