Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective


Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI 48109, USA
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(7), 1596; https://doi.org/10.3390/ijms18071596
Submission received: 19 June 2017
/
Revised: 12 July 2017
/
Accepted: 21 July 2017
/
Published: 23 July 2017
(This article belongs to the Special Issue Neuronal Protein Homeostasis in Health and Disease)
Abstract
:Neurodegenerative diseases in humans are frequently associated with prominent accumulation of toxic protein inclusions and defective organelles. Autophagy is a process of bulk lysosomal degradation that eliminates these harmful substances and maintains the subcellular environmental quality. In support of autophagy’s importance in neuronal homeostasis, several genetic mutations that interfere with autophagic processes were found to be associated with familial neurodegenerative disorders. In addition, genetic mutations in autophagy-regulating genes provoked neurodegenerative phenotypes in animal models. The Drosophila model significantly contributed to these recent developments, which led to the theory that autophagy dysregulation is one of the major underlying causes of human neurodegenerative disorders. In the current review, we discuss how studies using Drosophila enhanced our understanding of the relationship between autophagy and neurodegenerative processes.
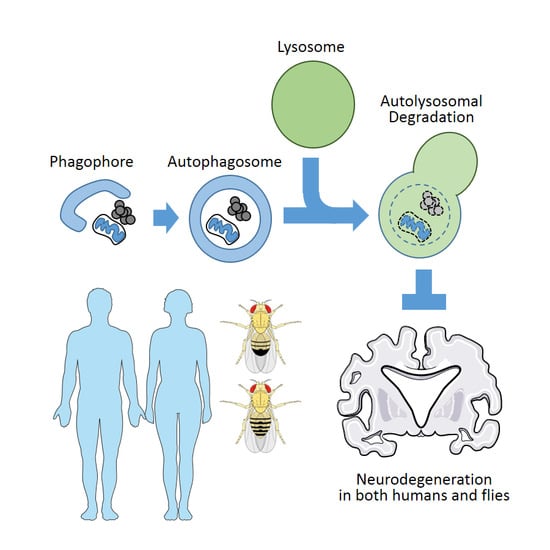
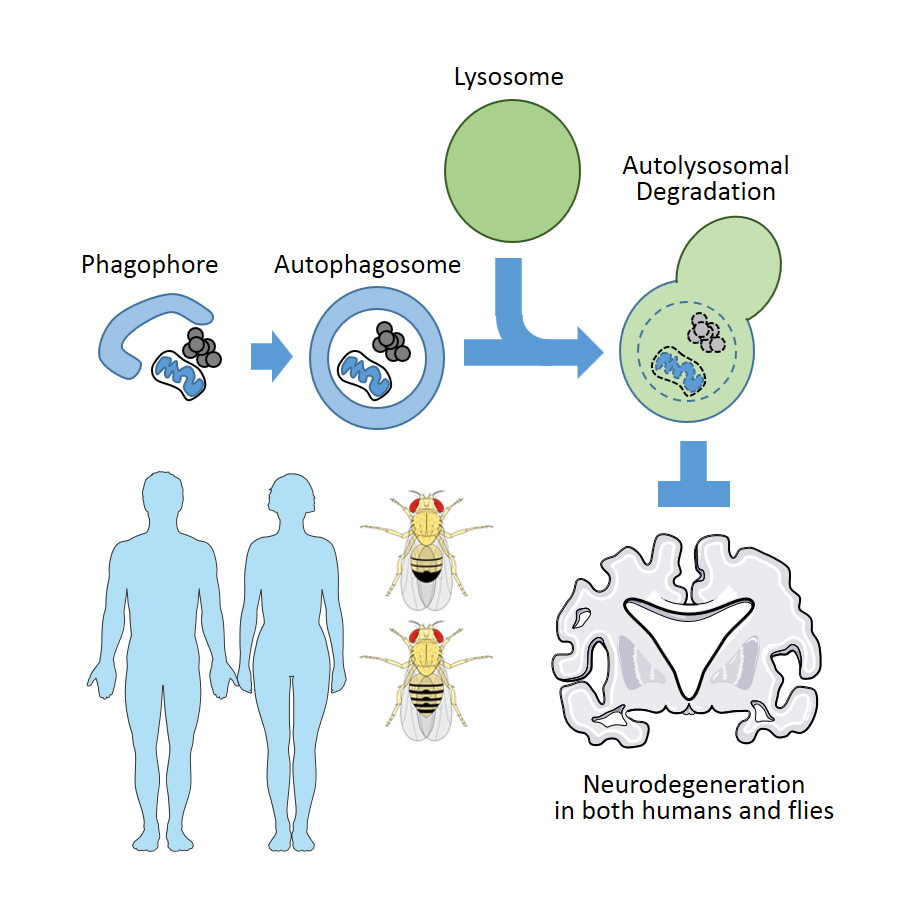
1. Autophagy in Human Neurodegenerative Diseases
1.1. Protein Inclusions and Mitochondrial Dysfunction: Hallmarks of Neurodegeneration
Neurodegeneration is a progressive loss of neuronal structure and function, resulting in an irreversible decline in cognitive abilities such as memory and decision-making, as well as bodily coordination and mobility. Of the many different types of neurodegeneration, the most notable and common forms include Alzheimer’s, Parkinson’s and Huntington’s diseases. Although these diseases manifest with distinct clinical features and affect different regions of the brain, they share very similar molecular pathologies at the cellular level, such as the accumulation of misfolded protein aggregates, which are toxic to the cells [1]. More specifically, Alzheimer’s disease is characterized by the extracellular accumulation of β-amyloid protein (Aβ) plaques as well as the intracellular accumulation of tau, a microtubule-associated protein. Parkinson’s disease is similarly characterized by intracellular aggregates of α-synuclein and ubiquitin, known as Lewy bodies. Huntington’s disease also exhibits protein inclusions, consisting of a mutant Huntingtin protein that has a polyglutamine expansion. In addition to the protein aggregates, these diseases also exhibit an accumulation of dysfunctional mitochondria, which produces excessive reactive oxygen species that damage cellular macromolecules such as DNA, lipids and proteins [2]. The accumulation of protein inclusions and damaged mitochondria are often considered histological hallmarks of neurodegeneration.
