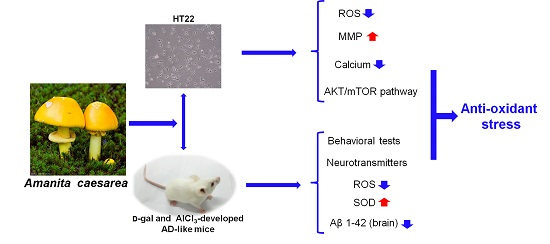
Anti-Oxidative Stress Activity Is Essential for Amanita caesarea Mediated Neuroprotection on Glutamate-Induced Apoptotic HT22 Cells and an Alzheimer’s Disease Mouse Model

Abstract
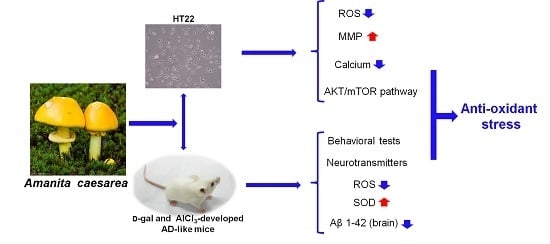
:
1. Introduction
2. Results
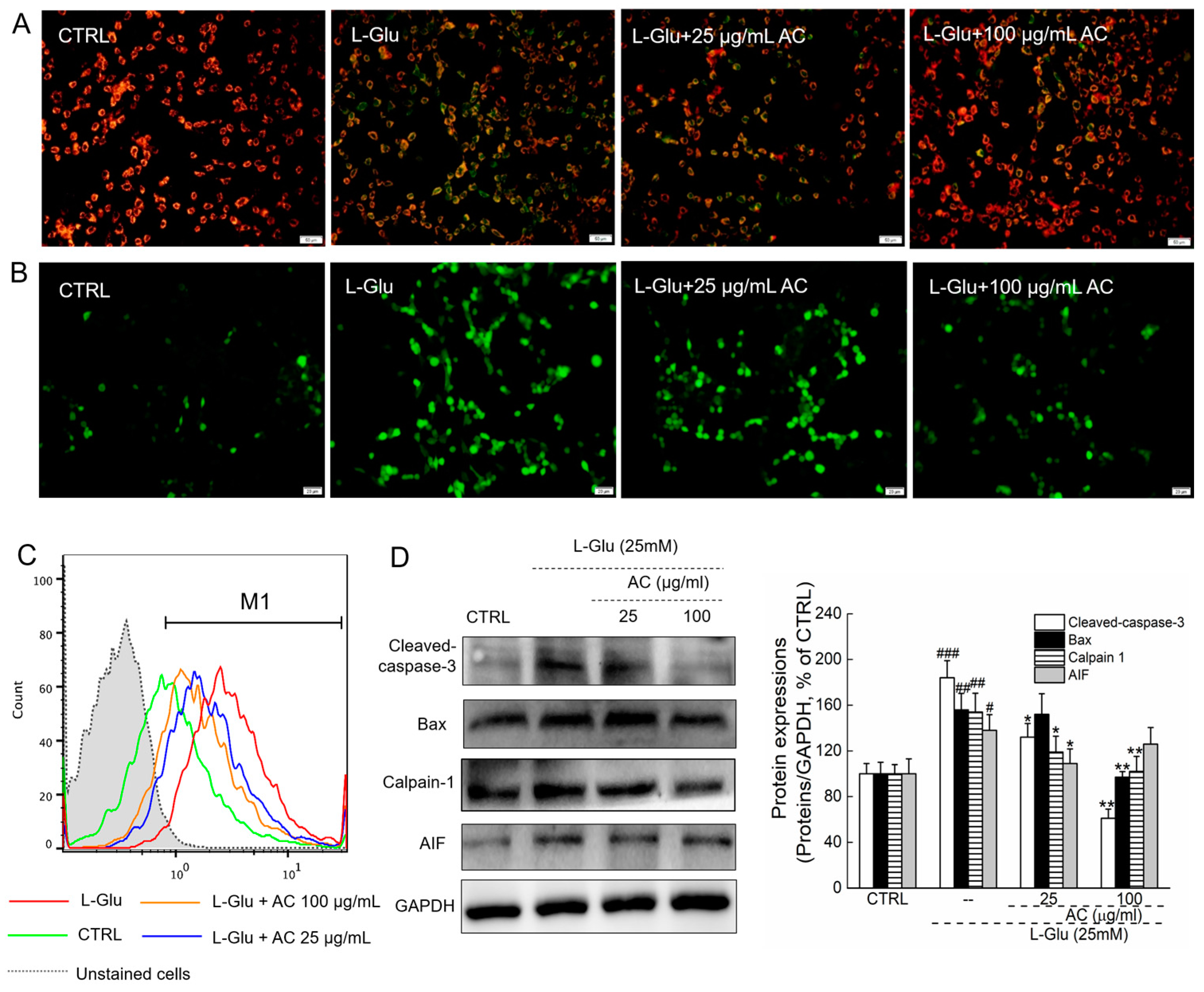
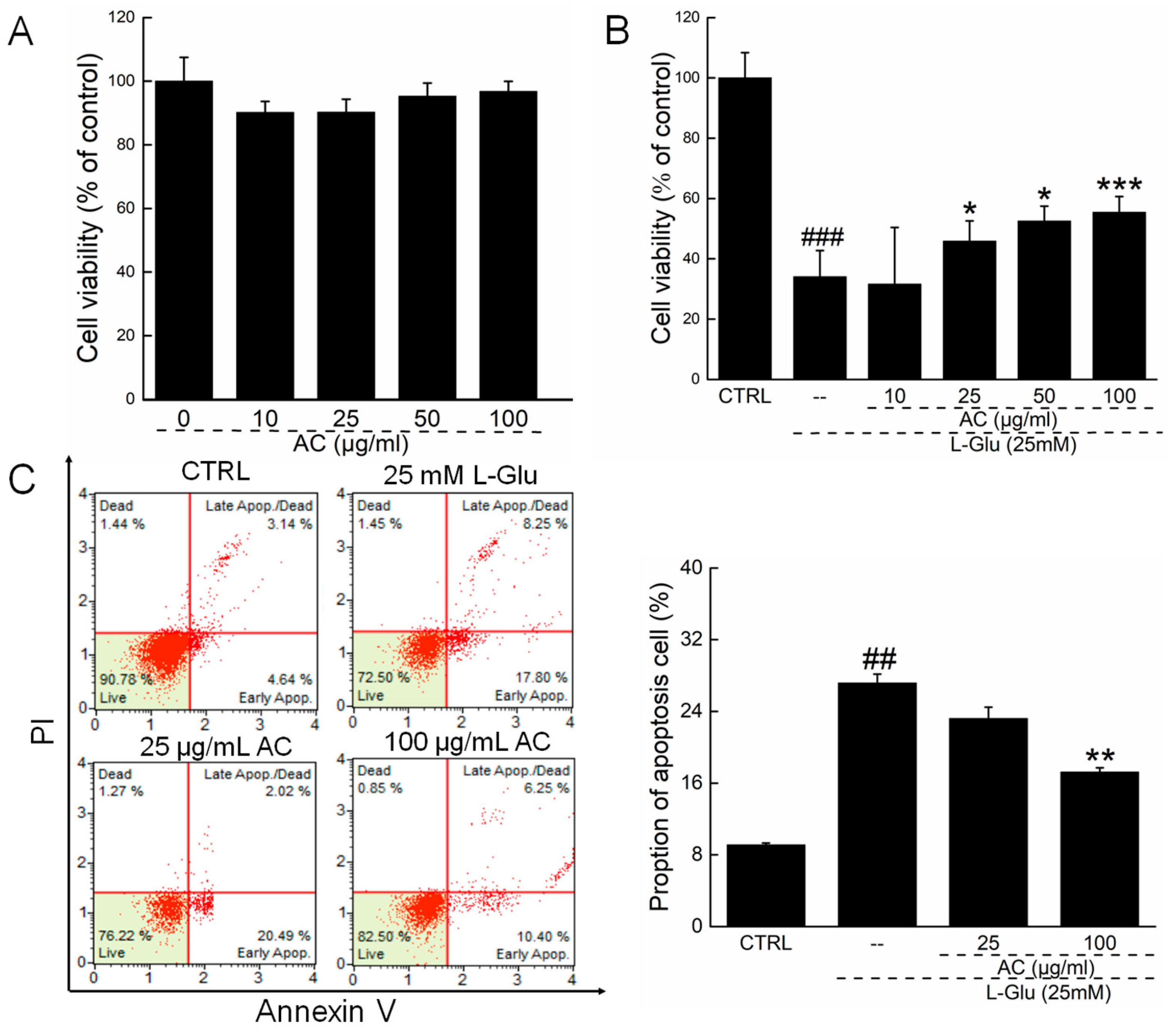
2.1. AC Ameliorated l-Glu-Induced Cytotoxicity and Apoptosis in HT22 Cells
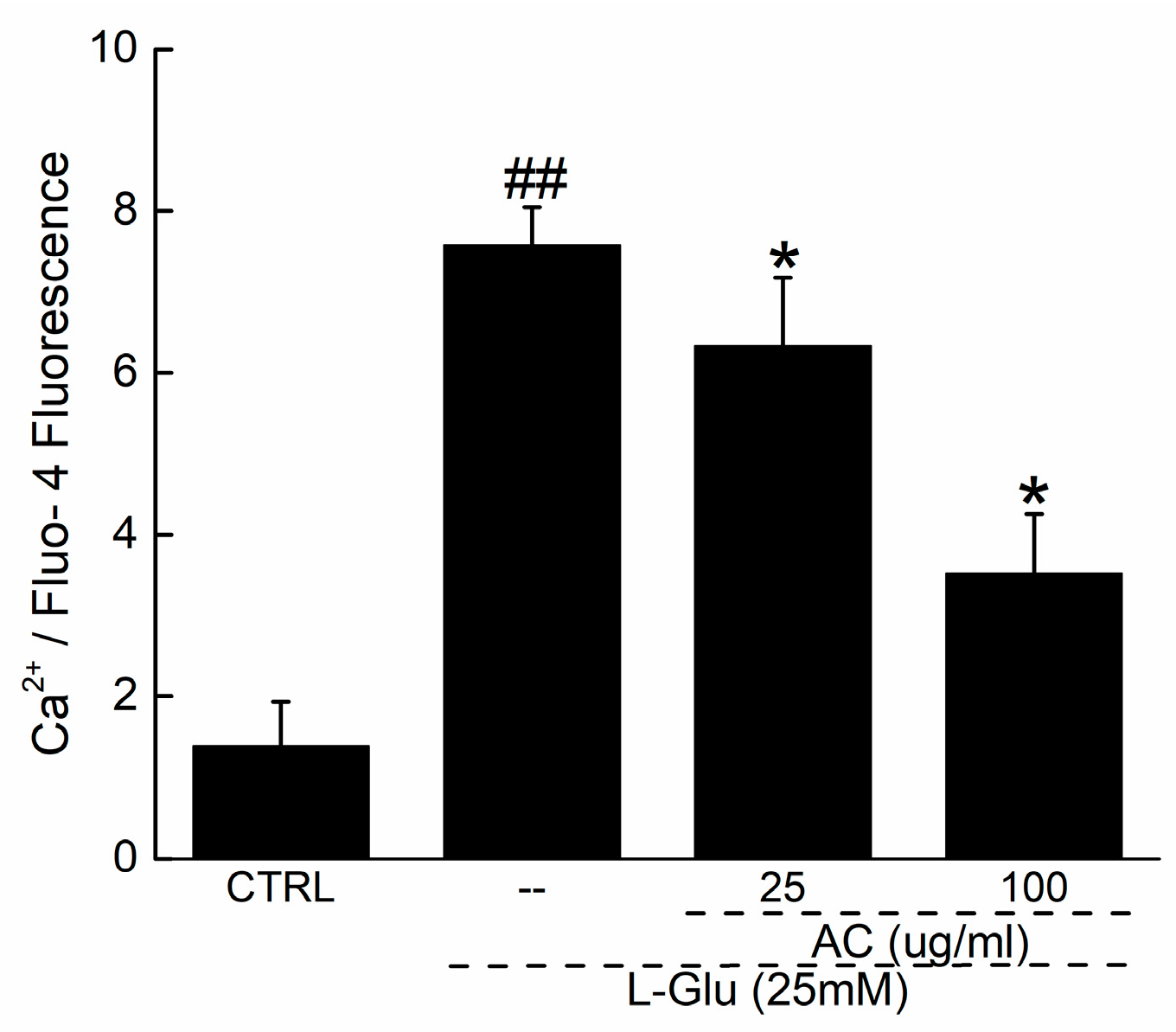
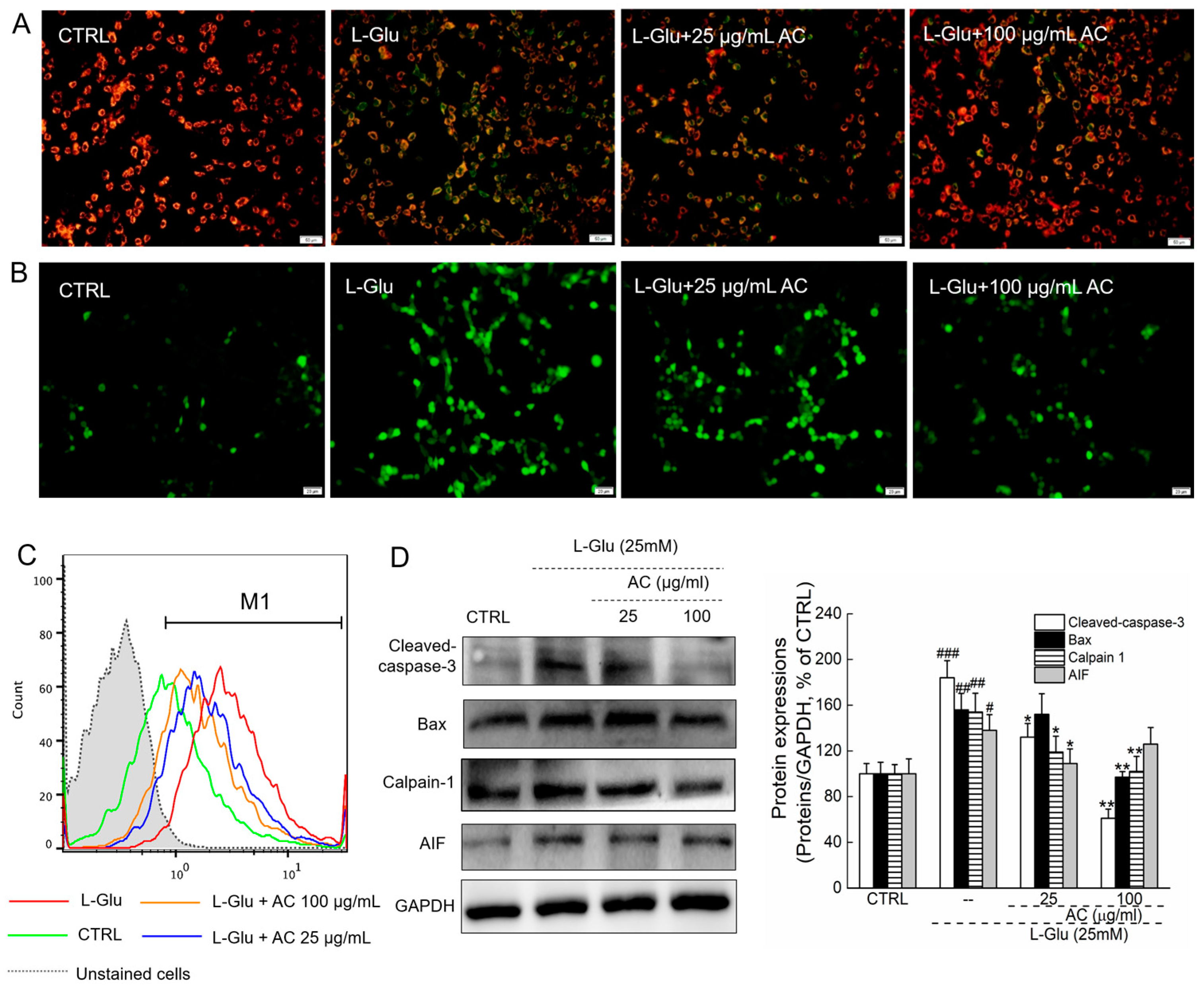
2.2. AC Ameliorated l-Glu-Caused Mitochondrial Dysfunction in HT22 Cells
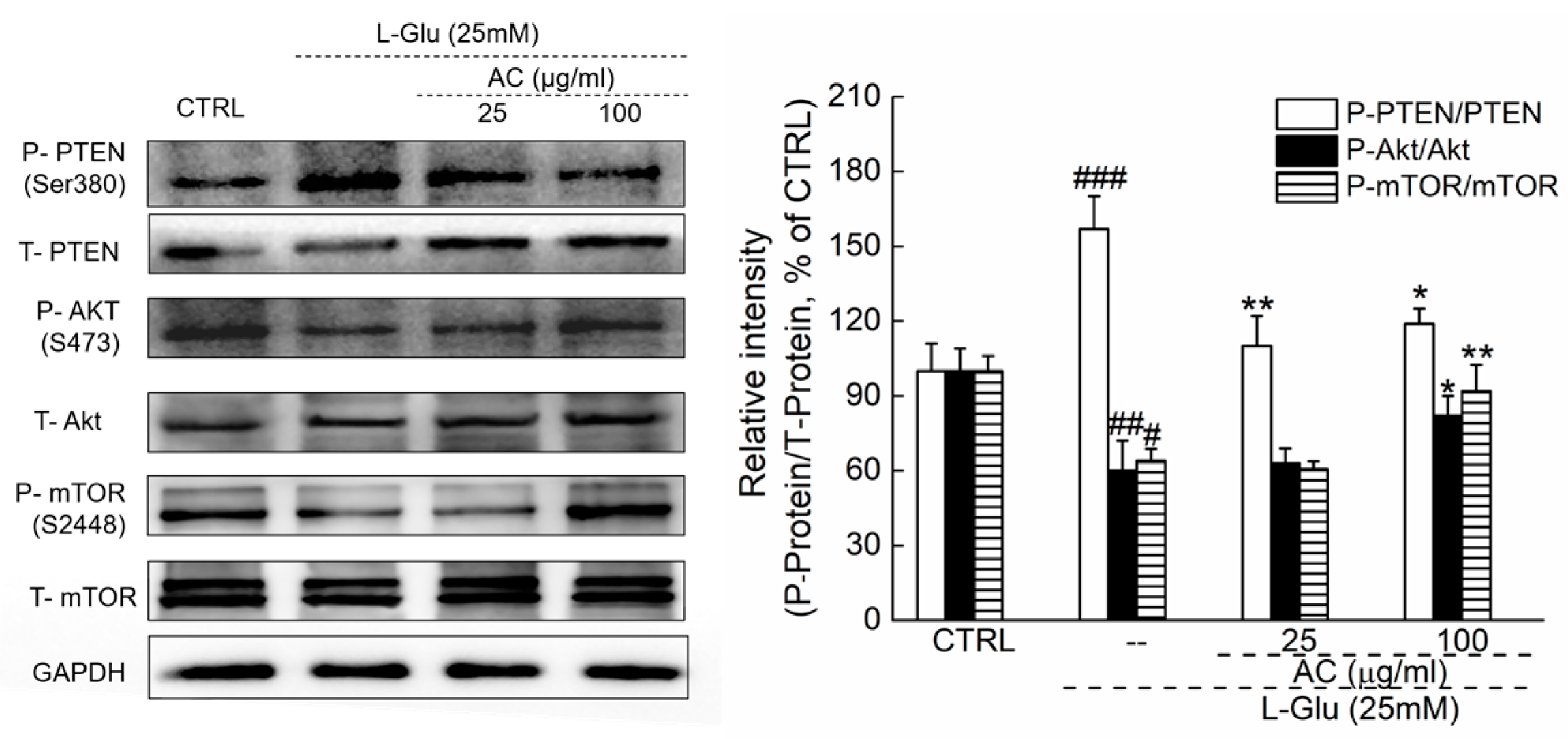
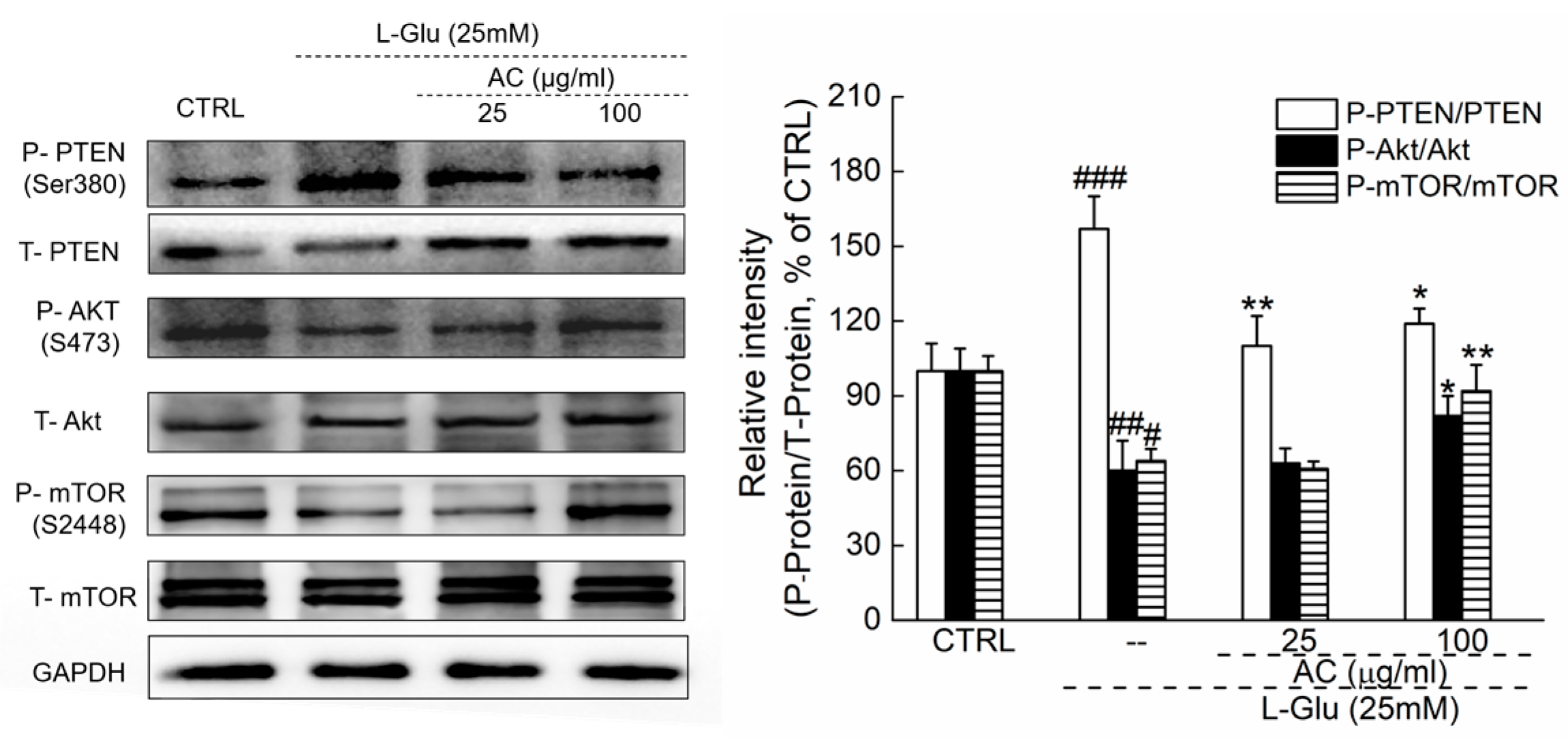
2.3. AC Ameliorated l-Glu-Induced Cell Injury by Akt/mTOR Pathway
2.4. AC Improved AD-Like Behaviors in d-gal and AlCl3 Induced AD Mice
2.5. AC Regulated the Levels of Acetylcholine (Ach), Acetyltransferase (ChAT), and Acetylcholine Esterase (AchE) in the Serum and Brain of AD Mice
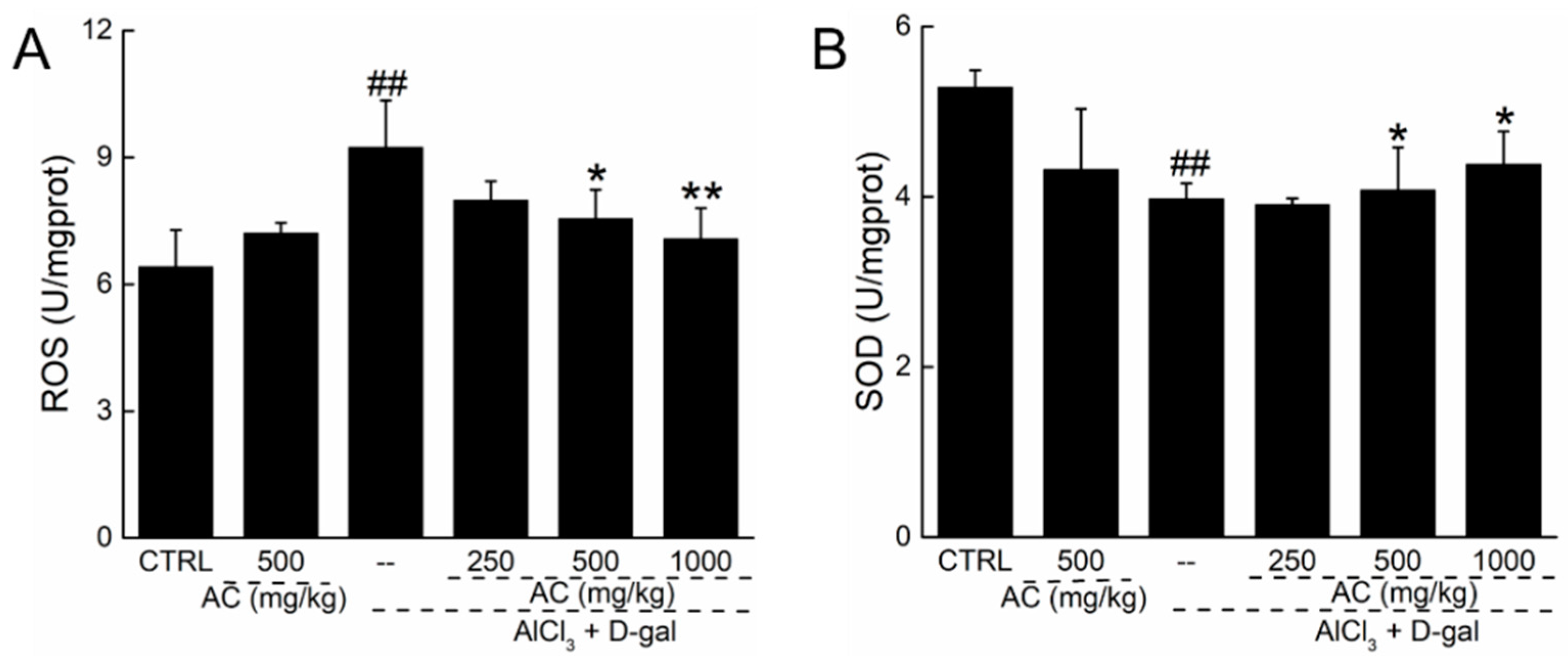
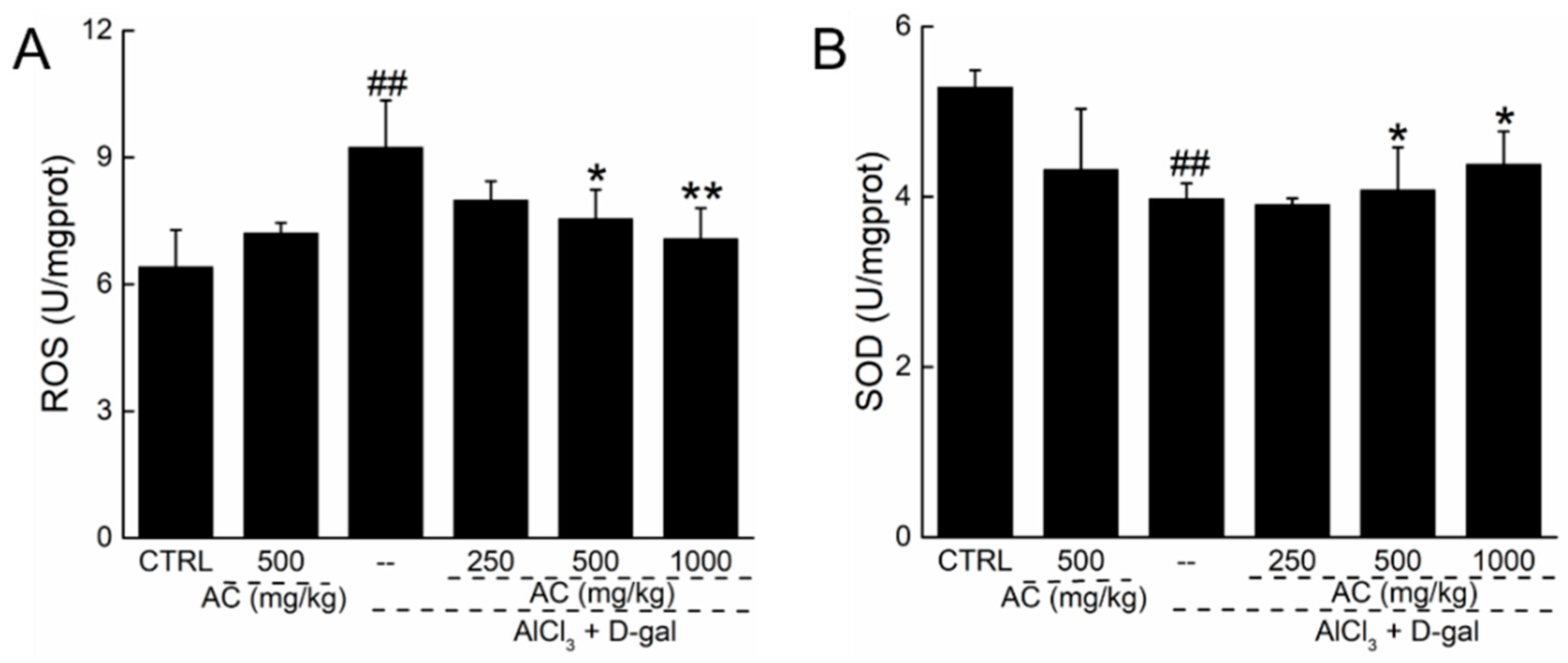
2.6. AC Modulated the Levels of Superoxide Dismutase (SOD) and ROS in the Brains of AD Mice
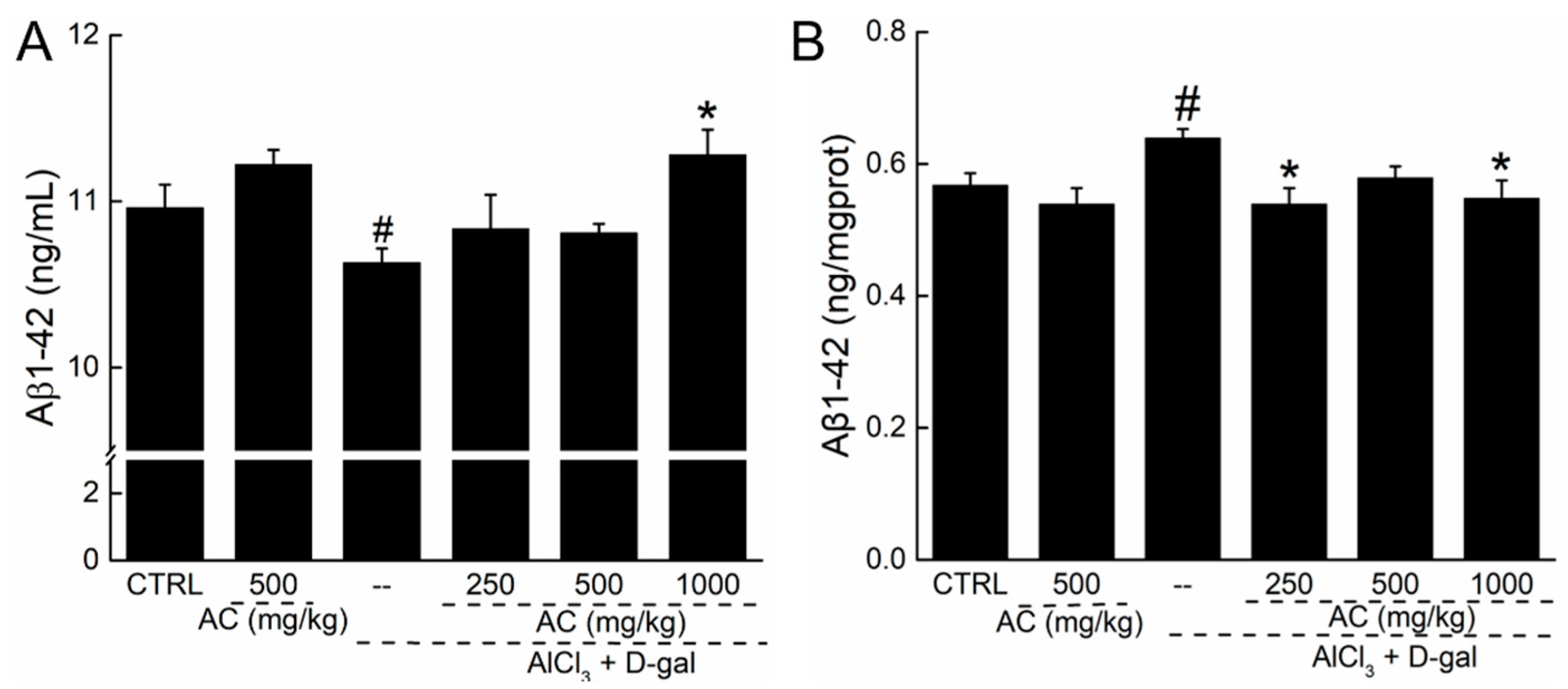
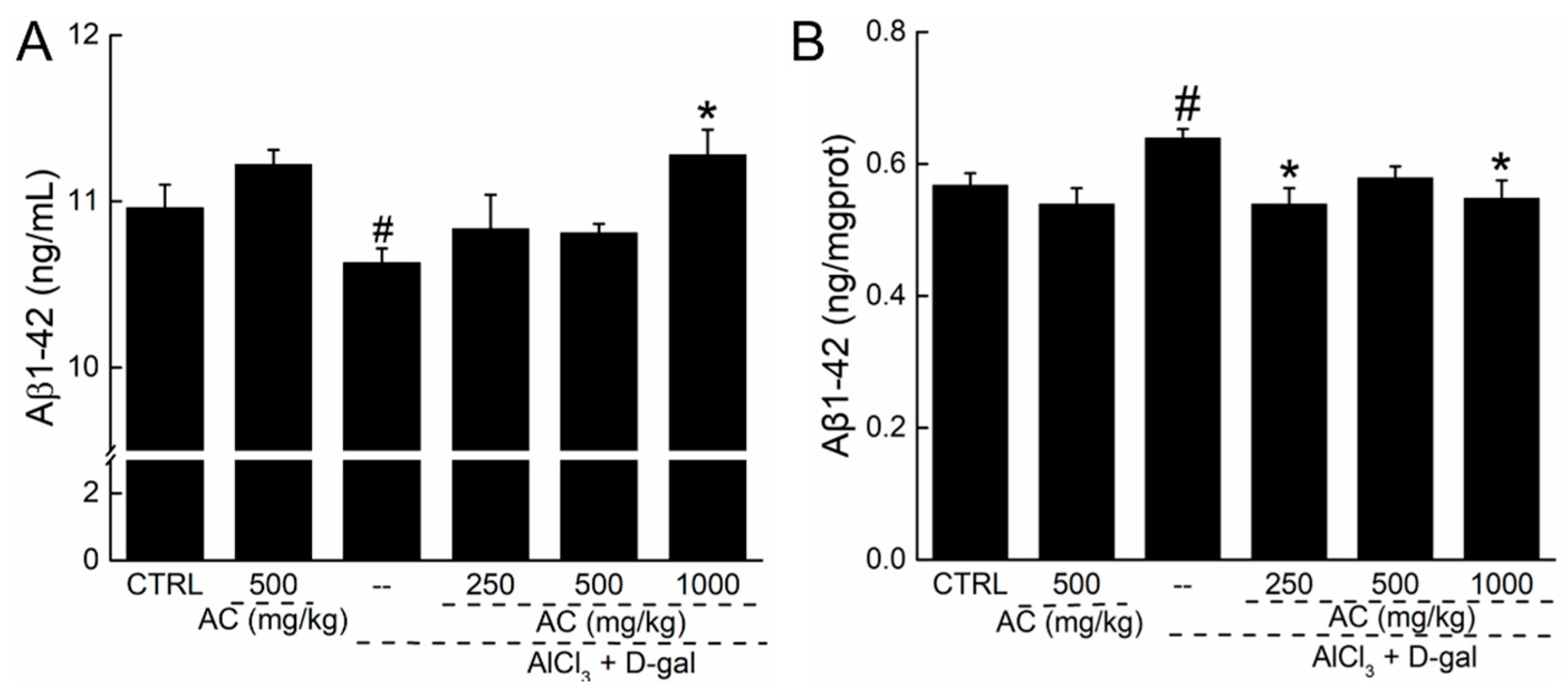
2.7. The Effect of AC on the Levels of Aβ 1-42 in Serum and Brain of AD Mice
3. Discussion
4. Materials and Methods
