LPS-Induced Low-Grade Inflammation Increases Hypothalamic JNK Expression and Causes Central Insulin Resistance Irrespective of Body Weight Changes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
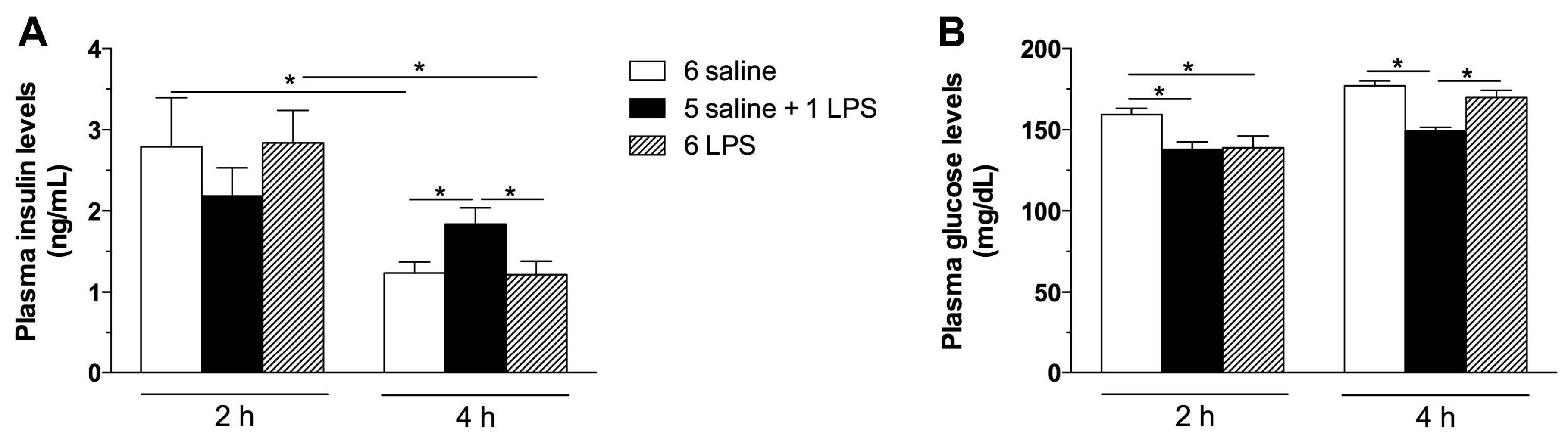
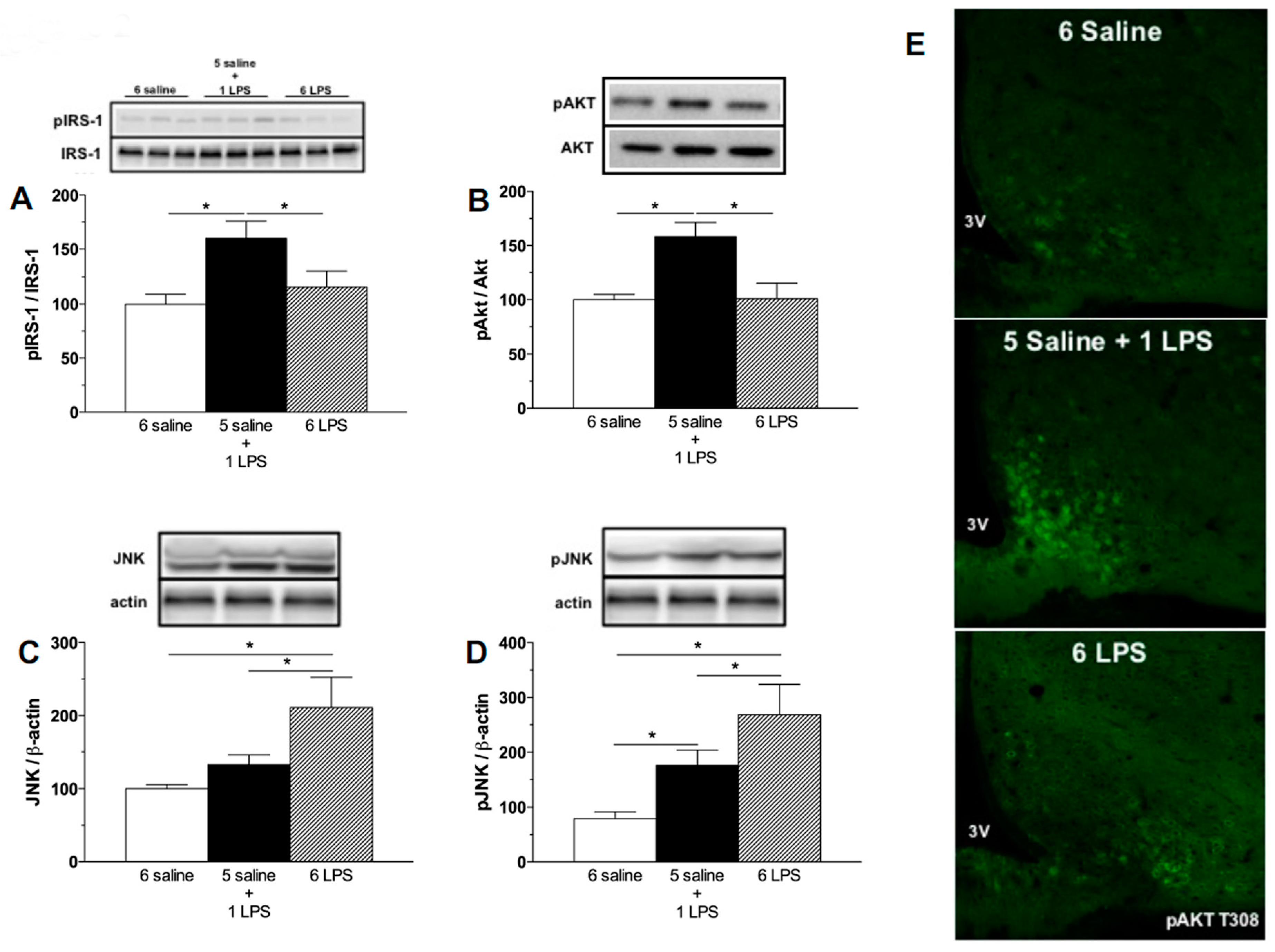
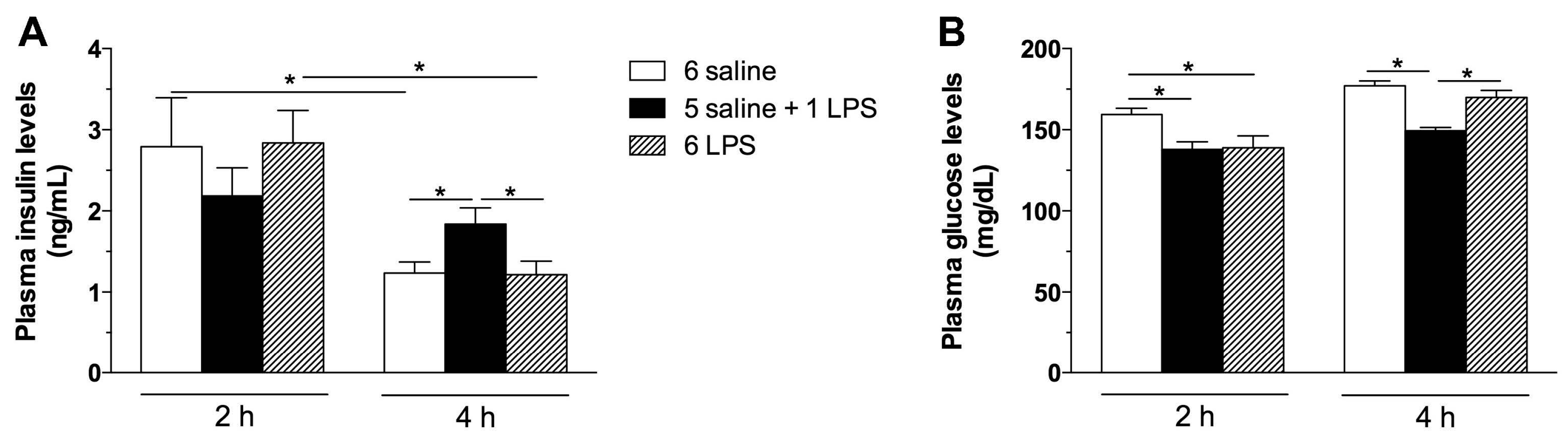
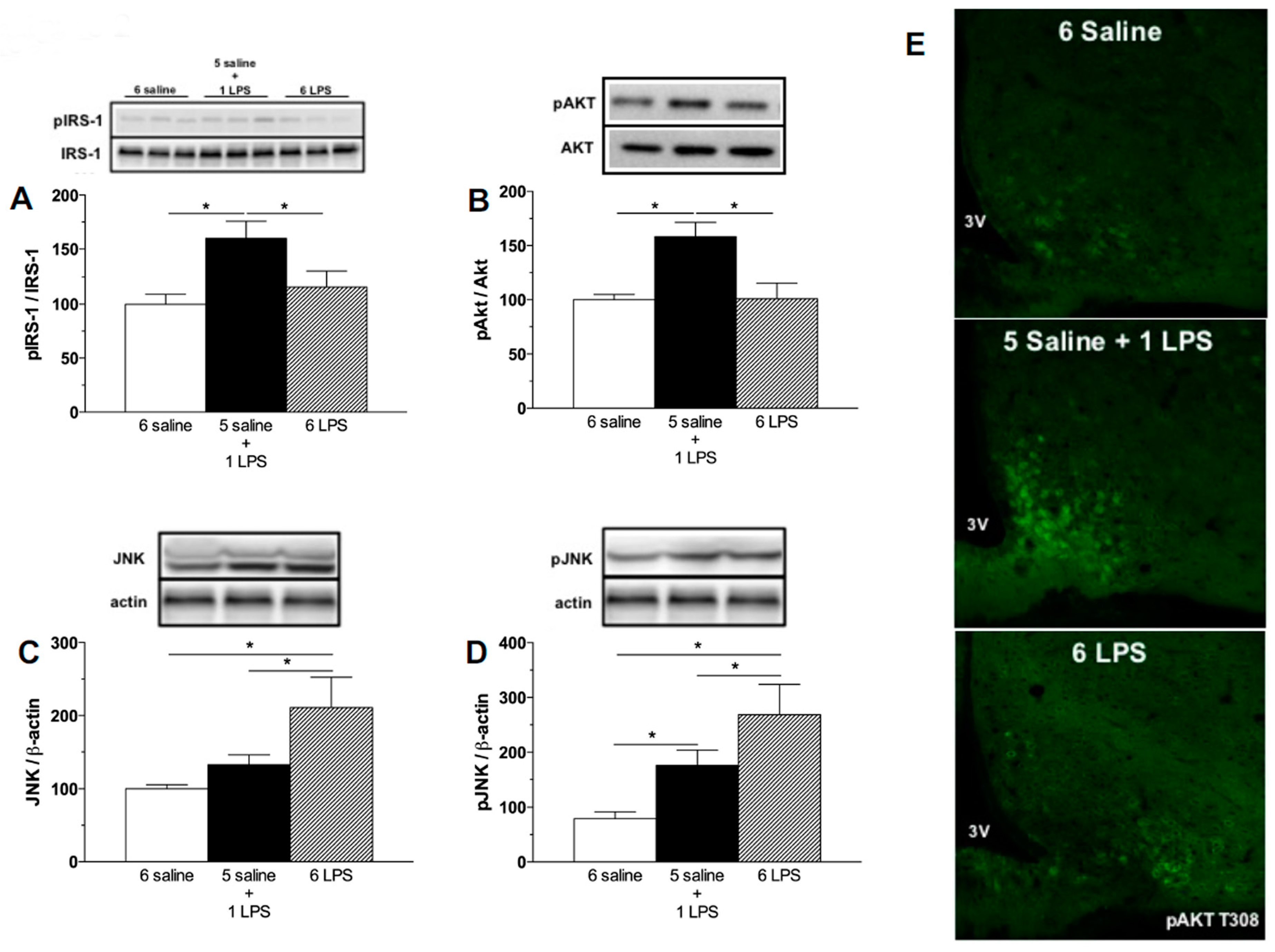
2.1. Acute, but Not Prolonged Endotoxemia, Promotes Insulin Secretion and Activates Insulin Signaling Cascades. Short and Long-Term LPS Exposure Increases N-Terminal Kinase (JNK) Phosphorylation in the Hypothalamus
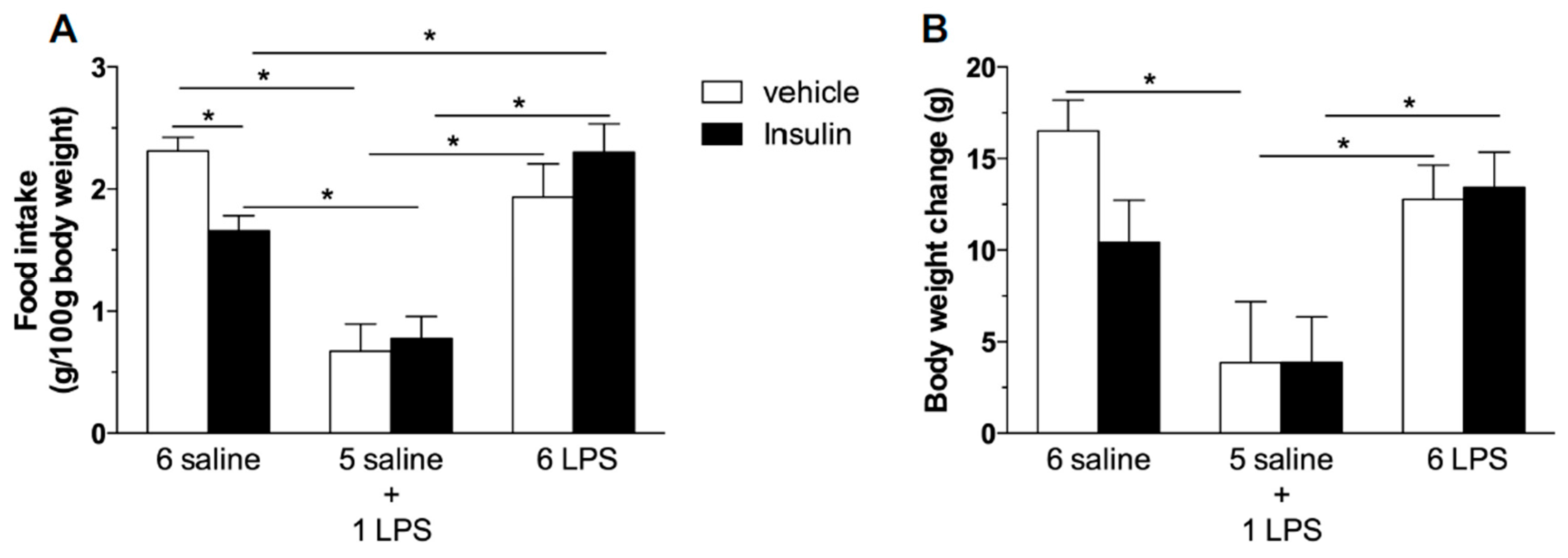
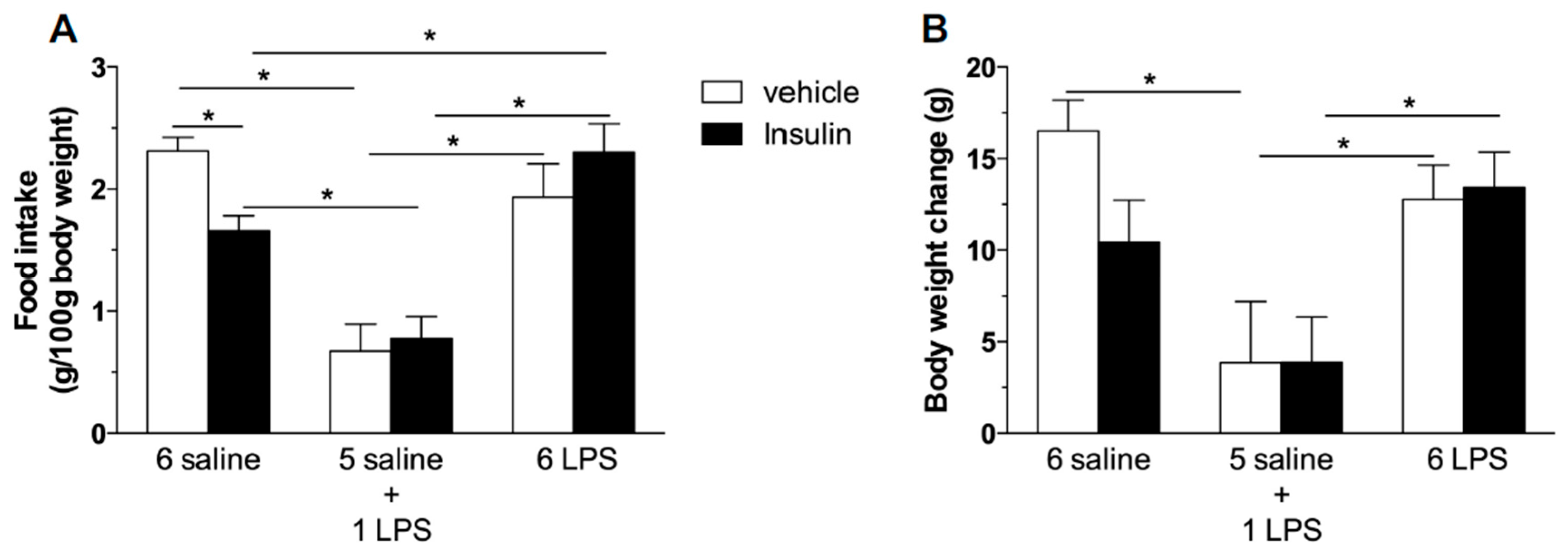
2.2. Hypophagic Effect of Central Insulin Injection Is Blunted in Acute and Prolonged Lipopolysaccharide—Treated Rats
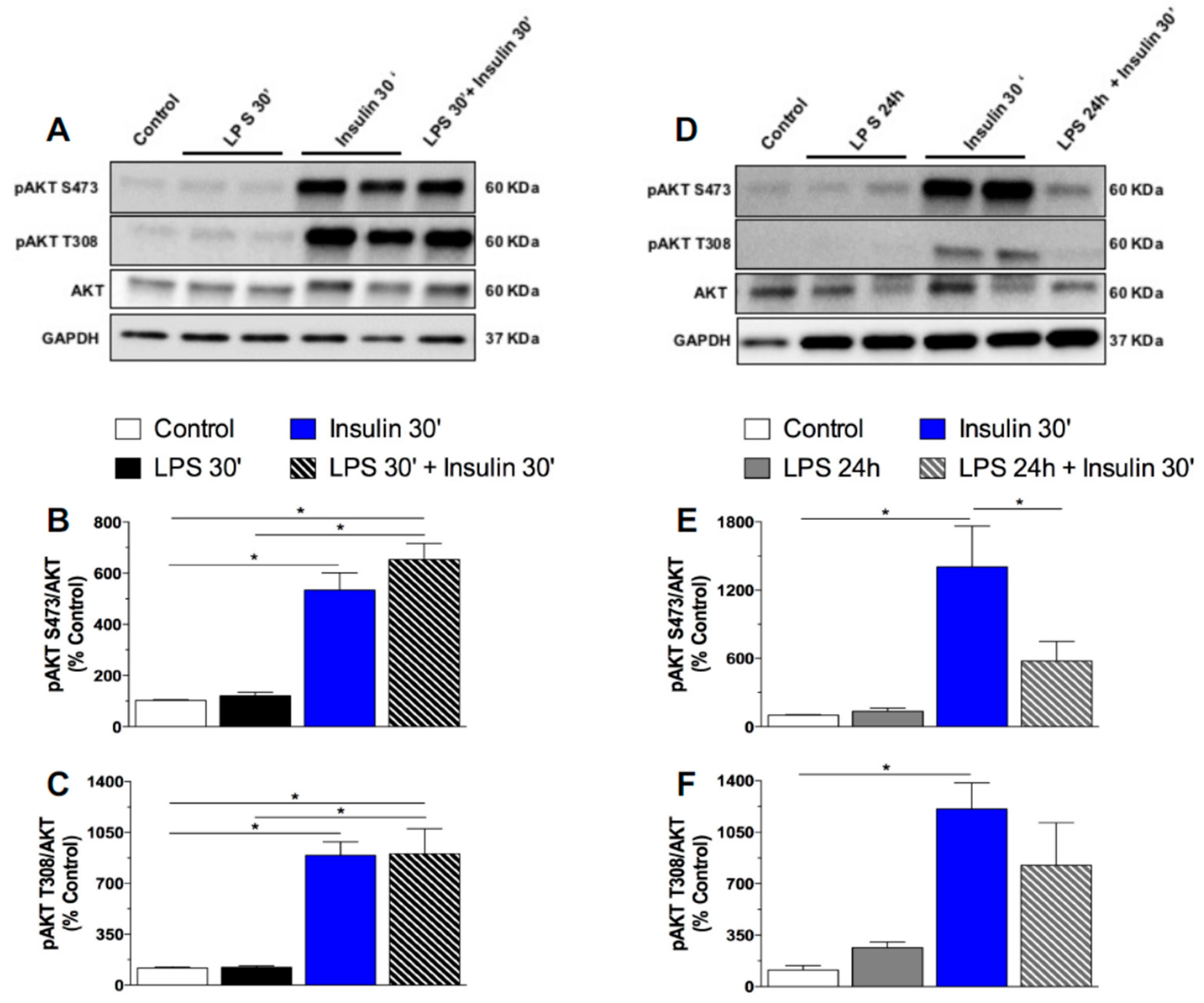
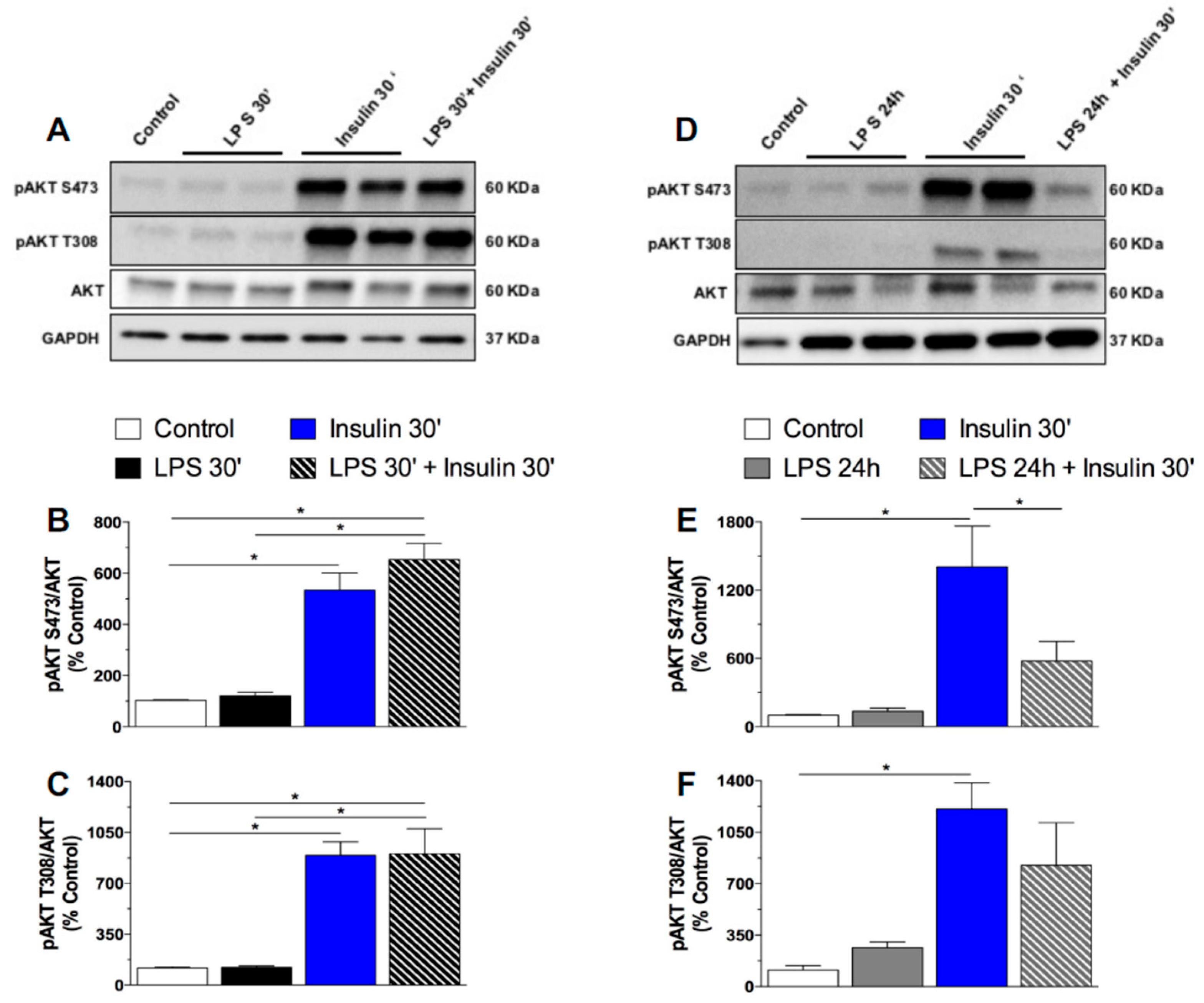
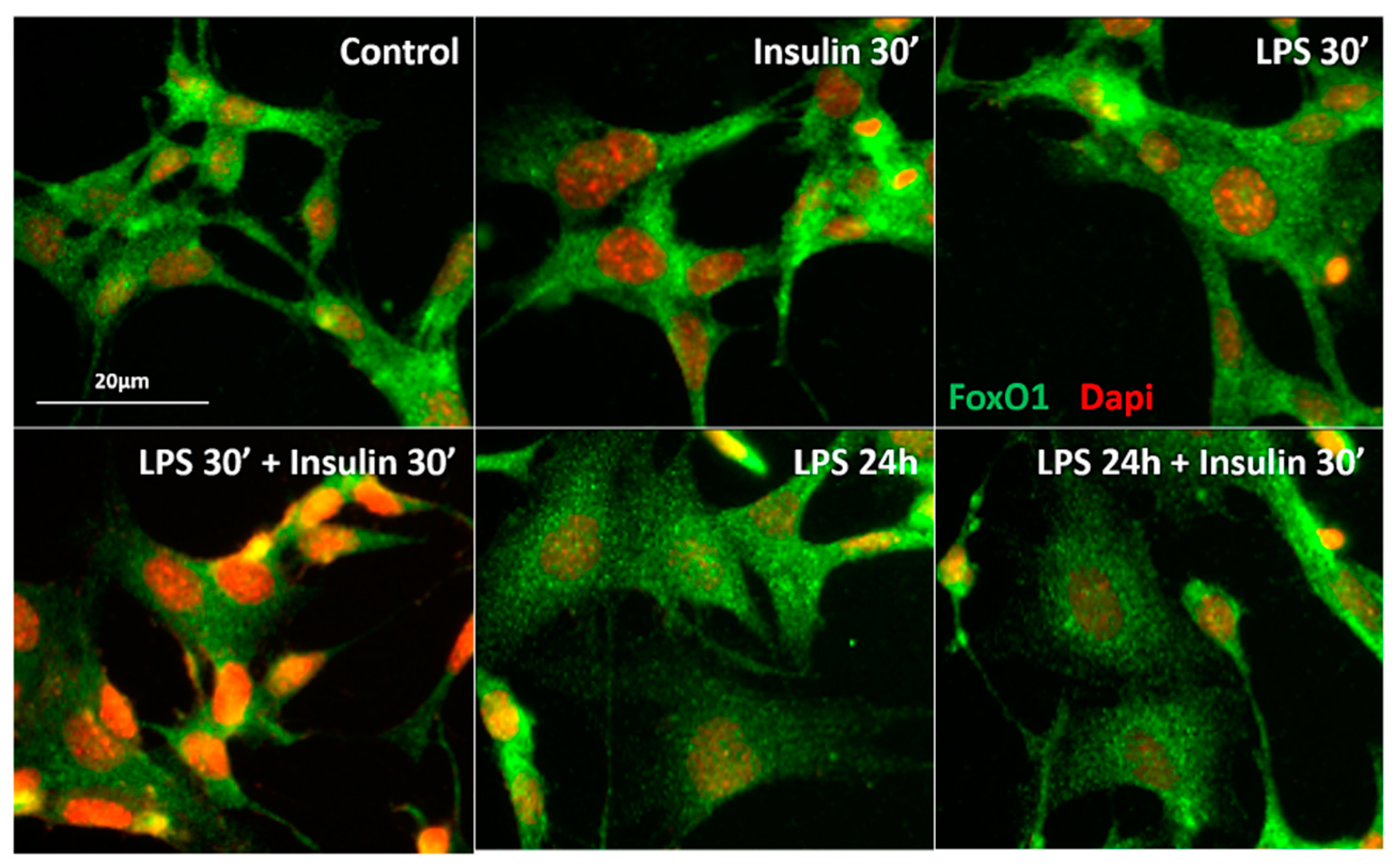
2.3. Prolonged LPS Treatment Blocks Insulin-Induced Protein Kinase B (AKT) Phosphorylation in the Culture of Neuronal Cells
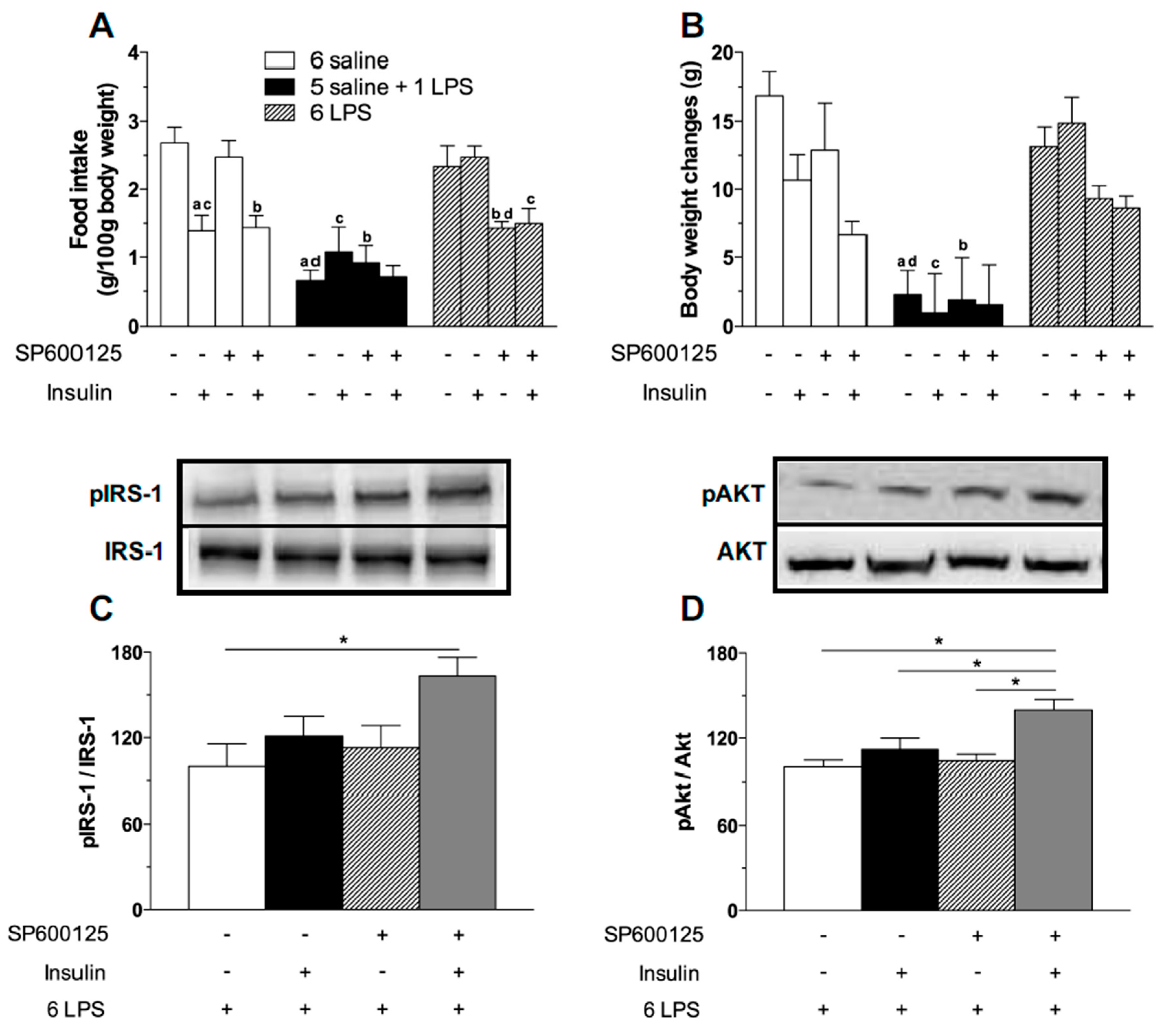
2.4. Central JNK Inhibition Restores the Hypothalamic Insulin Responsiveness in Rats Exposed to Repeated LPS Injections
3. Material and Methods
