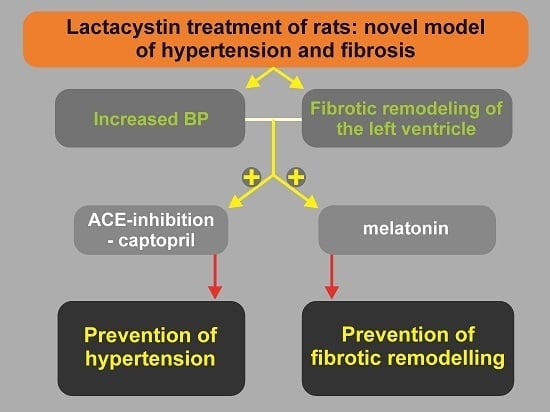
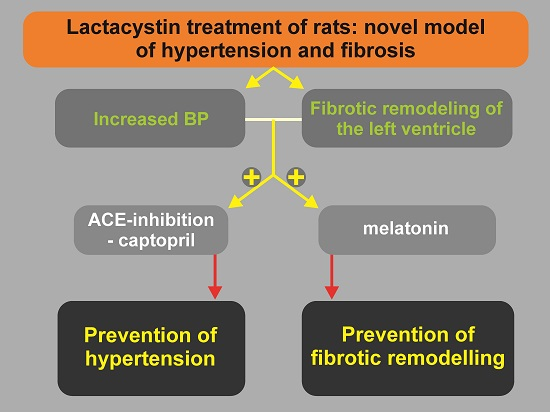
Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
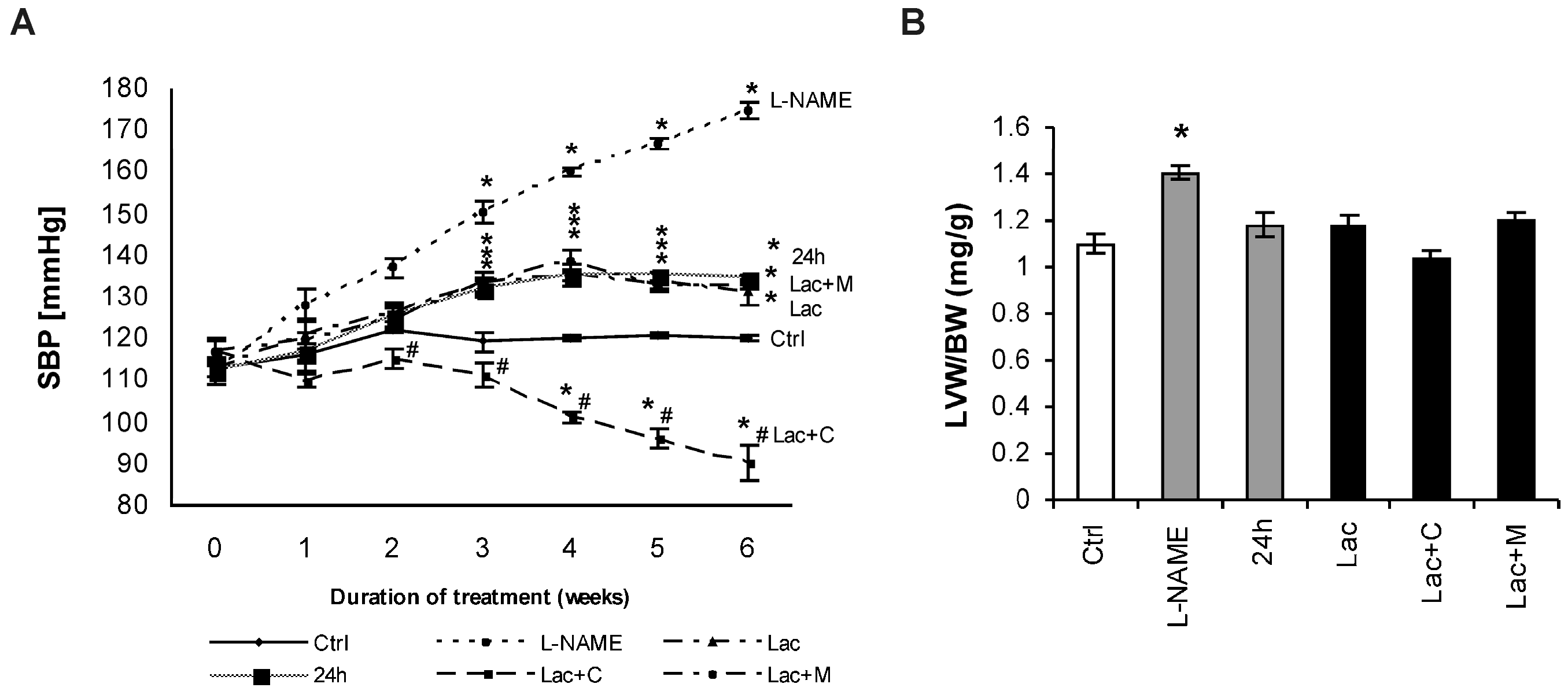
2.1. Cardiovascular Parameters
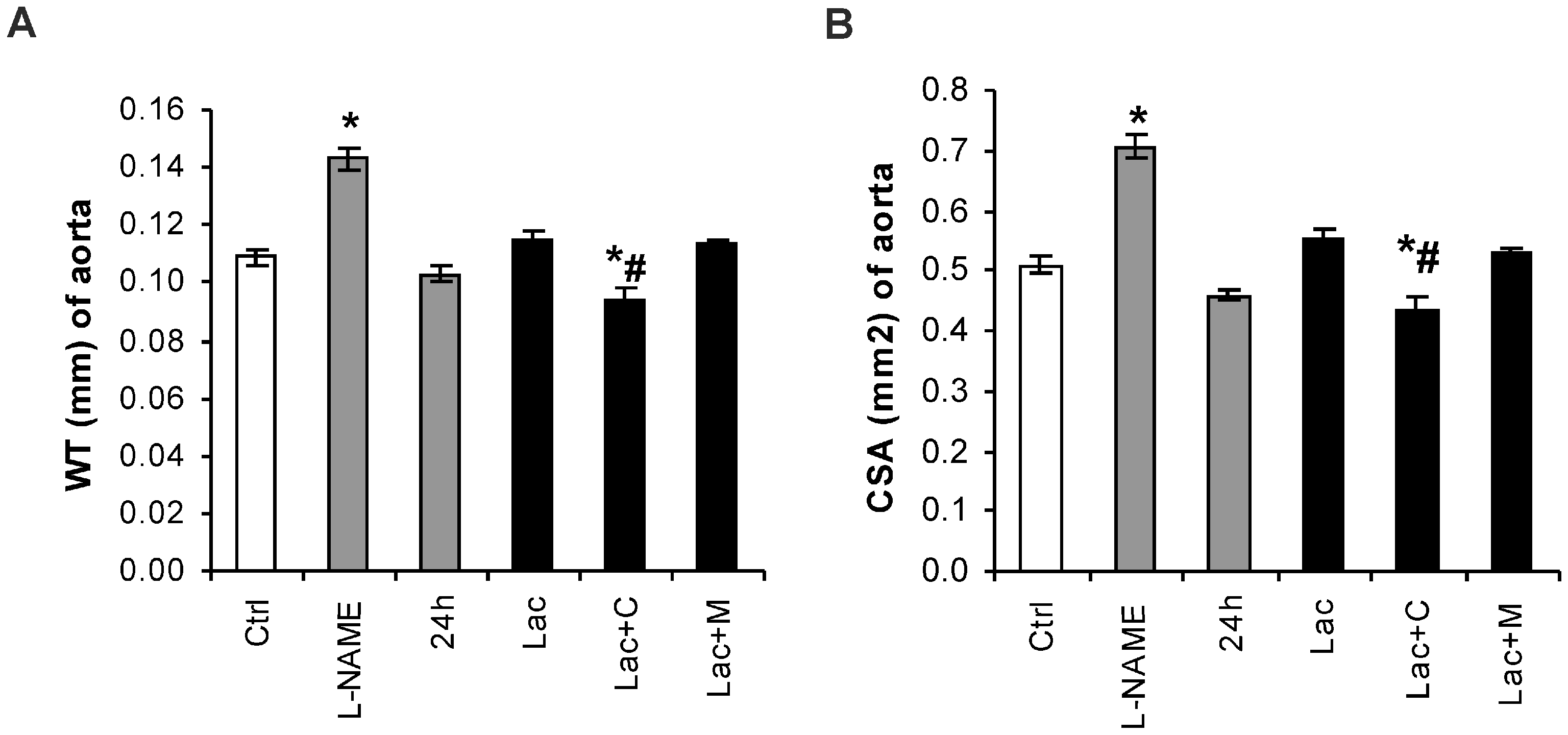
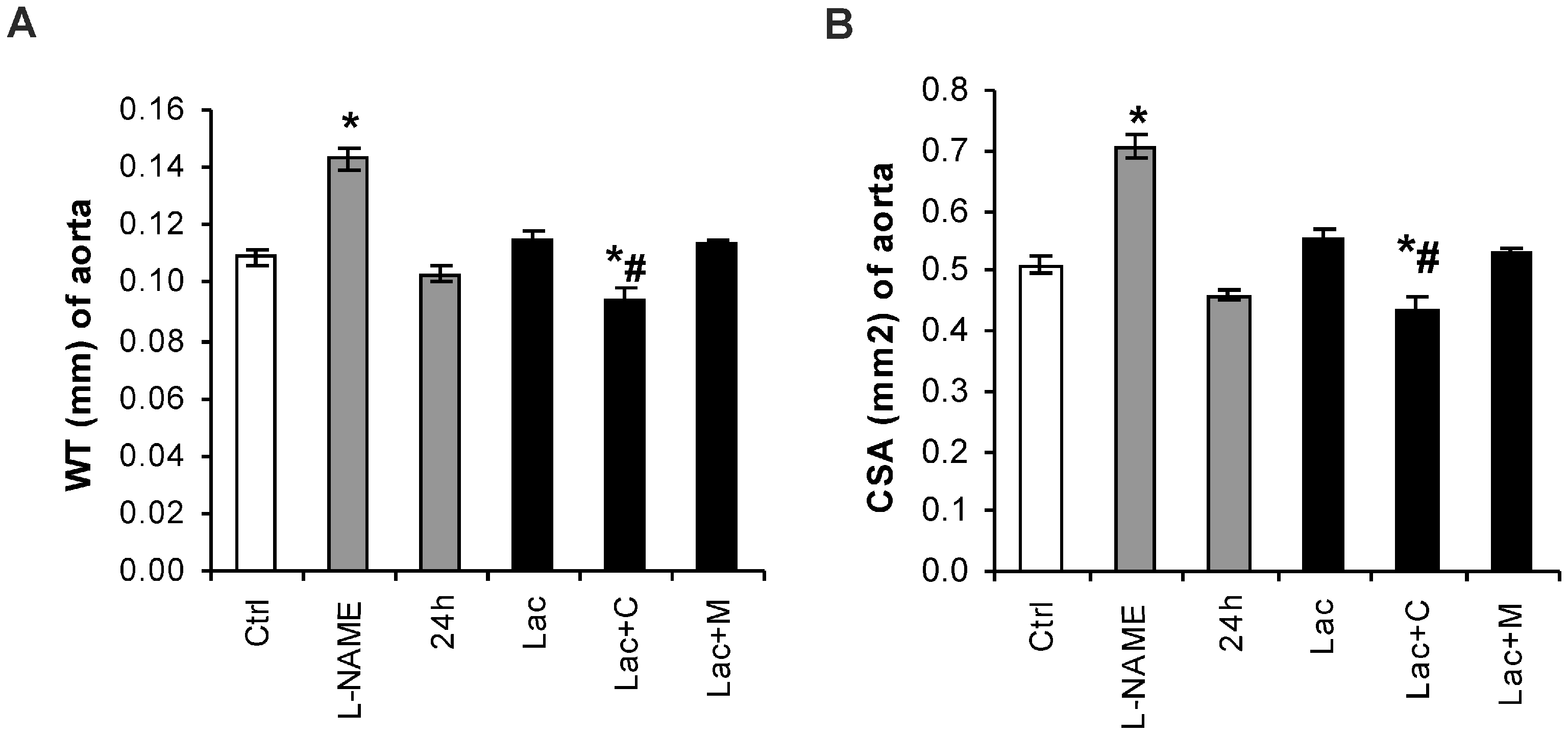
2.2. Morphometry of the Aorta
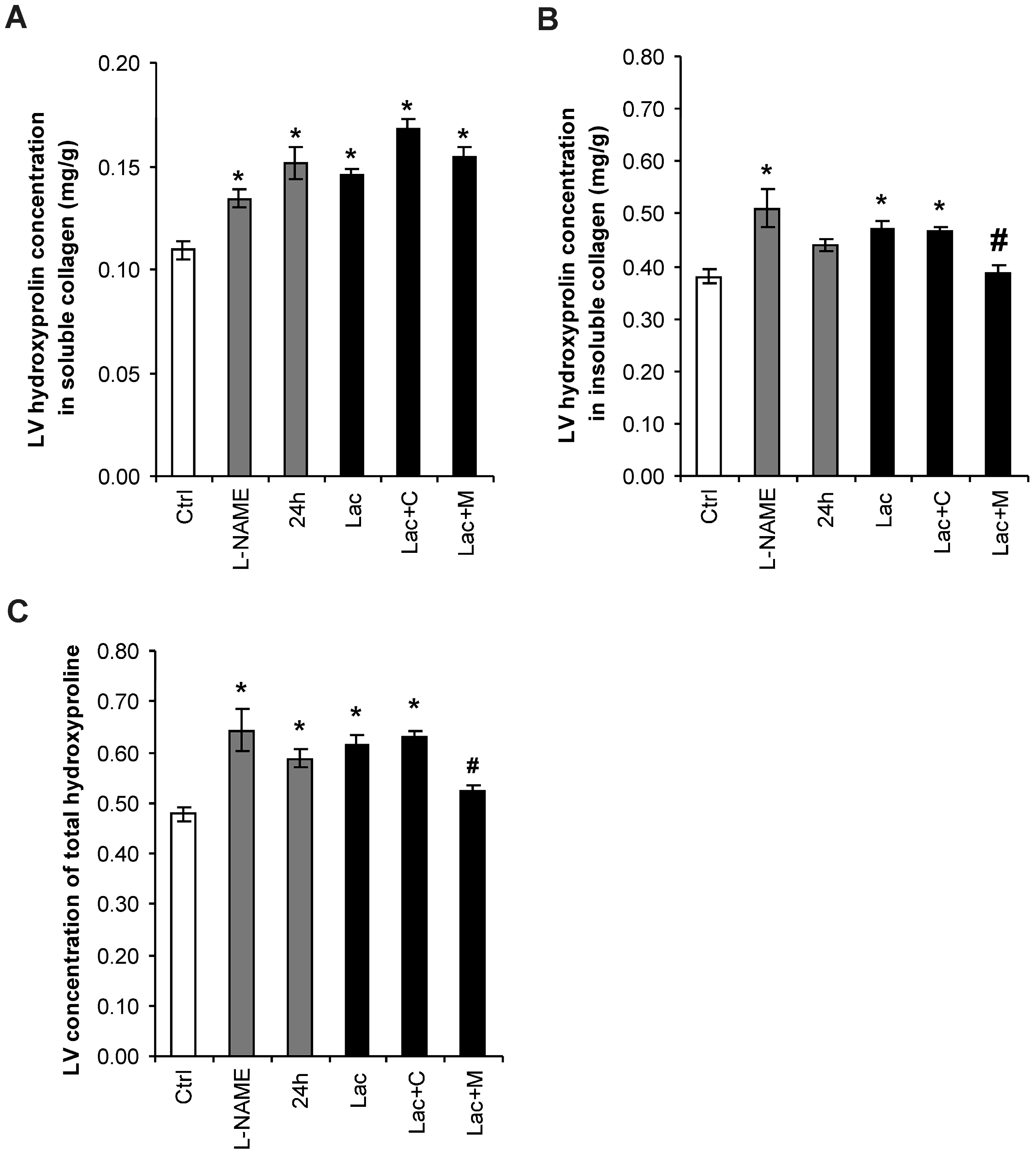
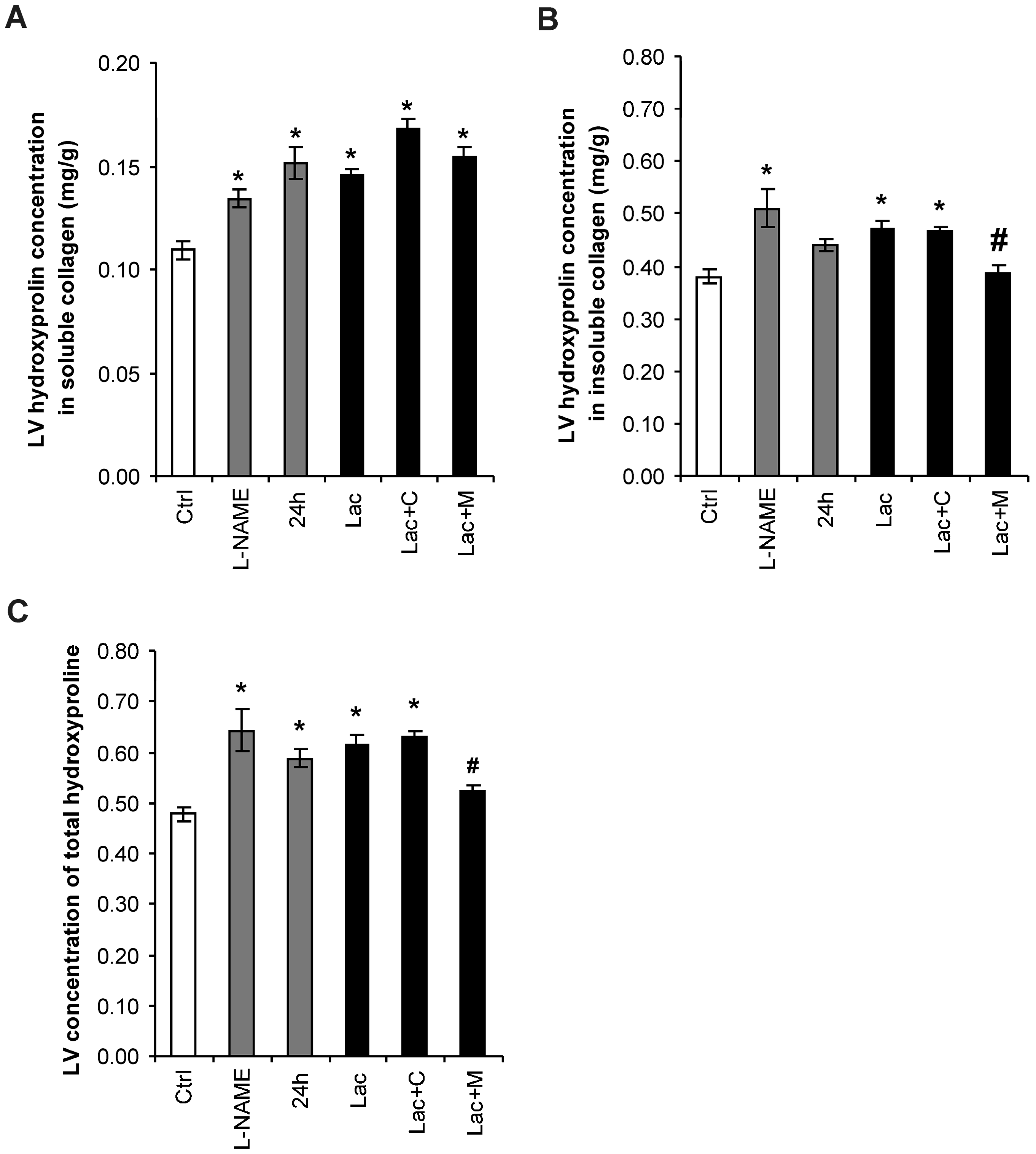
2.3. Hydroxyproline in Soluble and Insoluble Collagenous Fraction in the Left Ventricle (LV)
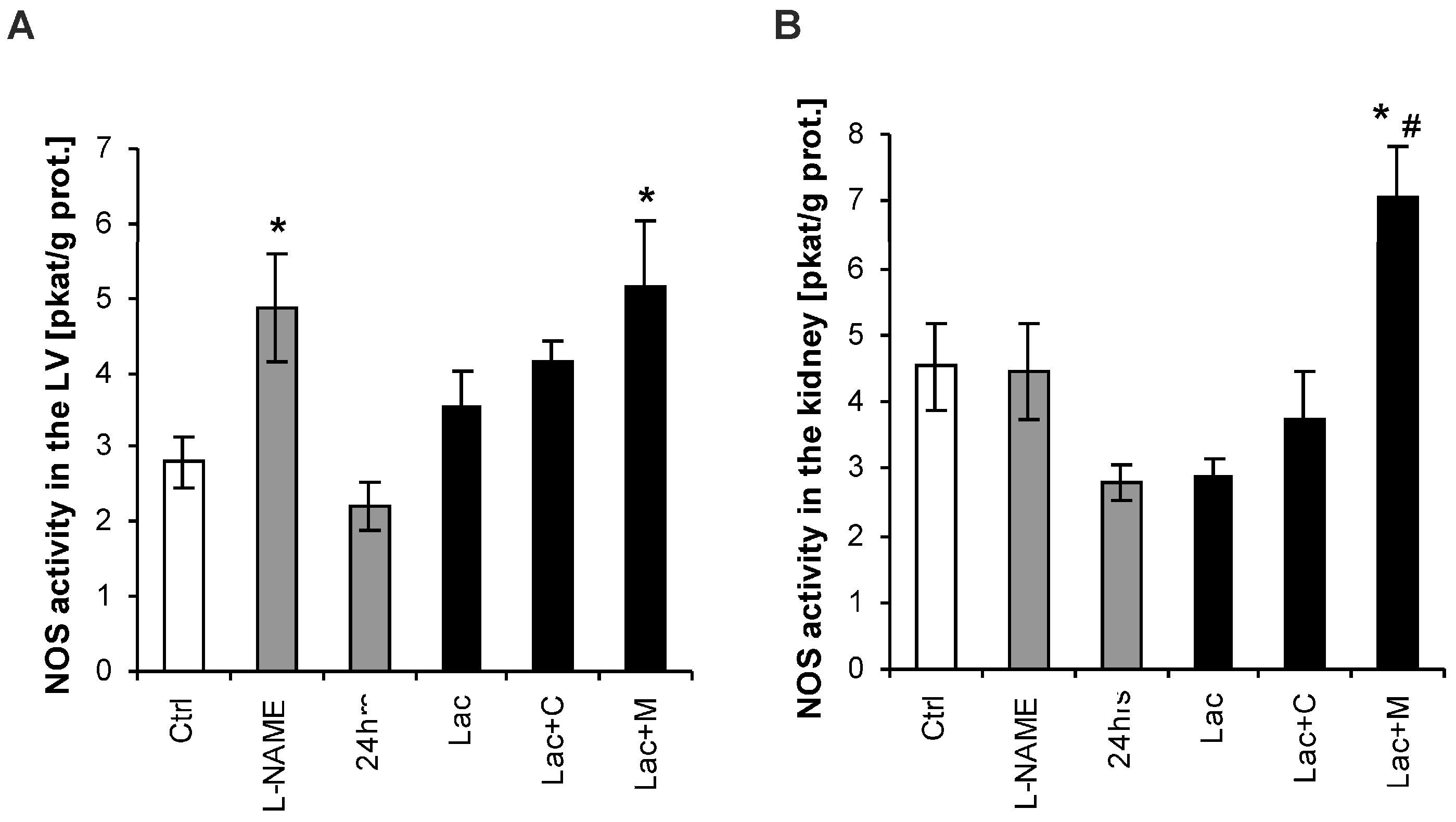
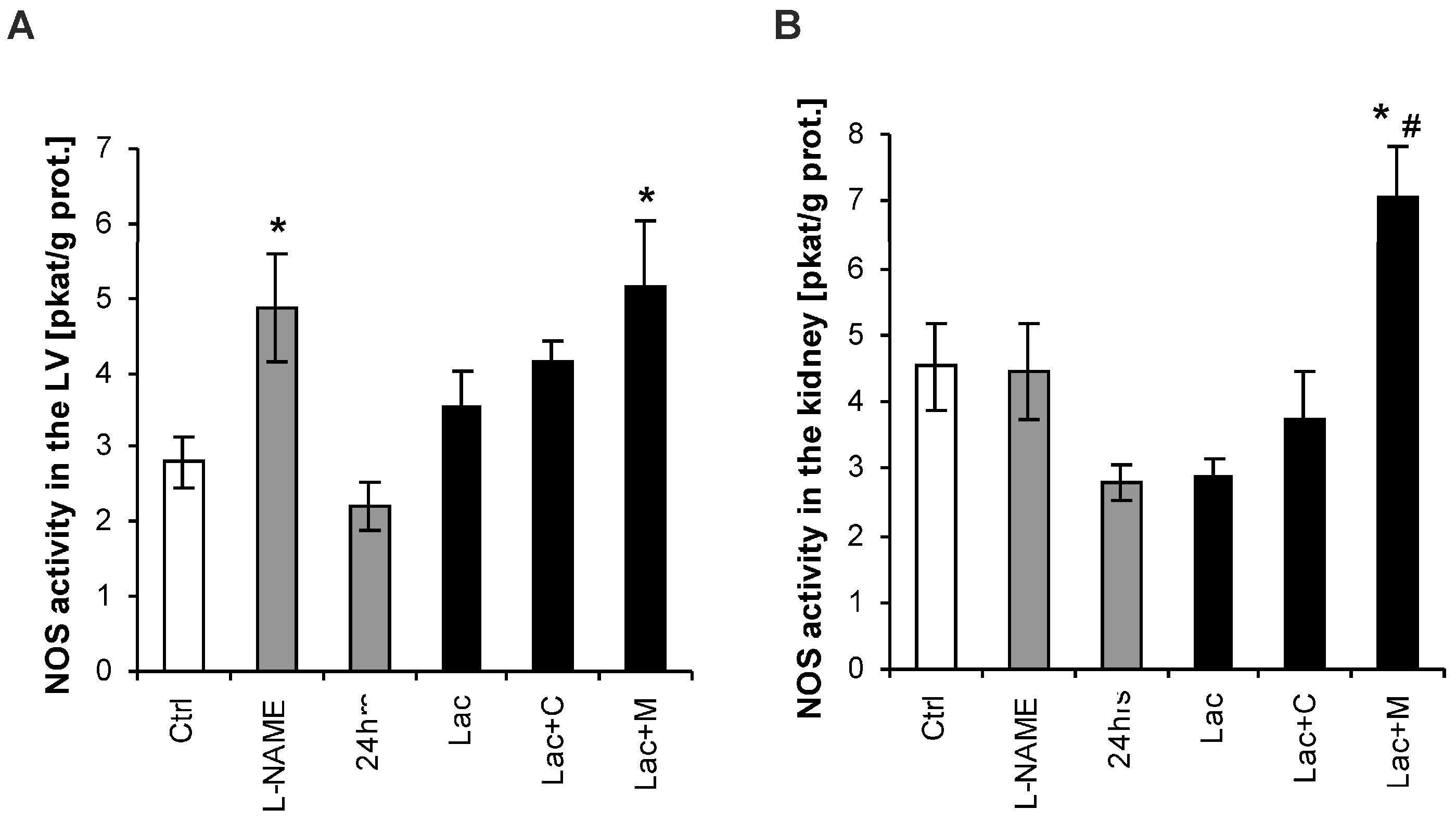
2.4. NO-Synthase (NOS) Activity in the LV and Kidney
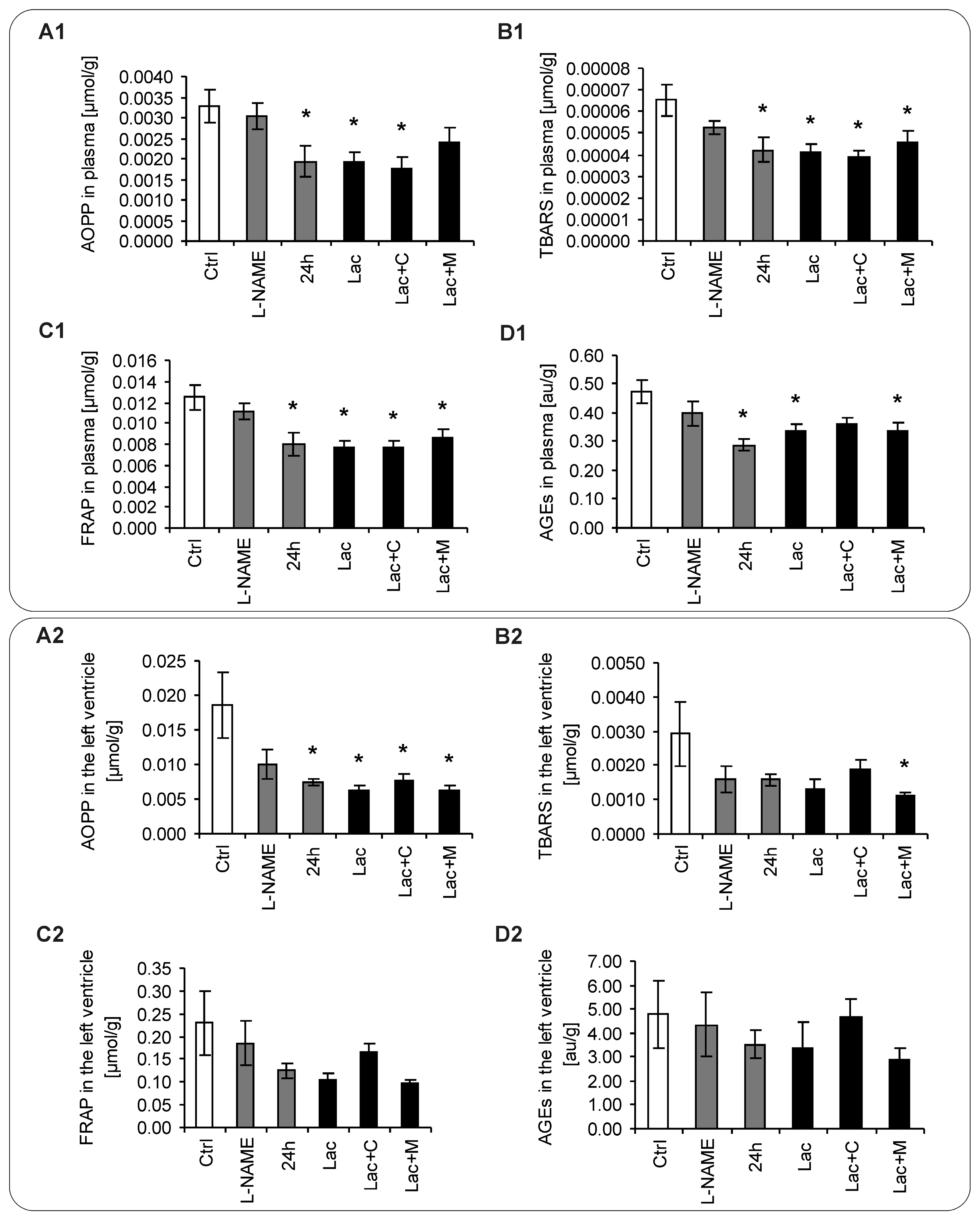
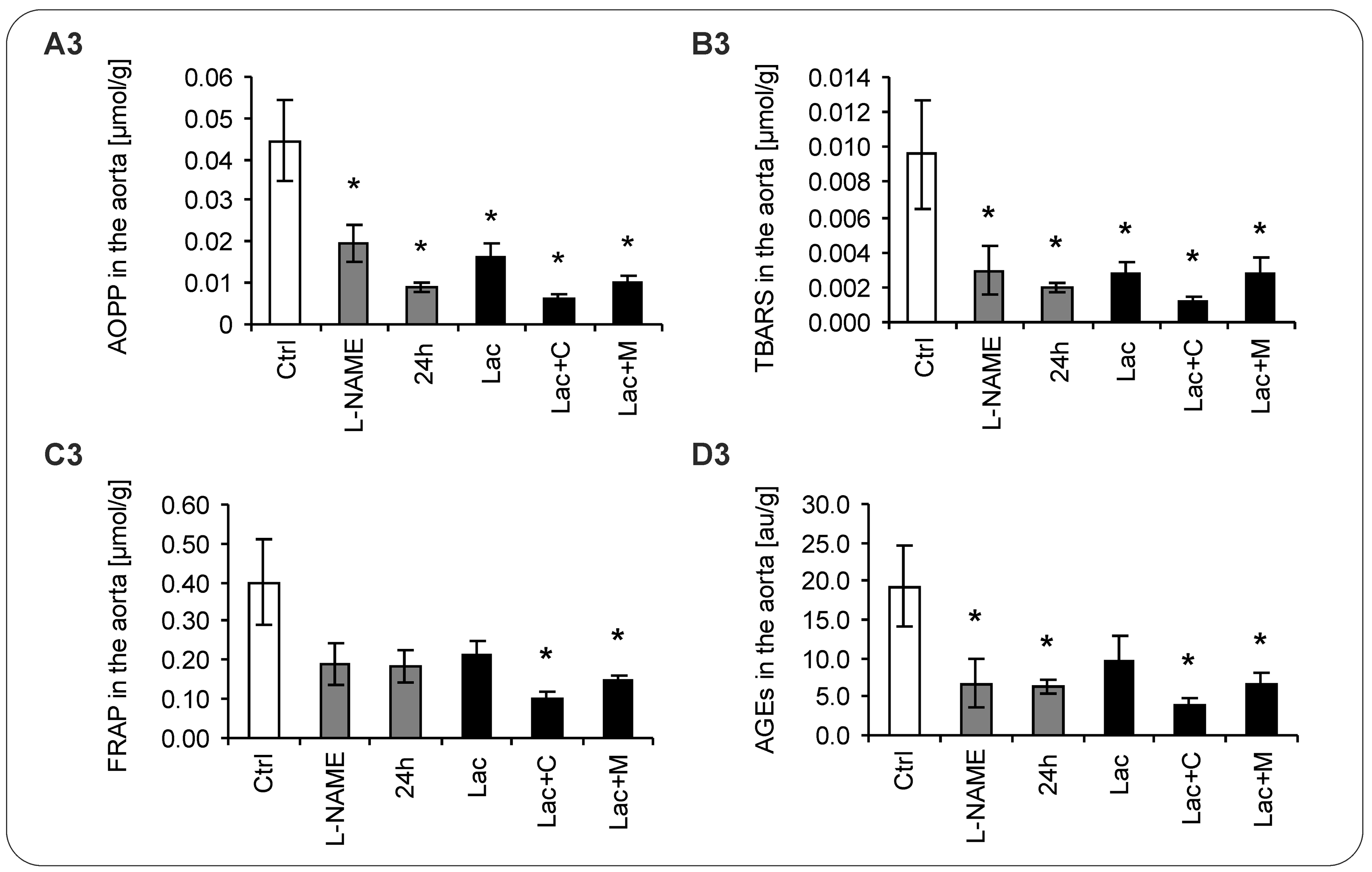
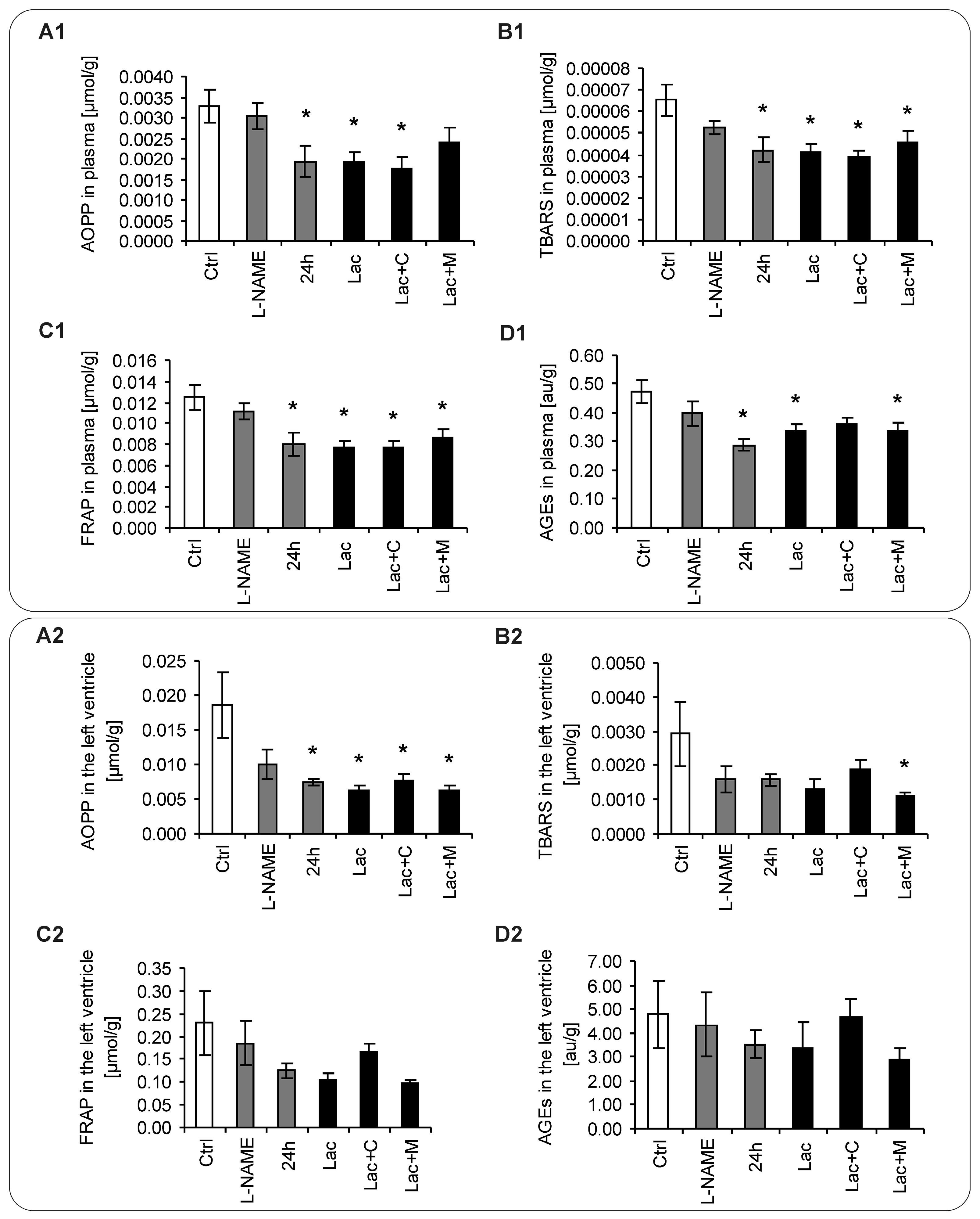
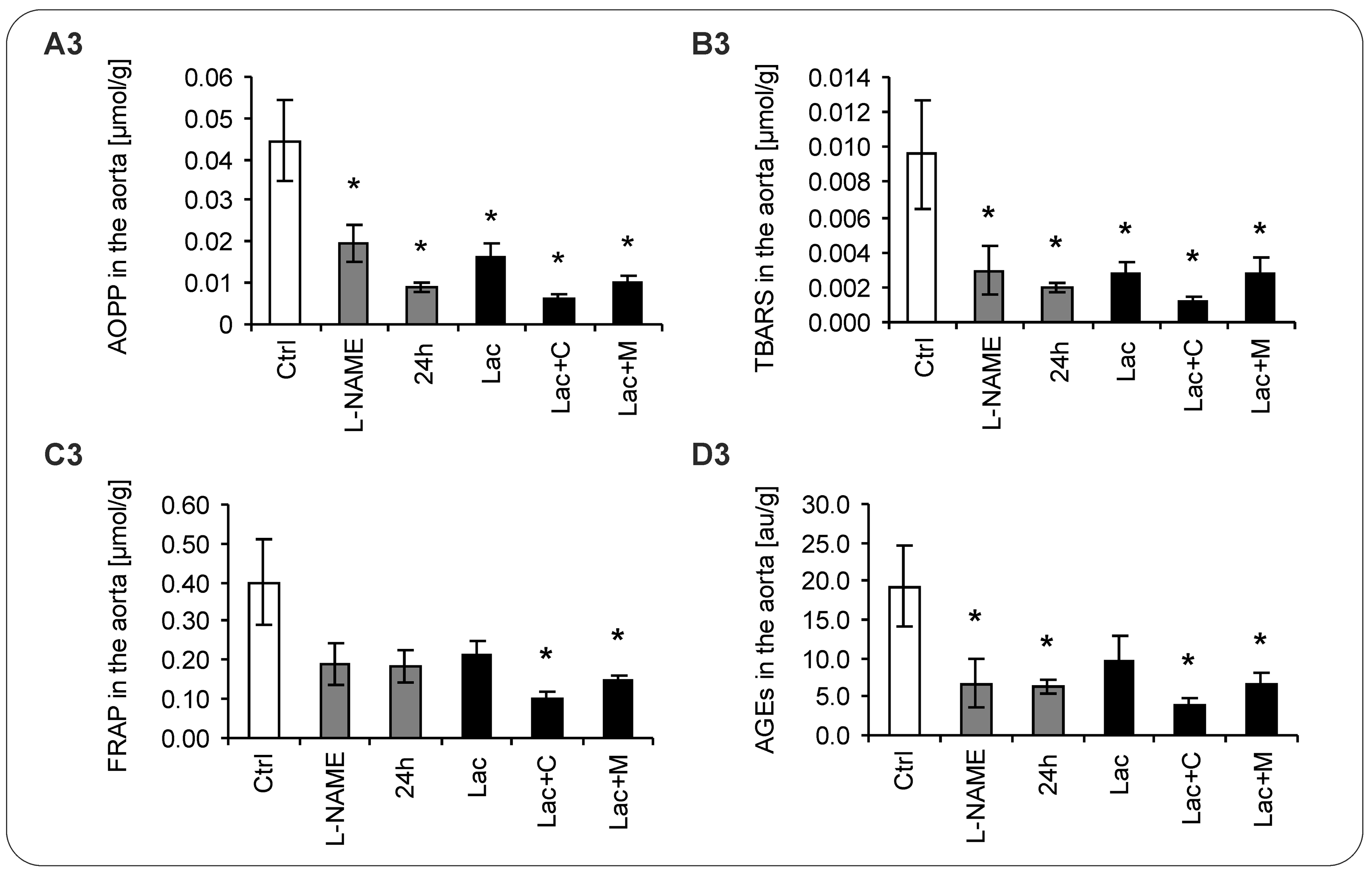
2.5. Oxidative Stress Parameters
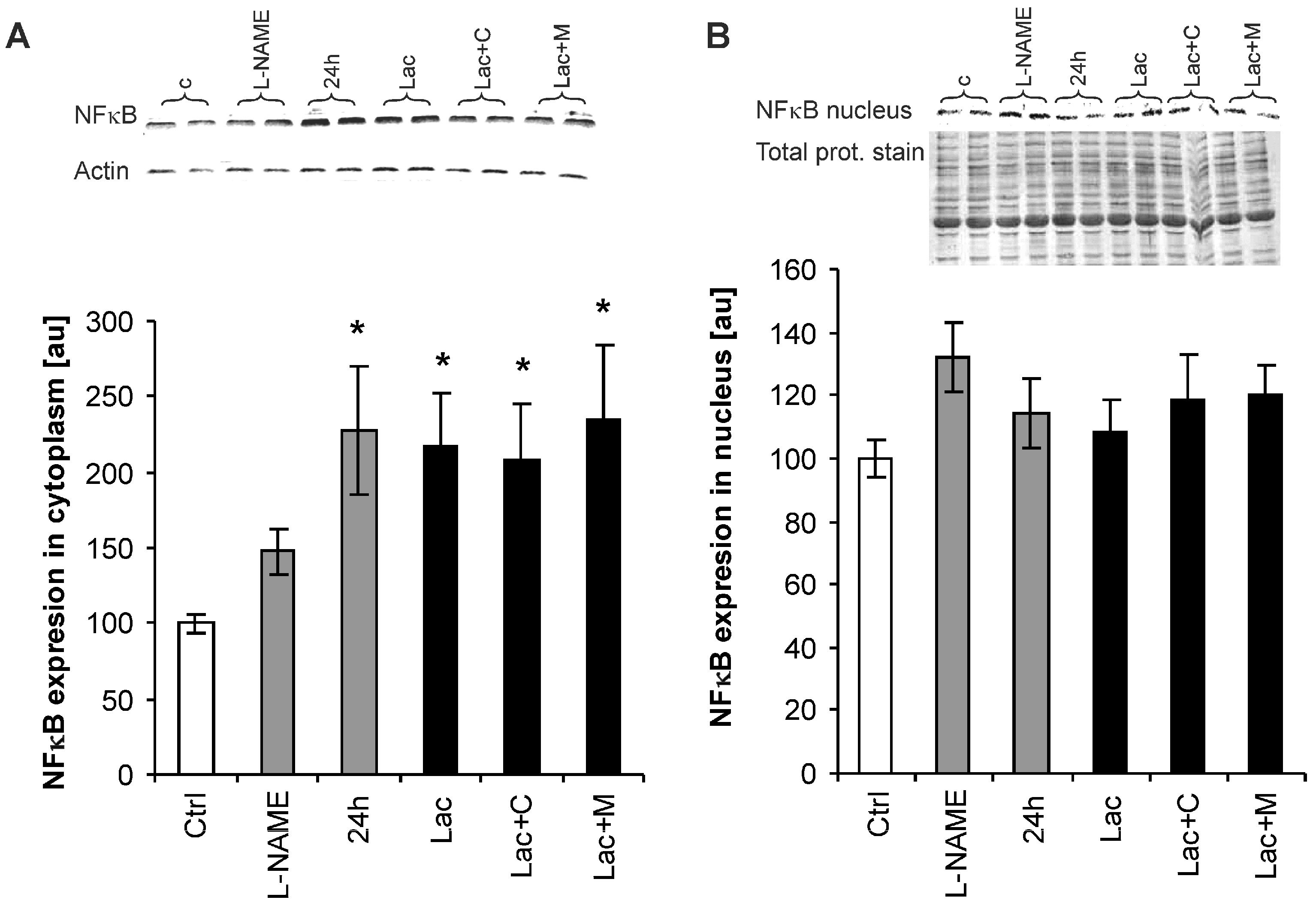
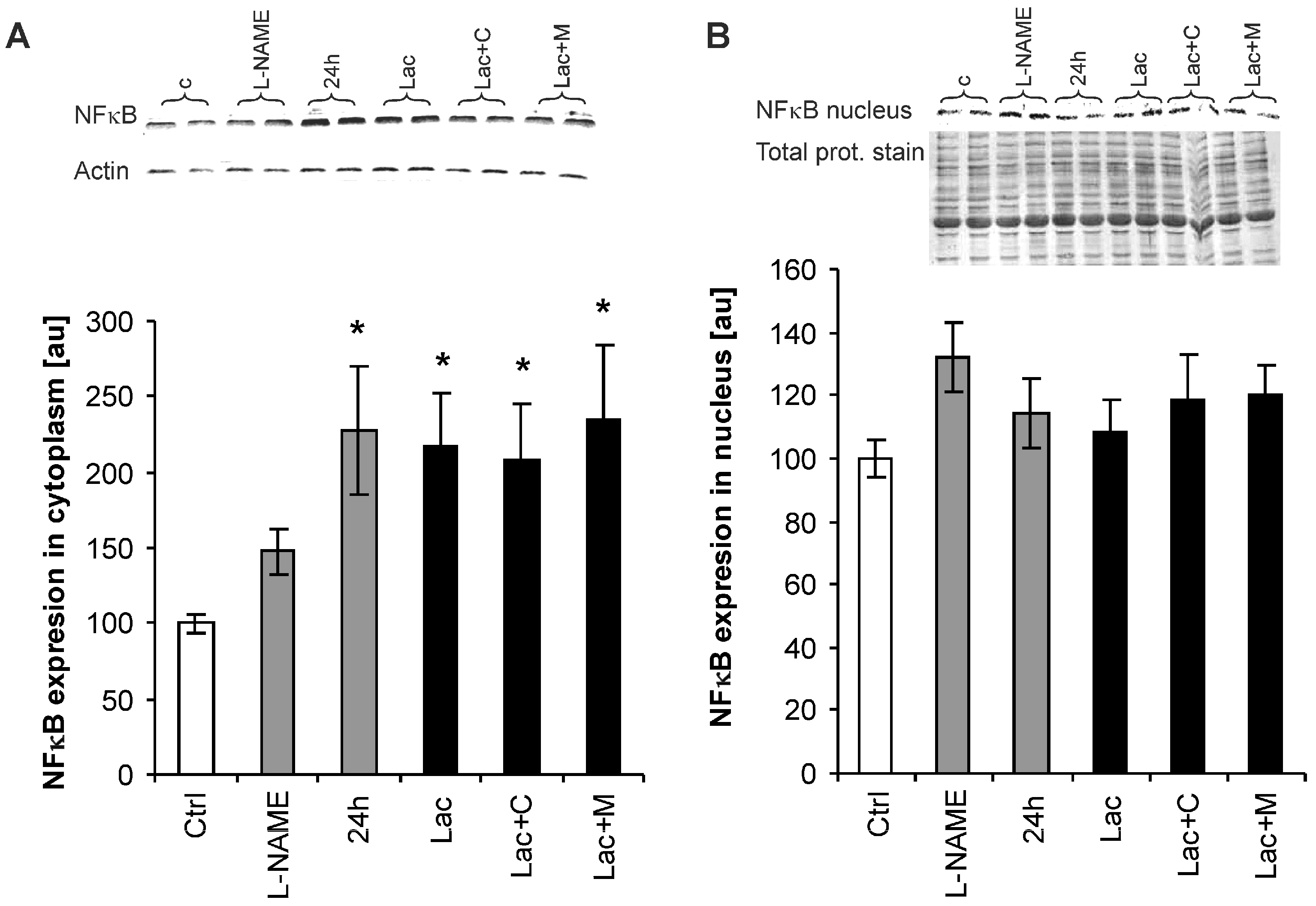
2.6. Nuclear Factor-Kappa B (NF-κB) Expression
3. Discussion
4. Experimental Procedures
4.1. Animals and Treatment
4.2. Morphometry of the Aorta
4.3. Determination of Hydroxyproline
4.4. Assay of NO-Synthase (NOS) Activity
4.5. Oxidative Load Measurement
4.6. Western Blotting of NF-κB
4.7. Statistical Analysis
5. Limitations
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klingbeil, A.U.; Schneider, M.; Martus, P.; Messerli, F.H.; Schmieder, R.E. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am. J. Med. 2003, 115, 41–46. [Google Scholar] [CrossRef]
- Simko, F. Pathophysiological principles of the relation between myocardial hypertrophy of the left ventricle and its regression. Physiol. Res. 1994, 43, 259–266. [Google Scholar] [PubMed]
- Simko, F. Left ventricular hypertrophy regression as a process with variable biological implications. Can. J. Cardiol. 1996, 12, 507–513. [Google Scholar] [PubMed]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Matuskova, J.; Luptak, I.; Krajcirovicova, K.; Kucharska, J.; Gvozdjakova, A.; Babal, P.; Pechanova, O. Effect of simvastatin on remodeling of the left ventricle and aorta in L-NAME-induced hypertension. Life Sci. 2004, 74, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Simko, F. Statins—A prespective for left ventricular hypertrophy treatment (review). Eur. J. Clin. Investig. 