1.2. Genetic Mutations that Lead to Protein Inclusions
How these molecular pathologies develop has remained a mystery for a long time. For some of these diseases, it was clearly shown that a genetic mutation is the primary cause of the disease. Huntington’s disease is caused by a polyglutamine expansion mutation in the gene encoding the Huntingtin protein; the mutant protein with a polyglutamine tract accumulates inside neurons as toxic protein inclusions [3]. Similar polyglutamine expansion mutations are found in other types of neurodegenerative diseases such as several forms of spinocerebellar ataxia [3]. Additionally, for a minor subset of Parkinson’s cases, several genetic mutations were identified as causing the disease by upregulating the α-synuclein-encoding genes, leading to the accumulation and aggregation of the protein [4]. Although these genetic mutations explain protein inclusion pathologies in a small number of patient populations, the predominant portion of Alzheimer’s and Parkinson’s cases do not clearly associate with genetic mutations. This obscures our complete understanding of the etiology of these diseases.
1.3. Autophagy—One of the Major Homeostatic Processes to Eliminate Protein Aggregates and Damaged Mitochondria
Autophagy is primarily defined as a bulk degradation system for various cytoplasmic components [5]; however, it can also be selectively directed towards a specific substrate such as aggregated proteins or dysfunctional mitochondria through specific adaptor proteins such as p62/SQSTM1 [6]. Autophagy is initiated by the formation of a phagophore, a double-membrane structure, around the target cargo. The phagophore engulfs the target, forming an autophagosome that sequesters its cargo from the cytosol. The autophagosome then fuses with a lysosome, which contains digestive enzymes to degrade the target cargo. Finally, degradation products are released from channels on the lysosomal membrane. Through this process, autophagy removes toxic protein aggregates, excessive nutrient deposits such as lipid droplets, and dysfunctional organelles such as damaged mitochondria [7,8].
Pioneering work in the yeast system has identified a number of genetic components that are critical for the formation of phagophores and autophagosomes [9]. These include the autophagy-regulating genes (ATGs). For example, ATG1 (ULK1), which complexes with ATG13 and ATG17 (FIP200), is a protein kinase whose activity is critical for the initiation of autophagy [10]. The Class III phosphatidylinositol 3-kinase (PI3K) complex, composed of Atg6 (BECN1), Vps15, and Vps34, is also important for autophagy initiation [11]. Atg18 (WIPIs) is one of the effectors of the class III PI3K activity in autophagy [12]. Upon ATG1 activation, class III PI3K activation and autophagy initiation, other ATG proteins are required for the formation and elongation of the phagophore [9]. Specifically, ATG9 is required for formation of the phagophore, and ATG5 plays a role in elongation and maturation of an autophagosome. During autophagy, ATG5 is covalently bound to ATG12, a ubiquitin-like protein, by the actions of the E1- and E2-like enzymes ATG7 and ATG10, respectively. ATG5–ATG12 conjugates, complexed with another protein ATG16, are also important for activating the lipidation process for ATG8, mediated by ATG7 and ATG3. Lipidated ATG8 is a marker for active autophagy because it is one of the major constituents of the autophagosomal membrane. All of these ATG proteins are strikingly well conserved throughout the eukaryotic kingdom, and they play a critical autophagy-regulating role in both animals and yeasts [5]. These mechanisms defined by using the yeast system exemplify the capability of genetic analyses in model organisms to approach complicated mechanisms in higher eukaryotes.
1.4. Role of Autophagy in Neuronal Homeostasis and Preventing Neurodegeneration
The autophagic removal of protein aggregates and dysfunctional mitochondria could be very critical for non-dividing cells of multicellular organisms, such as neurons and myocytes [7,13,14,15,16,17]. These long-lived cells are expected to accumulate unfolded or misfolded proteins as well as damaged organelles. However, autophagosomes are very scarcely found in neurons either in intact tissue or in primary culture; therefore, autophagy was initially thought to be inactive in neurons. It was later found that autophagosomes are not abundant in neurons because neurons have a highly efficient lysosomal degradation system, which removes autophagosomes rapidly. Accordingly, the inhibition of lysosomal activity in primary cultured neurons led to the accumulation of autophagosomes even in the absence of any autophagy-stimulating signals such as starvation, demonstrating that autophagy is highly active in neuronal cells [18]. Animal models with genetic ablation of core autophagy genes (e.g., ATG genes) also revealed a critical function of autophagy in maintaining neuronal health [19,20,21,22,23,24]. Autophagy gene mutations in flies and mice induced prominent deterioration of neuronal health [19,20,21,22,23,24]. Specifically, autophagy-deficient neurons resulted in the excessive accumulation of ubiquitin/p62-positive protein inclusions and damaged mitochondria, cellular hallmarks of neurodegenerative disease [19,20,21,22,23,24]. The presence of protein inclusions and damaged mitochondria in cells leads to neurodegenerative pathologies such as neuronal dysfunction and cell death, as well as subsequent deficits in cognitive function and mobility [19,20,21,22,23,24]. These results suggest that autophagy plays an important role in maintaining neuronal homeostasis and preventing neurodegenerative pathologies [15,16].