3.1. Animals
3.2. Experimental Procedures
3.2.1. Effect of Single or Repeated LPS Injections on Plasma Insulin and Glucose Levels, as Well as in the Hypothalamic Content of Insulin Signaling Proteins
3.2.2. Effect of Central Insulin Administration (ICV) Insulin Administration in Rats Treated with Single or Repeated LPS Injections on Food Intake and Body Weight
3.2.3. Effect of Short and Prolonged LPS Treatment on Insulin Signaling in GT1-7 Cells
3.2.4. Effect of JNK Inhibition on Food Intake and Body Weight Measurements after ICV Insulin Administration in Rats Treated with Single or Repeated LPS Injections.
3.2.5. Effects of JNK Inhibition on Hypothalamic Insulin Signaling in Rats Treated with Repeated LPS Injection
3.3. Cannula Implantation in the Lateral Ventricle
3.4. Western Blotting Analysis of Mediobasal Hypothalamus
3.5. Western Blotting Analysis in Cell Culture
3.6. Immunocytochemistry for FoxO1-Dapi
3.7. Plasma Insulin and Glucose Determination
3.8. Immunofluorescence for pAKT
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LPS | lipopolysaccharide |
| IRS-1 | insulin receptor substrate 1 |
| PI3K | phosphoinositide 3- kinase |
| JNK | Jun NH2-terminal kinase |