4.1. Preparation of A. caesarea Water Extract (AC)
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Apoptosis Assay
4.5. Mitochondrial Transmembrane Potential (MMP) Measurement
4.6. Intracellular ROS Measurement
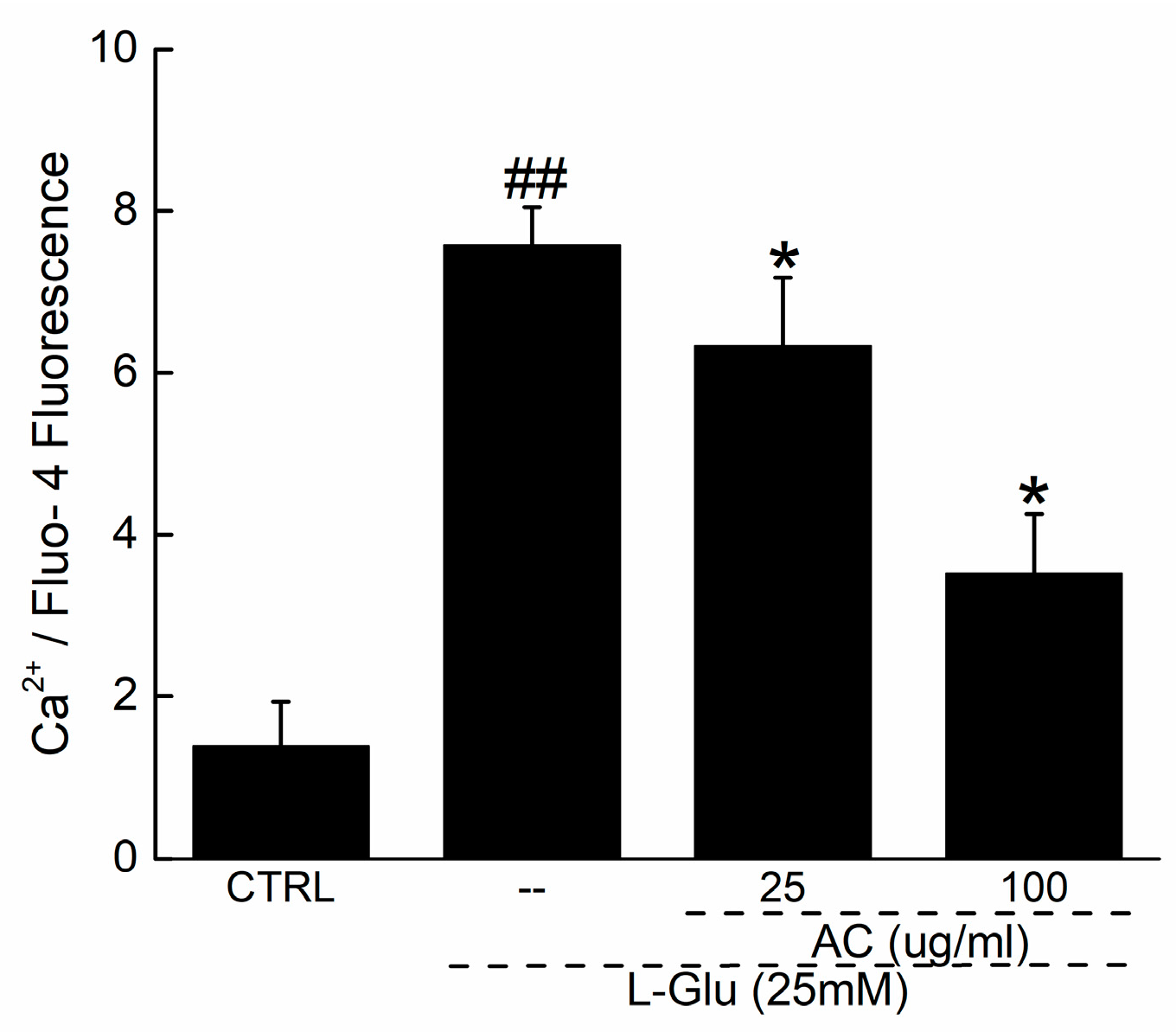
4.7. Intracellular Ca2+ Measurement
4.8. Western Blot
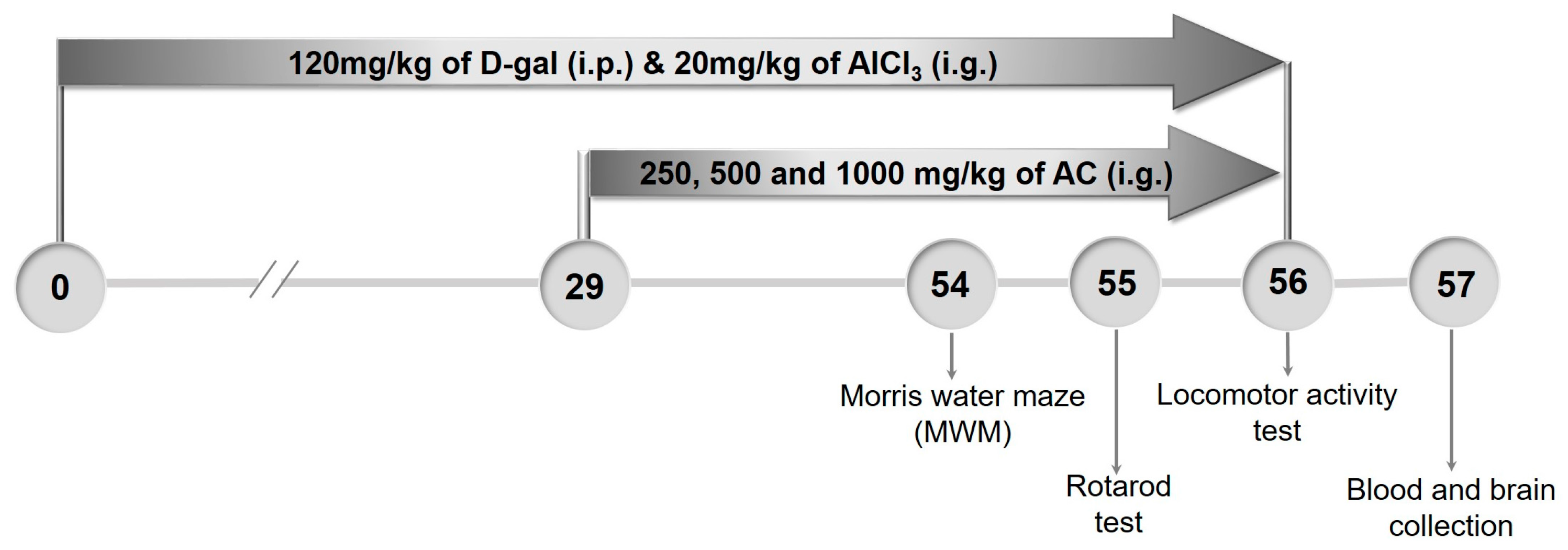
4.9. The Establishment of AD Mouse Model and Drug Treatment Process
4.10. Behavioral Tests
4.11. Biochemical Analysis
4.12. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ach | Acetylcholine |
| AchE | Acetylcholine esterase |
| AC | A. caesarea water extracts |
| AD | Alzheimer’s disease |
| Akt | Protein kinase B |
| ANOVA | A one-way analysis of variance |
| Aβ | Amyloid beta |
| ChAT | Choline acetyltransferase |
| d-gal | d-galactose |
| ELISA | Enzyme-linked immunosorbent assay |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| l-Glu | l-glutamic acid |
| MMP | Mitochondrial membrane potential |
| mTOR | Mammalian target of rapamycin |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
Appendix A
References
- Rygiel, K. Novel strategies for Alzheimer’s disease treatment: An overview of anti-amyloid beta monoclonal antibodies. Indian J. Pharmacol. 