1.5. Implication of Autophagy in Alzheimer’s Disease
Ultramicroscopic analysis of degenerating brain tissue from Alzheimer’s patients revealed the prominent accumulation of non-degraded autophagic vesicles, indicating that autophagic flux was arrested [25]. Cell or animal models for Alzheimer’s disease also frequently exhibited defective autophagic flux and the accumulation of autolysosomal vesicles. For example, the presenilin 1 (PS1) mutation, which is the most frequent cause of early onset familial Alzheimer’s disease, is known to cause autophagic arrest in both mouse [26,27] and cell culture models [28]. The expression of the amyloid precursor protein (APP) with mutations found in familial Alzheimer’s disease also triggered similar autophagic-lysosomal pathologies [29]. The expression of Aβ, the cleavage product of APP, also induced the accumulation of large autophagic vesicles in the Drosophila brain [30]. This evidence suggests that Alzheimer’s disease involves the defective regulation of autophagy at the autophagosomal degradation step [15].
1.6. Implication of Autophagy in Parkinson’s Disease
Parkinson’s disease is another prominent neurodegenerative disease with phenotypes indicative of defective autophagy. Analysis of familial Parkinson’s disease identified several causative genes called PARK genes [4]. Most of these genes, including PARK1 and PARK4 (SNCA), PARK2 (Parkin), PARK5 (UCHL1), PARK6 (PINK1), PARK7 (DJ-1), PARK8 (LRRK2) and PARK9 (ATP13A2), were found to play some role in the autophagic elimination of ubiquitinated proteins or damaged mitochondria [31,32,33,34]. In addition, diseased tissues from Parkinson’s patients present with protein aggregates and damaged mitochondria [31,32,33,34]. Therefore, autophagy also seems to prevent the progression of Parkinson’s disease pathologies. The Drosophila model has contributed significantly to understanding these PARK components, as well as other genetic components associated with neurodegeneration, in the regulation of autophagic processes.
2. Drosophila as a Model for Investigating the Autophagy-Neurodegeneration Relationship
2.1. Drosophila as a Genetically Tractable Model Organism
Drosophila has been extensively described as a useful genetic model of human diseases [35,36,37]. Historically, the Drosophila model has been proven to be valuable for genetic analyses including genome-scale screening studies. Drosophila has high genomic and physiological homology to mammals, and around 65% of human disease-causing genes are conserved [35,37]. Drosophila also have a rapid life cycle (~2 weeks per generation), high fecundity (one wild-type Drosophila female can lay up to 100 eggs in a day) and relatively low maintenance costs that allow for genome-scale genetic screening [36]. These attributes cannot be matched by any other vertebrate animal model. A number of genetic components in diverse signaling pathways, such as Ras-MAPK, Sonic Hedgehog (Hh), TGF-β (Dpp), Wnt (Wg) and Notch signaling pathways, were identified and characterized originally in the Drosophila model. Most of the signaling molecules identified from genetic screens in Drosophila were shown to be functionally conserved in mammalian organisms in subsequent studies, demonstrating the suitability of Drosophila for studying molecular signaling pathways in animals.
2.2. Drosophila as a Model for Investigating Autophagy
The first genetic study of autophagy in Drosophila was published in 2003 [38]. Soon, the Drosophila system proved to be highly efficient for analyzing the autophagy-related signaling network. Using Drosophila genetics, nutrient-regulated target of rapamycin (TOR) signaling [39], insulin/growth factor-regulated PI3K signaling [40,41] and nuclear hormone receptor signaling [41] were demonstrated to be part of the autophagy-regulating network that responds to both environmental and developmental cues. The genetic ablation of Atg7, a core autophagy gene, resulted in deterioration of neuronal health and longevity [21], revealing a role for autophagy in physiological homeostasis. Later, we showed that Atg1/ULK1 and Sestrin, another autophagy regulator [10,42,43], are critical for eliminating damaged mitochondria and maintaining functional and structural integrity of cardiac and skeletal muscle [44]. Mutations in other autophagy-related genes have also been shown to provoke neurodegeneration in Drosophila (see Table 1). Importantly, in mammalian systems, these autophagy components are also critical for metabolic (e.g., liver and muscle) and long-lived organs (e.g., neurons) [19,20,45,46]. This suggests that the Drosophila system is highly relevant for investigating the physiological role of autophagy in higher order animals.
2.3. Drosophila as a Model for Investigating Human Neurodegenerative Diseases
Completion of genome sequencing revealed that approximately 65% of human disease-causing genes are conserved in Drosophila [35,37]. Even in cases where a gene is not found in the Drosophila genome, the function of such a gene can still be investigated in the fly through ectopic expression of the human protein [36]. Genes associated with familial neurodegenerative diseases in humans and proteins associated with disease pathophysiology, such as Aβ, mutated tau (Alzheimer’s disease), α-synuclein, mutated Pink, Parkin and Dj-1 (Parkinson’s disease), mutated Huntingtin and a polyglutamine tract (Huntington’s disease and ataxia) caused neuropathologies in flies that are very similar to what was observed in human patients [52,53,54]. Notably, the mis-expression of some of these components resulted in apparent degeneration of eye photoreceptor cells, which can be non-invasively examined and easily scored for genetic interaction assays [52]. These studies establish Drosophila as a robust model for investigating neurodegenerative diseases, as well as their relationship with autophagy pathways (Figure 1).