2007, 37, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O. Remodelling of the heart and vessels in experimental hypertension: Advances in protection. J. Hypertens. 2010, 28, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Zanchetti, A. Hypertension: Cardiac hypertrophy as a target of antihypertensive therapy. Nat. Rev. Cardiol. 2010, 7, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J. Heart failure and angiotensin converting enzyme inhibition: Problems and perspectives. Physiol. Res. 1999, 48, 1–8. [Google Scholar] [PubMed]
- Simko, F.; Simko, J. The potential role of nitric oxide in the hypertrophic growth of the left ventricle. Physiol. Res. 2000, 49, 37–46. [Google Scholar] [PubMed]
- Mandarim-De-Lacerda, C.A.; Pereira, L.M. Effect of telmisartan on preexistent cardiac and renal lesions in spontaneously hypertensive mature rats. Histol. Histopathol. 2004, 19, 727–733. [Google Scholar] [PubMed]
- Mearini, G.; Schlossarek, S.; Willis, M.S.; Carrier, L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim. Biophys. Acta 2008, 1782, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Massaro, M.; de Caterina, R. Insulin potentiates cytokine-induced VCAM-1 expression in human endothelial cells. Biochim. Biophys. Acta 2008, 1782, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Carbajosa, N.A.; Corradi, G.; Verrilli, M.A.; Guil, M.J.; Vatta, M.S.; Gironacci, M.M. Tyrosine hydroxylase is short-term regulated by the ubiquitin-proteasome system in PC12 cells and hypothalamic and brainstem neurons from spontaneously hypertensive rats: Possible implications in hypertension. PLoS ONE 2015, 24, e0116597. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Perez-Giren, J.V.; Hernanz, R.; Palacios, R.; Briones, A.M.; Fortuño, A.; Zalba, G.; Salaices, M.; Alonso, M.J. Peroxisome proliferator-activated receptor-γ activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J. Hypertens. 2012, 30, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Eble, D.M.; Spragia, M.L.; Ferguson, A.G.; Samarel, A.M. Sarcomeric myosin heavy chain is degraded by the proteasome. Cell Tissue Res. 1999, 296, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; de la Torre-Hernandez, J.M.; Gonzalez-Gonzalez, J.; Garcia-Camarero, T.; Consuegra-Sanchez, L.; Garcia-Saiz, M.D.; Aldea-Perona, A.; Virgos-Aller, T.; Azpeitia, A.; et al. Effect of intravenous and intracoronary melatonin as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: Results of the melatonin adjunct in the acute myocaRdial infarction treated with angioplasty trial. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Hu, W.; Ma, Z.; Jiang, S.; Fan, C.; Deng, C.; Yan, X.; Di, S.; Lv, J.; Reiter, R.J.; Yang, Y. Melatonin: The dawning of a treatment for fibrosis? J. Pineal Res. 2016, 60, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, L.; Yang, Y.; Guo, Y.; Fan, Y.; Zhang, M.; Man, W.; Gao, E.; Hu, W.; Reiter, R.J.; et al. Melatonin alleviates postinfarction cardiac remodeling and dysfunction by inhibiting Mst1. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednárova, K.; Adamcova, M.; Paulis, L. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodeling in spontaneously hypertensive rats. J. Hypertens. 2009, 27, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova-Bednarova, K.; Krajcirovicova, K.; Celec, P.; Kamodyova, N.; Zorad, S.; Kucharska, J.; Gvozdjakova, A.; Adamcova, M.; et al. Hypertension and cardiovascular remodelling in rats exposed to continuous light: Protection by ACE-inhibition and melatonin. Mediat. Inflamm. 2014, 2014, 703175. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O. Recent trends in hypertension treatment: Perspectives from animal studies. J. Hypertens. 2009, 27, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: A physicochemical analysis. J. Pineal Res. 2015, 58, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; González-Candia, A.; Montt, C.; Ebensperger, G.; Chubretovic, M.; Serón-Ferré, M.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Melatonin reduces oxidative stress and improves vascular function in pulmonary hypertensive newborn sheep. J. Pineal Res. 2015, 58, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Chen, Y.; Tan, D.X.; Reiter, R.J.; Chan, Z.; He, C. Melatonin induces nitric oxide and the potential mechanisms relate to innate immunity against bacterial pathogen infection in Arabidopsis. J. Pineal Res. 2015, 59, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Sanchez-Sanchez, J.J.; Kaski, J.C.; Reiter, R.J. Melatonin and circadian biology in human cardiovascular disease. J. Pineal Res. 2010, 49, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. Melatonin feedback on clock genes: A theory involving the proteasome. J. Pineal Res. 2015, 58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Baka, T.; Paulis, L.; Reiter, R.J. Elevated heart rate and nondipping heart rate as potential targets for melatonin: A review. J. Pineal Res. 2016, 61, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. From inflammation to fibrosis: A stiff stretch of highway. Hypertension 2004, 43, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K. Oxidative shielding or oxidative stress? J. Pharmacol. Exp. Ther. 2012, 342, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Simko, F. Chronic antioxidant therapy fails to ameliorate hypertension: Potential mechanisms behind. J. Hypertens. 2009, 27, S32–S36. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E. Does increased oxidative stress cause hypertension? Diabetes Care 2008, 3, S185–S189. [Google Scholar] [CrossRef] [PubMed]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar]
- Ceriello, A. Possible role of oxidative stress in the pathogenesis of hypertension. Diabetes Care 2008, 31, S181–S184. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D. Type 2 diabetes as a redox disease. Lancet 2014, 383, 841. [Google Scholar] [CrossRef]
- Lalu, M.M.; Xu, H.; Sankaralingam, S.; Davidge, S.T. Proteasome inhibition decreases inflammation in human endothelial cells exposed to lipopolysaccharide. J. Cardiovasc. Pharmacol. 2012, 60, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Simko, F. LA419, a novel nitric oxide donor, prevents cardiac remodeling via the endothelial nitric oxide pathway. NO donors as a means of antiremodeling. Hypertension 2007, 50, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Pechanova, O.; Zicha, J.; Krajcirovicova, K.; Barta, A.; Pelouch, V.; Adamcova, M.; Simko, F. Melatonin prevents fibrosis but not hypertrophy development in the left ventricle of NG-nitro-l-arginine-methyl ester hypertensive rats. J. Hypertens. 2009, 27, S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Celec, P.; Palffy, R.; Bednarova, K.; Vrankova, S.; Adamcova, M.; Paulis, L. Continuous light and L-NAME—Induced left ventricular remodelling: Different protection by melatonin and captopril. J. Hypertens. 