2016, 48, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Rosello, A.; Warnes, G.; Meier, U.C. Cell death pathways and autophagy in the central nervous system and its involvement in neurodegeneration, immunity and central nervous system infection: To die or not to die--that is the question. Clin. Exp. Immunol. 2012, 168, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J. Vascular dementia and alzheimer’s disease: A waning dichotomy. J. Alzheimer’s Dis. 2007, 12, 343–344. [Google Scholar] [CrossRef]
- Faizi, M.; Seydi, E.; Abarghuyi, S.; Salimi, A.; Nasoohi, S.; Pourahmad, J. A search for mitochondrial damage in Alzheimer’s disease using isolated rat brain mitochondria. Iran. J. Pharm. Res. 2016, 15, 185–195. [Google Scholar] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Fei, F.; Zhang, L.; Qu, Y.; Fei, Z. The role of glutamate receptors in traumatic brain injury: Implications for postsynaptic density in pathophysiology. Brain Res. Bull. 2011, 85, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Luo, Y.; Niu, F.; Sun, Z.; Cao, W.; Zhang, X.; Guan, D.; Lv, Z.; Zhang, B.; Xu, Y. Altered expression of abeta metabolism-associated molecules from d-galactose/ALCL3) induced mouse brain. Mech. Ageing Dev. 2009, 130, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Lindequist, U.; Niedermeyer, T.H.; Julich, W.D. The pharmacological potential of mushrooms. Evid.