2.4. Drosophila Models of Alzheimer’s Disease: β-amyloid and Tau Flies and Their Autophagic Phenotypes
β-amyloid (Aβ), a cleavage product of amyloid precursor protein (APP), is the major component of extracellular plaques found in the brain tissue of patients with Alzheimer’s disease. Recent evidence demonstrates that intracellular accumulation of Aβ could precede extracellular accumulation and promote the early pathogenesis of Alzheimer’s disease [55]. Consistent with this intraneuronal role, the expression of Aβ in Drosophila neurons produced prominent degenerative phenotypes [30,56]. These include visible external degeneration of the ommatidia structure in the eyes [30], as well as dramatically reduced lifespan and mobility [56]. Of note, Aβ neurotoxicity is accompanied by a strong arrest of autophagic flux, which was assayed by degradation of exogenously expressed green fluorescent protein (GFP) [30]. Neurons expressing Aβ accumulate large amounts of non-degraded and oftentimes damaged autophagosomes, which are visible through Atg8-GFP reporters as well as through a transmission electron microscope (TEM) [30]. These histological pathologies bear a great resemblance to what is observed in human Alzheimer’s patients [25] and suggest that intracellular Aβ interferes with autophagosome degradation.
In addition to Aβ, Alzheimer’s disease is also characterized by intracellular accumulation of tau, a microtubule-associated protein, suggesting that tau neurotoxicity could be another important pathogenetic mechanism for the disease [57]. Tau neurotoxicity was also successfully modelled in Drosophila by overexpressing human tau protein [58,59]. Like the Aβ model, tau overexpression produced strong neurodegeneration that provoked ommatidia malformation [59], brain tissue degeneration and reduced lifespan [58]. Several genetic modifier screens were performed using either an overexpression library (enhancer and promoter (EP) lines) [60] or a lethal mutant library (P-lethal lines) [61], which isolated signaling proteins that post-translationally modify tau. Some autophagy components, such as Atg6, were also isolated from the screens [61]. Importantly, upregulation of autophagy significantly reduced tau toxicity [62], suggesting that autophagy also protects neuronal health in this fly model.
For both models, disease-associated pathogenic variants, such as the Arctic mutation (E22G) of Aβ [56] and frontotemporal dementia-associated mutations (V337M and R406W) of tau [58] produced stronger pathogenetic effects in the flies. Furthermore, the expression of both Aβ and tau synergistically accelerated neurodegeneration in Drosophila [63,64]. These findings indicate that the Aβ and tau models in Drosophila are relevant for investigating Alzheimer’s disease and suggest that these neurodegenerative pathologies are closely associated with dysfunctional or dysregulated autophagy.
2.5. Drosophila Models of Parkinson’s Disease: α-Synuclein, PARK Genes and How Autophagic Processes are Compromised in These Models
Flies expressing the human α-synuclein (α-syn) gene were one of the first transgenic models in Drosophila to focus on investigating a major human neurodegenerative disease [65]. α-syn is the primary structural component of a Lewy body, an insoluble protein aggregate found in brain tissues of Parkinson’s patients, as well as some other neurodegenerative diseases. Of note, several mutations in α-syn-encoding genes, such as point mutations and gene amplifications, are associated with familial Parkinson’s disease [4]. As in humans, the overexpression of α-syn in Drosophila provoked prominent neurodegeneration associated with Lewy body-like structures, neuronal cell death and mobility impairment [65]. α-syn overexpression also caused neurodegeneration in the Drosophila eye, including degeneration of the rhabdomere structure. Interestingly, α-syn overexpression did not lead to externally visible eye defects, suggesting a much milder phenotype than the models discussed above. Still, α-syn in the Drosophila model was shown to directly interfere with autophagic flux, resulting in a pronounced accumulation of other autophagy substrates, such as mutant Huntingtin protein with a polyglutamine tract (see below) [66].
There are also fly models that reproduce genetic mutations found in familial Parkinson’s disease. These include flies with loss-of-function mutations in PARK2/Parkin [67,68,69], PARK6/Pink1 [70,71], PARK7/Dj1 [72,73,74] and PARK8/Lrrk2 [75,76,77], as well as PARK11/Gigyf [78] that we recently characterized. Original reports of Drosophila Parkin and Pink1 mutants linked the Parkin and Pink1 proteins with mitochondrial homeostasis [67,68,70,71,79] and later cell biological studies established that these two proteins are essential components for eliminating dysfunctional mitochondria through a selective autophagic process called mitophagy [80,81]. In this process, damaged mitochondria with defective membrane potential recruit Pink1 to their surface. Pink1 then recruits Parkin, which ubiquitinates mitochondrial surface proteins. This ubiquitination then induces the formation of a phagophore around the damaged mitochondria and targets the defective mitochondria for autophagic degradation. This is considered one of the major quality control mechanisms for mitochondria, not only in neurons but also in other tissues such as the liver [82] and heart [83].
Other genes, such as Dj1, Lrrk2 and Gigyf were also shown to be critical for autophagic elimination of ubiquitinated proteins or damaged mitochondria, but the mechanism still needs further clarification [31,32,33,34,78]. Although currently uncharacterized, Drosophila also has a single ortholog of PARK9/Atp13A2, which in mammals is required for efficient lysosomal degradation of autophagosomes [84,85].