2010, 28, S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Bednarova, K.; Krajcirovicova, K.; Hrenak, J.; Celec, P.; Kamodyova, N.; Gajdosechova, L.; Zorad, S.; Adamcova, M. Melatonin reduces cardiac remodeling and improves survival in rats with isoproterenol-induced heart failure. J. Pineal Res. 2014, 57, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Paulis, L. Antifibrotic effect of melatonin—Perspective protection in hypertensive heart disease. Int. J. Cardiol. 2013, 168, 2876–2877. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Reiter, R.J.; Pechanova, O.; Paulis, L. Experimental models of melatonin-deficient hypertension. Front. Biosci. 2013, 18, 616–625. [Google Scholar] [CrossRef]
- Gupta, S.; Young, D.; Maitra, R.K.; Gupta, A.; Popovic, Z.B.; Yong, S.L.; Mahajan, A.; Wang, Q.; Sen, S. Prevention of cardiac hypertrophy and heart failure by silencing of NF-κB. J. Mol. Biol. 2008, 375, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, F.M.; Ehrlich, B.E. Calcium, calpains, and cardiac hypertrophy: A new link. Circ. Res. 2009, 104, e19–e20. [Google Scholar] [CrossRef] [PubMed]
- Timmers, L.; van Keulen, J.K.; Hoefer, I.E.; Meijs, M.F.; van Middelaar, B.; den Ouden, K.; van Echteld, C.J.; Pasterkamp, G.; de Kleijn, D.P. Targeted deletion of nuclear factor κB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ. Res. 2009, 104, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Chen, H.; Jiang, J.; Kong, W.; Wang, Y. Silencing MR-1 attenuates inflammatory damage in mice heart induced by Ang II. Biochem. Biophys. Res. Commun. 2010, 391, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Hingtgen, S.D.; Li, Z.; Kutschke, W.; Tian, X.; Sharma, R.V.; Davisson, R.L. Superoxide scavenging and Akt inhibition in myocardium ameliorate pressure overload-induced NF-κB activation and cardiac hypertrophy. Physiol. Genom. 2010, 41, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zelarayan, L.; Renger, A.; Noack, C.; Zafiriou, M.P.; Gehrke, C.; van der Nagel, R.; Dietz, R.; de Windt, L.; Bergmann, M.W. NF-κB activation is required for adaptive cardiac hypertrophy. Cardiovasc. Res. 2009, 84, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Jumnongprakhon, P.; Govitrapong, P.; Tocharus, C.; Tocharus, J. Melatonin promotes blood-brain barrier integrity in