| AKT | Protein kinase B |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| β-actin | Beta-actin |
| FoxO1 | Forkhead box protein O1 |
| SP600125 | JNK inhibitor |
References
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Borges, B.C.; Rorato, R.; Avraham, Y.; da Silva, L.E.; Castro, M.; Vorobiav, L.; Berry, E.; Antunes-Rodrigues, J.; Elias, L.L. Leptin resistance and desensitization of hypophagia during prolonged inflammatory challenge. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E858–E869. [Google Scholar] [CrossRef] [PubMed]
- Borges, B.C.; Rorato, R.C.; Uchoa, E.T.; Marangon, P.B.; Elias, C.F.; Antunes-Rodrigues, J.; Elias, L.L. Protein tyrosine phosphatase-1B contributes to LPS-induced leptina resistance in male rats. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E40–E50. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Schwartz, M.W. Insulin and leptin revisited: Adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol. 2003, 24, 1–10. [Google Scholar] [CrossRef]
- Woods, S.C.; Lotter, E.C.; McKay, L.D.; Porte, J.R.D. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979, 282, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Brief, D.J.; Davis, J.D. Reduction of food intake and body weight by chronic intraventricular insulin infusion. Brain Res. Bull. 1984, 12, 571–575. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, M.; Posner, B.I.; Kopriwa, B.M.; Brawer, J.R. Insulin binding sites localized to nerve terminals in rat median eminence and arcuate nucleus. Science 1980, 207, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G.; Seeley, R.J.; Schwartz, M.W. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: A key mediator of insulin-induced anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Belgardt, B.F.; Brüning, J.C. Central insulin action in energy and glucose homeostasis. J. Clin. Investig. 2006, 116, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.Z.; Huan, J.N.; Gupta, S.; Pal, R.; Sahu, A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat. Neurosci. 2002, 5, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Borges, B.C.; Garcia-Galiano, D.; Rorato, R.; Elias, L.L.; Elias, C.F. PI3K p110 β subunit in leptin receptor expressing cells is required for the acute hypophagia induced by endotoxemia. Mol. Metab. 