2.6. Drosophila Models of Polyglutamine Toxicity: Polyglutamine Flies and Their Genetic Interaction with the Autophagic Pathway
In several inheritable neurodegenerative diseases, known as polyglutamine diseases, genetic mutations include a long polyglutamine (pQ) tract in the coding sequence of a specific gene [3]. For example, Huntington’s disease is caused by a pQ extension in the Huntingtin (HTT) gene, while spinocerebellar ataxia (SCA) is caused by a similar pQ extension in various genes including Ataxins (e.g., ATXN1-3). The accumulation of proteins with a polyglutamine tract often causes protein aggregation and inclusion, which pathogenetically produces proteotoxicity. Because protein aggregates containing pQ can be eliminated through autophagic machinery, autophagy is important for preventing the development of pQ pathologies. In the Drosophila model, pQ toxicity can be easily modeled by its drastic effects on eye morphology [53]. Genetic modifiers of pQ toxicity in Drosophila include critical autophagy components such as Atg6, Atg12 and Ref(2)P, a p62/SQSTM1 homolog [86,87]. Chemical enhancers of autophagy also alleviated pQ-induced retinal degeneration [88]. Recent cell biological studies showed that pQ also interferes with autophagic flux by downregulating class III PI3K through inhibiting its interaction with Ataxin-3, the gene mutated in SCA3 [89]. Importantly, HTT, whose expression is abrogated by the pQ expansion in Huntington’s disease, was recently found to facilitate selective autophagy by acting as a scaffold between autophagy adaptor p62/SQSTM1 and core autophagy molecules such as Atg1/ULK1 and Atg8/LC3 proteins [90,91]. The role of HTT in autophagy was initially identified from Drosophila genetics, and later confirmed in knockout mice and human cells [90,91]. In other cell biological studies, pQ aggregates were found to interact with ALFY, another scaffolding protein for p62/SQSTM1 [92,93,94]. Mutations in the Drosophila homolog of ALFY, known as blue cheese (bchs), also provoked progressive neurodegeneration associated with prominent accumulation of insoluble protein inclusions [95,96,97]. Eye-based genetic screening in Drosophila further identified many autophagic components in the bchs pathology including: Atg1; Atg2; Atg6; Atg8a, and; Atg18 [95]. All of this information acquired from HTT, pQ and bchs flies shows how Drosophila genetics enhances our understanding of the relationship between autophagy and neurodegeneration.
2.7. Using Drosophila for Investigating the Pathogenetic Role of Human Autophagy Mutations
Extensive studies established that genetic disruption of autophagy provokes neurodegenerative phenotypes in model animals such as Drosophila and mice, and many mutations that cause neurodegenerative disease in humans compromise the autophagic process. The direct evidence indicating that autophagic defect causes neurodegeneration in humans was also recently provided: a mutation in ATG5, a core autophagy-regulating gene, was discovered in two ataxia patients from a consanguineous family [22]. The mutation (E122D) is a subtle amino acid change, but assays in patient cell lines indicated that the mutant ATG5 (ATG5–E122D) has a defect conjugating with ATG12, which is necessary for its autophagy-initiating activity. In addition, the mutant cells exhibit dramatic impairment in autophagic flux, monitored by an LC3-II conversion assay. Furthermore, the mutant cells accumulate higher amounts of p62/SQSTM1, an autophagy substrate. Still, it was unknown whether this subtle mutation indeed produces pathogenesis.
Here, the power of Drosophila genetics was again proven to be extremely useful. From a Drosophila model, the effect of the ATG5–E122D mutation on neuronal autophagy and homeostasis was successfully reproduced [22]. Flies in which Atg5 is substituted with the human mutant form (ATG5–E122D) were found to exhibit severe movement disorder, while flies expressing the wild-type human protein (ATG5-WT) were protected from such degeneration. ATG5–E122D was also inferior to ATG5-WT in preventing protein aggregation and neurodegeneration in the brain tissue of Atg5-deficient mutant flies. The comparable mutation in yeast also produced strong deterioration of autophagic flux [22], suggesting that the role of E122 is critical across different animal species. These results together established the pathogenetic role of the ATG5–E122D mutation, and provided an example of how the impairment of core autophagy machinery directly causes neurodegenerative disease in humans.
It was also previously shown that heterozygotic mutations in Atg18/WDR45/WIPI4 are associated with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA) [98]. The mutation of Atg18 in Drosophila was shown to impair autophagy through Atg9 mis-regulation [49], although its role in neuronal homeostasis has yet to be examined. Considering that Atg18 family proteins have important roles in mediating the effect of the class III PI3K complex [12], this is another example that highlights the role of autophagy in human neurodegeneration.
3. Conclusions and Future Directions
The Drosophila model has been successful in modelling human neurodegenerative disease and investigating physiological roles and regulation of autophagy. These findings, combined with studies in other model systems, such as cultured cells and mice, established the theory that autophagic homeostasis is critical for preventing neurodegenerative phenotypes. Still, there are many different areas where the Drosophila model can be used for future studies in investigating the relationship between autophagy and neurodegeneration.
First, it is not currently known why genetic mutations in autophagy components preferentially deteriorate cerebellar motor function compared to other cognitive functions of the brain. It is most likely that cerebellar neurons are somehow hypersensitive to neurotoxic insults caused by autophagy dysregulation. The preferential motor deficit phenotypes were observed in autophagy-defective human [22], mouse [19,20,24] and Drosophila [21,22,23]. Therefore, the Drosophila model could again prove to be useful for discovering the mechanistic basis of this pathophysiology. How different molecular pathologies in autophagy affect different areas of the brain, causing distinct clinical presentations of neurodegeneration, should also be clarified. As reviewed above, different neurotoxic proteins compromise autophagy through distinct mechanisms in both Drosophila and mammalian cells, which may be the reason why they affect different regions of the brain.