methamphetamine-induced inflammation in primary rat brain microvascular endothelial cells. Brain Res. 2016, 1, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Li, H.; Tian, T.; Lu, Y.X.; Bai, L.; Chen, L.Z.; Sheng, H.; Mo, H.Y.; Zeng, J.B.; Deng, W.; et al. Melatonin overcomes gemcitabine resistance in pancreatic ductal adenocarcinoma by abrogating nuclear factor-κB activation. J. Pineal Res. 2016, 60, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Lee, L.M.; Lee, W.J.; Chu, C.Y.; Tan, P.; Yang, Y.C.; Chen, W.Y.; Yang, S.F.; Hsiao, M.; Chien, M.H. Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-κB DNA-binding activity. J. Pineal Res. 2016, 60, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Lin, W.Y.; Shen, C.C.; Pan, H.C.; Keh-Bin, W.; Chen, Y.C.; Jan, Y.J.; Lai, D.W.; Tang, S.C.; Tien, H.R.; et al. Melatonin set out to ER stress signaling thwarts epithelial mesenchymal transition and peritoneal dissemination via calpain-mediated C/EBPβ and NF-κB cleavage. J. Pineal Res. 2016, 60, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Acuña-Castroviejo, D.; Tresguerres, J.A. Beneficial effects of melatonin on cardiological alterations in a murine model of accelerated aging. J. Pineal Res. 2010, 49, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. Melatonin as a proteasome inhibitor. Is there any clinical evidence? Life Sci. 2014, 12, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. The KEAP1-Nrf2-antioxidant response element pathway: A review of its regulation by melatonin and the proteasome. Mol. Cell. Endocrinol. 2015, 5, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Brasier, A.R. Angiotensinogen gene activation by angiotensin II is mediated by the Rel A (nuclear factor-κB p65) transcription factor: One mechanism for the renin angiotensin system positive feedback loop in hepatocytes. Mol. Endocrinol. 1996, 10, 252–264. [Google Scholar] [PubMed]
- Sekiguchi, K.; Li, X.; Coker, M.; Flesch, M.; Barger, P.M.; Sivasubramanian, N.; Mann, D.L. Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc. Res. 2004, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Presa, M.; Bustos, C.; Ortego, M.; Tuñon, J.; Renedo, G.; Ruiz-Ortega, M.; Egido, J. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-κB activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997, 18, 1532–1541. [Google Scholar] [CrossRef]