2016, 5, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Kim, Y.B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.C.; Air, E.L.; Coolen, L.M.; Strauss, R.; Jackman, A.; Clegg, D.J.; Seeley, R.J.; Woods, S.C. The catabolic action of insulin in the brain is mediated by melanocortins. J. Neurosci. 2002, 22, 9048–9052. [Google Scholar] [PubMed]
- Belgardt, B.F.; Mauer, J.; Wunderlich, F.T.; Ernst, M.B.; Pal, M.; Spohn, G.; Brönneke, H.S.; Brodesser, S.; Hampel, B.; Schauss, A.C.; et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 6028–6033. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kim, K.H.; Ahn, D.K.; Kim, H.S.; Kim, J.Y.; Lee, D.C.; Park, S.Y. Time-course changes of hormones and cytokines by lipopolysaccharide and its relation with anorexia. J. Physiol. Sci. 2007, 57, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Laviano, A.; Meguid, M.M.; Chen, C.; Rossi-Fanelli, F.; Hatakeyama, K. Involvement of plasma leptin, insulin and free tryptophan in cytokine-induced anorexia. Clin. Nutr. 2003, 22, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Zawalich, W.S.; Zawalich, K.C. Interleukin 1 is a potent stimulator of islet insulin secretion and phosphoinositide hydrolysis. Am. J. Physiol. 1989, 256, E19–E24. [Google Scholar] [PubMed]