In addition, future studies aimed at isolating additional autophagy-regulating components, through genome-level functional screening in Drosophila, may lead to the construction of a systems-level view of the autophagy network that fine tunes neuronal homeostasis. The construction of this network may enable us to approach the molecular etiology of sporadic Alzheimer’s and Parkinson’s disease, which cover a predominant portion of neurodegeneration cases in humans, yet are not associated with clear genetic mutations. It is possible that age-dependent deterioration of neuronal homeostasis, associated with some subtle genetic predisposition and environmental influence, contribute to the dysregulation of autophagic machinery. The Drosophila model was also recently found to be useful for investigating the effect of aging [99], external insults [100], and subtle genetic alteration [22] on autophagic pathway and neurodegeneration. Therefore, it will provide an efficient platform for investigating the interaction between aging, environments and genetics in the context of neurodegeneration.
These lines of study are expected to enhance our understanding of how autophagic defects contribute to neurodegenerative pathologies. These advancements may ultimately lead to the discovery of novel therapeutics that can restore autophagic flux in diseased neurons and their associated cells. For instance, several chemicals such as rapamycin and calcium channel blockers were shown to enhance autophagy and attenuate neurodegeneration in the Drosophila model [47,62,86,88,101], and similar beneficial effects of these chemicals were observed in different mouse models [101,102,103]. Therefore, further exploration of this autophagy-neurodegeneration network in flies might uncover important pathogenetic mechanisms that can be used for developing clinically-relevant therapeutic strategies to treat human neurodegenerative disease.
Acknowledgments
Work was supported by grants from the NIH (R21OD018265 to Myungjin Kim and Jun Hee Lee and T32GM008322 and T32AG000114 to Allison Ho). Some graphics in the graphical abstract were obtained and modified from Servier Medical Art (http://www.servier.com/Powerpoint-image-bank) and Wikimedia commons (https://commons.wikimedia.org).
Author Contributions
Myungjin Kim, Allison Ho and Jun Hee Lee wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| α-syn | α-synuclein |
| Aβ | β-amyloid |
| APP | amyloid precursor protein |
| ATG | autophagy-related |
| FIP200 | FAK family kinase-interacting protein of 200 kDa |
| MAPK | mitogen-activated protein kinases |
| PARK | Parkinson Disease |
| PI3K | phosphatidylinositol 3-kinase |
| TOR | target of rapamycin |
| ULK1 | Unc51-like protein kinase 1 |
References
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. Polyglutamine pathogenesis: Emergence of unifying mechanisms for huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagic processes in yeast: Mechanism, machinery and regulation. Genetics 2013, 194, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. The incredible ulks. Cell Commun. Signal. 2012, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Vps15 is required for stress induced and developmentally triggered autophagy and salivary gland protein secretion in Drosophila. Cell Death Differ. 2015, 22, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Takacs, Z.; Donnes, P.; Kohlbacher, O. WIPI proteins: Essential Ptdins3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin.Cell Dev. Biol. 2010, 21, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.L.; Klionsky, D.J. How to live long and prosper: Autophagy, mitochondria, and aging. Physiology 2008, 23, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Erdi, B.; Sass, M.; Neufeld, T.P. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007, 21, 3061–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.H.; et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5, e12245. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.L.; Park, H.W.; Ro, S.H.; Nam, S.G.; Reed, J.M.; Guan, J.L.; Lee, J.H. Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy 2013, 9, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.L. Neural-specific deletion of Fip200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Esselens, C.; Oorschot, V.; Baert, V.; Raemaekers, T.; Spittaels, K.; Serneels, L.; Zheng, H.; Saftig, P.; De Strooper, B.; Klumperman, J.; et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 2004, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Murphy, D.D.; Giasson, B.I.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Degradative organelles containing mislocalized α-and β-synuclein proliferate in presenilin-1 null neurons. J. Cell Biol. 2004, 165, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Song, H.J.; Garza, D.; Neufeld, T.P.; Salvaterra, P.M. Aβ42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE 2009, 4, e4201. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Kabuta, T.; Wada, K. Insights into links between familial and sporadic parkinson’s disease: Physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 2008, 4, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Models Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Csikos, G.; Sinka, R.; Erdelyi, M.; Sass, M. The Drosophila homolog of Aut1 is essential for autophagy and development. FEBS Lett. 2003, 543, 154–158. [Google Scholar] [CrossRef]