-Based Complement. Altern. Med. 2005, 2, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, D.; Zhang, J.; Du, M.; Cheng, Y.; Liu, Y.; Zhang, N.; Wang, D.; Wu, Y. Mitochondria related pathway is essential for polysaccharides purified from sparassis crispa mediated neuroprotection against glutamate-induced toxicity in differentiated pc12 cells. Int. J. Mol. Sci. 2016, 17, 133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; An, S.; Hu, W.; Teng, M.; Wang, X.; Qu, Y.; Liu, Y.; Yuan, Y.; Wang, D. The neuroprotective properties of hericium erinaceus in glutamate-damaged differentiated pc12 cells and an Alzheimer’s disease mouse model. Int. J. Mol. Sci. 2016, 17, 1810. [Google Scholar] [CrossRef] [PubMed]
- Dogan, H.H.; Akbas, G. Biological activity and fatty acid composition of caesar’s mushroom. Pharm. Biol. 2013, 51, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ding, X.; Wang, M.; Hou, Y.; Hou, W.; Yue, C. Structure and antioxidant activity of a novel polysaccharide derived from Amanita caesarea. Mol. Med. Rep. 2016, 14, 3947–3954. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Swaminathan, K.; Ramesh, A.; Kurian, G.A. Nicorandil attenuates neuronal mitochondrial dysfunction and oxidative stress associated with murine model of vascular calcification. Acta Neurobiol. Exp. 2017, 77, 57–67. [Google