- Grumbach, I.M.; Chen, W.; Mertens, S.A.; Harrison, D.G. A negative feedback mechanism involving nitric oxide and nuclear factor κB modulates endothelial nitric oxide synthase transcription. J. Mol. Cell. Cardiol. 2005, 39, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Simko, F. The role of nuclear factor κB and nitric oxide interaction in heart remodelling. J. Hypertens. 2010, 28, S39–S44. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Matuskova, J.; Adamcova, M.; Pelouch, V.; Simko, J.; Krajcirovicova, K.; Potacova, A.; Hulin, I.; Janega, P.; Pechanova, O.; et al. Regression of left ventricular hypertrophy and aortic remodelling in NO-deficient hypertensive rats: Effect of l-arginine and spironolactone. Acta Physiol. 2008, 194, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Pelouch, V.; Milerova, M.; Ostadal, B.; Samánek, M.; Hucín, B. Protein profiling of human atrial and ventricular musculature: The effect of normoxaemia and hypoxaemia in congenital heart diseases. Physiol. Res. 1993, 42, 235–242. [Google Scholar] [PubMed]
- Pelouch, V.; Milerova, M.; Ostadal, B.; Hucín, B.; Samánek, M. Differences between atrial and ventricular protein profiling in children with congenital heart disease. Mol. Cell. Biochem. 1995, 147, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.K.; Enwemeka, C.S. A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 1996, 29, 225–229. [Google Scholar] [CrossRef]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Munch, G. Determination of advanced glycation end products in serum by fluorescence spectroscopy and competitive ELISA. Eur. J. Clin. Chem. Clin. Biochem. 1997, 35, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Bhatwadekar, A.D.; Ghole, V.S. Rapid method for the preparation of an AGE-BSA standard calibrator using thermal glycation. J. Clin. Lab. Anal. 2005, 19, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Behuliak, M.; Palffy, R.; Gardlik, R.; Hodosy, J.; Halcak, L.; Celec, P. Variability of thiobarbituric acid reacting substances in saliva. Dis. Mark. 2009, 26, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Helenius, M.; Hänninen, M.; Lehtinen, S.K.; Salminen, A. Aging-induced upregulation of nuclear binding activities of oxidative stress responsive NF-κB transcription factor in mouse cardiac muscle. J. Mol. Cell. Cardiol. 