- Scott, R.C.; Schuldiner, O.; Neufeld, T.P. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 2004, 7, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.L.; Baehrecke, E.H. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007, 131, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Lindmo, K.; Juhasz, G.; Sass, M.; Seglen, P.O.; Brech, A.; Stenmark, H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell 2004, 7, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Semple, I.A.; Park, H.; Park, H.; Park, H.W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes Unc-51-like kinase 1 (ULK1)-mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a feedback inhibitor of tor that prevents age-related pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lao, U.; Edgar, B.A. Tor-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J. Cell Biol. 2009, 186, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Hegedus, K.; Pircs, K.; Varga, A.; Juhasz, G. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in Drosophila. FEBS Lett. 2014, 588, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Nagy, P.; Arsikin Csordas, K.; Kovacs, A.L.; Hegedus, K.; Juhasz, G. Loss of Atg16 delays the alcohol-induced sedation response via regulation of corazonin neuropeptide production in Drosophila. Sci. Rep. 2016, 6, 34641. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Karpati, M.; Varga, A.; Pircs, K.; Venkei, Z.; Takats, S.; Varga, K.; Erdi, B.; Hegedus, K.; Juhasz, G. Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila. Autophagy 2014, 10, 453–467. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an in vivo model for human neurodegenerative disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef] [PubMed]
- Krench, M.; Littleton, J.T. Neurotoxicity pathways in drosophila models of the polyglutamine disorders. Curr. Top. Dev. Biol. 2017, 121, 201–223. [Google Scholar] [PubMed]
- Hewitt, V.L.; Whitworth, A.J. Mechanisms of parkinson’s disease: Lessons from Drosophila. Curr. Top. Dev. Biol. 2017, 121, 173–200. [Google Scholar] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.; Gubb, D.C.; Lomas, D.A. Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-type Tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [PubMed]
- Ambegaokar, S.S.; Jackson, G.R. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from Tau phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Folwell, J.; Cowan, C.M.; Ubhi, K.K.; Shiabh, H.; Newman, T.A.; Shepherd, D.; Mudher, A. Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of alzheimer’s disease. Exp. Neurol. 2010, 223, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Feany, M.B.; Bender, W.W. A Drosophila model of parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Andrews, L.A.; Parker, T.J.; Pallanck, L.J. Genetic and genomic studies of drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum. Mol. Genet. 2005, 14, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Cha, G.H.; Kim, S.; Park, J.; Lee, E.; Kim, M.; Lee, S.B.; Kim, J.M.; Chung, J.; Cho, K.S. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc. Natl. Acad. Sci. USA 2005, 102, 10345–10350. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila pink1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, S.Y.; Cha, G.H.; Lee, S.B.; Kim, S.; Chung, J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Yenisetti, S.C.; Min, K.T. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr. Biol. 2005, 15, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, W.; Lee, S.; Chung, J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in drosophila. Biochem. Biophys. Res. Commun. 2007, 358, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, X.; Yu, Y.; Li, X.; Wang, T.; Jiang, H.; Ren, Q.; Jiao, Y.; Sawa, A.; Moran, T.; et al. A drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. USA 2008, 105, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Semple, I.; Kim, B.; Kiers, A.; Nam, S.; Park, H.W.; Park, H.; Ro, S.H.; Kim, J.S.; Juhasz, G.; et al. Drosophila Gyf/Grb10 interacting GYF protein is an autophagy regulator that controls neuron and muscle homeostasis. Autophagy 2015, 11, 1358–1372. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.K.; Lee, S.H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.X. Targeting PINK1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Finkel, T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic. Biol. Med. 2016, 109, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type atpase ATP13a2/PARK9 function induces general lysosomal deficiency and leads to parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Krainc, D. Lysosomal dysfunction in neurodegeneration: The role of ATP13A2/PARK9. Autophagy 2012, 8, 987–988. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Fujikake, N.; Okamoto, Y.; Popiel, H.A.; Hatanaka, Y.; Ueyama, M.; Suzuki, M.; Gaumer, S.; Murata, M.; Wada, K.; et al. P62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J. Biol. Chem. 2015, 290, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for huntington’s disease in an mtor-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. P62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Cumming, R.C.; Lindmo, K.; Galaviz, V.; Cheng, S.; Rusten, T.E.; Finley, K.D. Genetic modifiers of the drosophila blue cheese gene link defects in lysosomal transport with decreased life span and altered ubiquitinated-protein profiles. Genetics 2007, 176, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Finley, K.D.; Edeen, P.T.; Cumming, R.C.; Mardahl-Dumesnil, M.D.; Taylor, B.J.; Rodriguez, M.H.; Hwang, C.E.; Benedetti, M.; McKeown, M. Blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J. Neurosci. 2003, 23, 1254–1264. [Google Scholar] [PubMed]
- Lim, A.; Kraut, R. The Drosophila BEACH family protein, blue cheese, links lysosomal axon transport with motor neuron degeneration. J. Neurosci. 2009, 29, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, E.P.; Mauntz, R.E.; Kotzebue, R.W.; Gonzalez, A.; Achal, M.; Barekat, A.; Finley, K.A.; Sparhawk, J.M.; Robinson, J.E.; Herr, D.R.; et al. Aging and autophagic function influences the progressive decline of adult drosophila behaviors. PLoS ONE 2015, 10, e0132768. [Google Scholar] [CrossRef] [PubMed]
- Barekat, A.; Gonzalez, A.; Mauntz, R.E.; Kotzebue, R.W.; Molina, B.; El-Mecharrafie, N.; Conner, C.J.; Garza, S.; Melkani, G.C.; Joiner, W.J.; et al. Using Drosophila as an integrated model to study mild repetitive traumatic brain injury. Sci. Rep. 2016, 6, 25252. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mtor induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed]
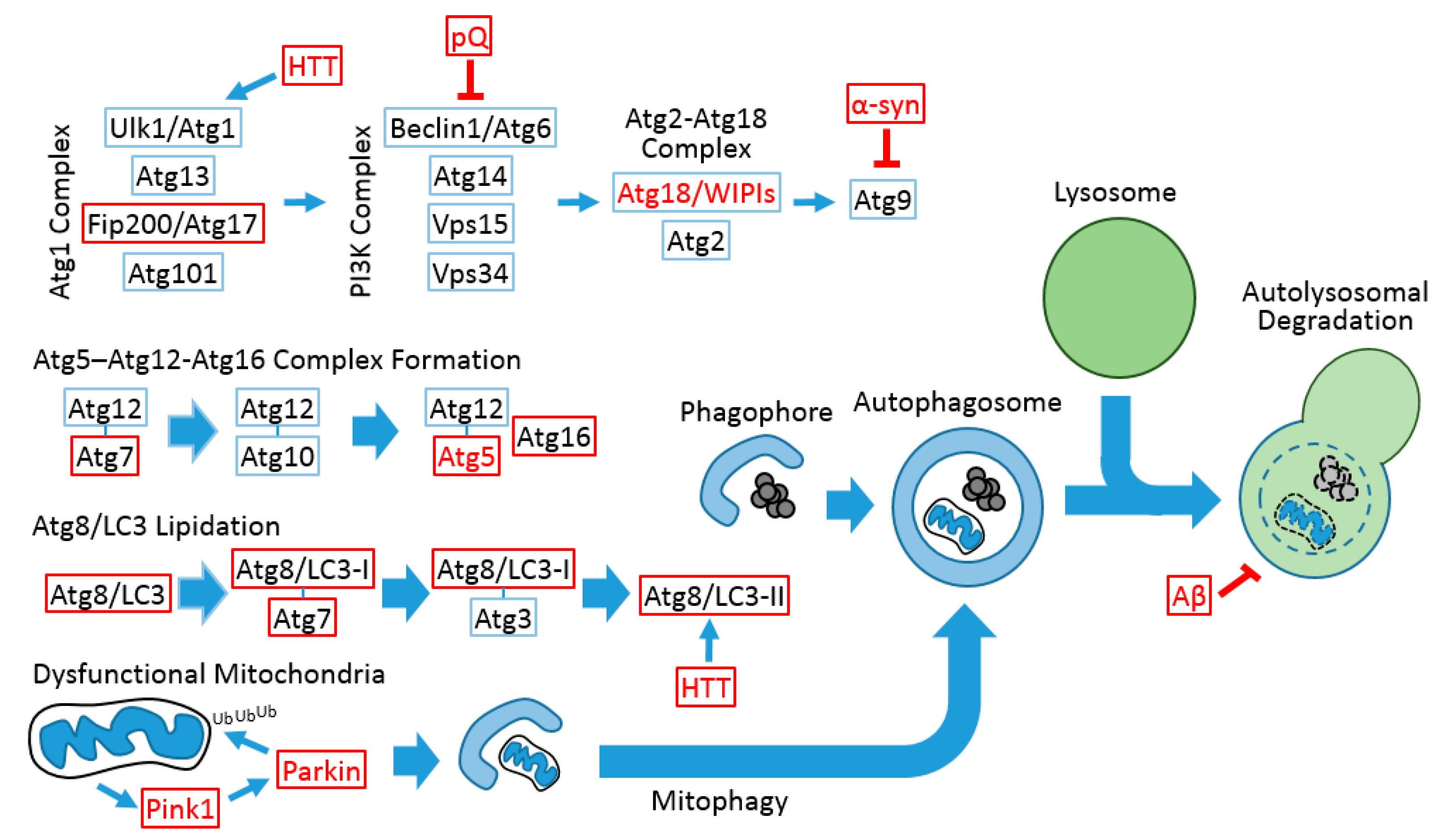
Figure 1.
Conserved autophagic processes between Drosophila and mammals. Red letters indicate that the gene and protein are associated with neurodegenerative diseases in humans. Red boxes indicate that the gene mutation and/or transgenic modulation can provoke neurodegeneration or neuronal dysfunction in the Drosophila system. Blue boxes indicate that, although the Drosophila gene is implicated in the autophagic process, its function in neuronal homeostasis has not been assessed. Thin arrows indicate signal flow, and thick arrows indicate the modification of complexes and vesicles. Blunted arrows indicate inhibition signals. The autophagosomal inner membrane and its contents are degraded after lysosomal fusion (dashed circle). pQ, polyglutamine tract; α-syn, α-synuclein; HTT, Huntingtin; Aβ, β-amyloid.
Figure 1.
Conserved autophagic processes between Drosophila and mammals. Red letters indicate that the gene and protein are associated with neurodegenerative diseases in humans. Red boxes indicate that the gene mutation and/or transgenic modulation can provoke neurodegeneration or neuronal dysfunction in the Drosophila system. Blue boxes indicate that, although the Drosophila gene is implicated in the autophagic process, its function in neuronal homeostasis has not been assessed. Thin arrows indicate signal flow, and thick arrows indicate the modification of complexes and vesicles. Blunted arrows indicate inhibition signals. The autophagosomal inner membrane and its contents are degraded after lysosomal fusion (dashed circle). pQ, polyglutamine tract; α-syn, α-synuclein; HTT, Huntingtin; Aβ, β-amyloid.


{kind=link}
{kind=link}
Table 1.
Autophagy gene mutations in Drosophila that provokes neurodegenerative phenotypes.
| Gene | Neurodegenerative Phenotype(s) Exhibited by Null Mutants or Hypomorphs |
|---|---|
| Atg5 | Defective locomoter activity, p62/Ref(2)P accumulation in brain, neuronal cell death [22] |
| Atg7 | Defective locomotor activity, ubiquitin accumulation in brain, neuronal inclusion body formation, neuronal cell death, brain vacuolization [21] |
| Retinal degeneration [47] | |
| Atg8a | Ubiquitin accumulation in brain [48] |
| p62/Ref(2)P inclusion formation in neurons [49] | |
| Atg16 | Neuronal inclusion body formation, p62/Ref(2)P accumulation in brain [50] |
| Atg17/Fip200 | Defective locomotor activity, defective wing posture, ubiquitin accumulation in brain, neuronal inclusion body formation, mitochondrial dysfunction, brain vacuolization [23,51] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, M.; Ho, A.; Lee, J.H. Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective. Int. J. Mol. Sci. 2017, 18, 1596. https://doi.org/10.3390/ijms18071596
AMA Style
Kim M, Ho A, Lee JH. Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective. International Journal of Molecular Sciences. 2017; 18(7):1596. https://doi.org/10.3390/ijms18071596
Chicago/Turabian StyleKim, Myungjin, Allison Ho, and Jun Hee Lee. 2017. "Autophagy and Human Neurodegenerative Diseases—A Fly’s Perspective" International Journal of Molecular Sciences 18, no. 7: 1596. https://doi.org/10.3390/ijms18071596
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.