Scholar]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M. Aif, reactive oxygen species, and neurodegeneration: A “complex” problem. Neurochem. Int. 2013, 62, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of pi3k/akt/mtor pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Gais, S.; Schonauer, M. Untangling a cholinergic pathway from wakefulness to memory. Neuron 2017, 94, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Losada-Barreiro, S.; Bravo-Diaz, C. Free radicals and polyphenols: The redox chemistry of neurodegenerative diseases. Eur. J. Med. Chem. 2017, 133, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of beta-amyloid in alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Guell-Bosch, J.; Villegas, S. Mouse models of alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Sarikurkcu, C.; Tepe, B.; Semiz, D.K.; Solak, M.H. Evaluation of metal concentration and antioxidant activity of three edible mushrooms from mugla, turkey. Food Chem. Toxicol. 2010, 48, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef]
- Cregan, S.P.; Dawson, V.L.; Slack, R.S. Role of aif in caspase-dependent and caspase-independent cell death. Oncogene 2004, 23, 2785–2796. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Sub-Cell. Biochem. 2007, 45, 481–506. [Google Scholar]
- Tang, X.-Q.; Feng, J.-Q.; Chen, J.; Chen, P.-X.; Zhi, J.-L.; Cui, Y.; Guo, R.-X.; Yu, H.-M. Protection of oxidative preconditioning against apoptosis induced by H2O2 in pc12 cells: Mechanisms via mmp, ros, and bcl-2. Brain Res. 2005, 1057, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Katz, S.G. Non-apoptotic functions of bcl-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S.D. Pathogenic role of mitochondrial amyloid-beta peptide. Expert Rev. Neurother. 2007, 7, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Romero-Leguizamon, C.R.; Ramirez-Latorre, J.A.; Mora-Munoz, L.; Guerrero-Naranjo, A. Signaling pathways mtor and akt in epilepsy. Rev. Neurol. 2016, 63, 33–41. [Google Scholar] [PubMed]
- Ricciardi, M.R.; Mirabilii, S.; Licchetta, R.; Piedimonte, M.; Tafuri, A. Targeting the akt, gsk-3, bcl-2 axis in acute myeloid leukemia. Adv. Biol. Regul. 2017. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.R.; Hattori, H.; Hossain, M.A.; Hester, L.; Huang, Y.; Lee-Kwon, W.; Donowitz, M.; Nagata, E.; Snyder, S.H. Akt as a mediator of cell death. Proc. Natl. Acad. Sci. USA 2003, 100, 11712–11717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Lin, S.L.; Huang, C.N.; Lu, F.J.; Chiu, P.Y.; Huang, W.N.; Lai, T.J.; Lin, C.L. Mir-302 attenuates amyloid-beta-induced neurotoxicity through activation of akt signaling. J. Alzheimer’s Dis. 2016, 50, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.B.; Zhang, B.L. Biochemical changes in d-galactose induced subacute toxicity and mimetic aging in mice. Chin. J. Pharm. Toxicol. 