1996, 28, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Welinder, C.; Ekblad, L. Coomassie staining as loading control in Western blot analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Racchumi, G.; Iadecola, C. cis-Acting, element-specific transcriptional activity of differentially phosphorylated nuclear factor-κB. J. Biol. Chem. 2005, 7, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene 2006, 30, 6717–6730. [Google Scholar] [CrossRef] [PubMed]
- Mikenberg, I.; Widera, D.; Kaus, A.; Kaltschmidt, B.; Kaltschmidt, C. Transcription factor NF-κB is transported to the nucleus via cytoplasmic dynein/dynactin motor complex in hippocampal neurons. PLoS ONE 2007, 11, e589. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.N.; Bae, J.W.; Gao, E.; Koch, W.J. Regulation of nuclear factor κB (NF-κB) in the nucleus of cardiomyocytes by G protein-coupled receptor kinase 5 (GRK5). J. Biol. Chem. 2013, 13, 35683–35689. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Pechanova, O.; Zicha, J.; Barta, A.; Gardlik, R.; Celec, P.; Kunes, J.; Simko, F. Melatonin interactions with blood pressure and vascular function during L-NAME-induced hypertension. J. Pineal Res. 2010, 48, 102–108. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simko, F.; Pechanova, O.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Celec, P.; Tothova, L.; Vrankova, S.; Balazova, L.; Zorad, S.; et al. Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril. Int. J. Mol. Sci. 2017, 18, 1612. https://doi.org/10.3390/ijms18081612
Simko F, Pechanova O, Repova K, Aziriova S, Krajcirovicova K, Celec P, Tothova L, Vrankova S, Balazova L, Zorad S, et al. Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril. International Journal of Molecular Sciences. 2017; 18(8):1612. https://doi.org/10.3390/ijms18081612
Chicago/Turabian StyleSimko, Fedor, Olga Pechanova, Kristina Repova, Silvia Aziriova, Kristina Krajcirovicova, Peter Celec, Lubomira Tothova, Stanislava Vrankova, Lucia Balazova, Stefan Zorad, and et al. 2017. "Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril" International Journal of Molecular Sciences 18, no. 8: 1612. https://doi.org/10.3390/ijms18081612