- Cavaillon, J.M.; Adrie, C.; Fitting, C.; Adib-Conquy, M. Endotoxin tolerance: Is there a clinical relevance? J. Endotoxin Res. 2003, 9, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.R.; Chuang, J.C.; Xu, Y.; Choi, M.; et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Katsuki, H.; Okawara, M.; Kume, T.; Akaike, A. c-Jun N-terminal kinase inhibition and alpha-tocopherol protect midbrain dopaminergic neurons from interferon-gamma/lipopolysaccharide-induced injury without affecting nitric oxide production. J. Neurosci. Res. 2006, 83, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Tsaousidou, E.; Paeger, L.; Belgardt, B.F.; Pal, M.; Wunderlich, C.M.; Brönneke, H.; Collienne, U.; Hampel, B.; Wunderlich, F.T.; et al. Distinct Roles for JNK and IKK Activation in Agouti-Related Peptide Neurons in the Development of Obesity and Insulin Resistance. Cell Rep. 2014, 9, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.C.; Kemp, C.J.; Elias, C.F.; Abplanalp, W.; Herman, J.P.; Migrenne, S.; Lefevre, A.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Yu, F.; et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J. Clin. Investig. 2009, 119, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Marks, J.L.; Sipols, A.J.; Baskin, D.G.; Woods, S.C.; Kahn, S.E.; Porte, D. Central insulin administration reduces neuropeptide Y mRNA expression in the arcuate nucleus of food-deprived lean (Fa/Fa) but not obese (fa/fa) Zucker rats. Endocrinology 1991, 128, 2645–2647. [Google Scholar] [CrossRef] [PubMed]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 2002, 5, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Kim, F.; Schwartz, M.W.; Wisse, B.E. Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1122–E1130. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Howard, S.; LoGrass, P.V. Pharmacological Inhibition of c-Jun N-terminal Kinase Reduces Food Intake and Sensitizes Leptin’s Anorectic Signaling Actions. Sci. Rep. 2017, 7, 41795. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.K.; Piper, M.L.; Olofsson, L.E.; Xu, A.W. Functional role of c-Jun-N-terminal kinase in feeding regulation. Endocrinology 2010, 151, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Milanski, M.; Coope, A.; Torsoni, A.S.; Ropelle, E.; Carvalho, D.P.; Carvalheira, J.B.; Velloso, L.A. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 2011, 152, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Thomas, D.R.; Wilson, M.M. Cachexia: Pathophysiology and clinical relevance. Am. J. Clin. Nutr. 2006, 83, 735–743. [Google Scholar] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rorato, R.; Borges, B.d.C.; Uchoa, E.T.; Antunes-Rodrigues, J.; Elias, C.F.; Elias, L.L.K. LPS-Induced Low-Grade Inflammation Increases Hypothalamic JNK Expression and Causes Central Insulin Resistance Irrespective of Body Weight Changes. Int. J. Mol. Sci. 2017, 18, 1431. https://doi.org/10.3390/ijms18071431
Rorato R, Borges BdC, Uchoa ET, Antunes-Rodrigues J, Elias CF, Elias LLK. LPS-Induced Low-Grade Inflammation Increases Hypothalamic JNK Expression and Causes Central Insulin Resistance Irrespective of Body Weight Changes. International Journal of Molecular Sciences. 2017; 18(7):1431. https://doi.org/10.3390/ijms18071431
Chicago/Turabian StyleRorato, Rodrigo, Beatriz de Carvalho Borges, Ernane Torres Uchoa, José Antunes-Rodrigues, Carol Fuzeti Elias, and Lucila Leico Kagohara Elias. 2017. "LPS-Induced Low-Grade Inflammation Increases Hypothalamic JNK Expression and Causes Central Insulin Resistance Irrespective of Body Weight Changes" International Journal of Molecular Sciences 18, no. 7: 1431. https://doi.org/10.3390/ijms18071431