1990, 4, 309–310. [Google Scholar]
- Banks, W.A.; Niehoff, M.L.; Drago, D.; Zatta, P. Aluminum complexing enhances amyloid beta protein penetration of blood-brain barrier. Brain Res. 2006, 1116, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, Y.L.; Luo, L.; Wu, D.M.; Sun, D.X.; Feng, Y.J. Quercetin reverses d-galactose induced neurotoxicity in mouse brain. Behav. Brain Res. 2006, 171, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M. Aluminum modulates brain amyloidosis through oxidative stress in app transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Li, X.G.; Zhang, X.Y.; Hou, J.D.; Lin, L.F.; Gao, Q.; Luo, H.M. Combined administration of d-galactose and aluminium induces alzheimer-like lesions in brain. Neurosci. Bull. 2011, 27, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dogra, S.; Prakash, A. Protective effect of curcumin (curcuma longa), against aluminium toxicity: Possible behavioral and biochemical alterations in rats. Behav. Brain Res. 2009, 205, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G. Oxidative stress induced-metabolic imbalance, mitochondrial failure, and cellular hypoperfusion as primary pathogenetic factors for the development of alzheimer disease which can be used as a alternate and successful drug treatment strategy: Past, present and future. CNS Neurol. Disord. Drug Targets 2011, 10, 147–148. [Google Scholar] [PubMed]
- Christen, Y. Oxidative stress and alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621s–629s. [Google Scholar] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Fei, M.; Jianghua, W.; Rujuan, M.; Wei, Z.; Qian, W. The relationship of plasma abeta levels to dementia in aging individuals with mild cognitive impairment. J. Neurol. Sci. 2011, 305, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Gao, Z.; Zheng, L.; Zhang, C.; Liu, Z.; Yang, Y.; Teng, H.; Hou, L.; Yin, Y.; Zou, X. Protective effects of fucoidan on aβ 25–35 and d-gal-induced neurotoxicity in pc12 cells and d-gal-induced cognitive dysfunction in mice. Mar. Drugs 2017, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Paliga, K.; Durrwang, U.; Reinhard, F.B.; Schuckert, O.; Evin, G.; Masters, C.L. Proteolytic processing of the alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J. Biol. Chem. 1999, 274, 5823–5829. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G.M. Cerebral microvascular pathology in aging and alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Neurotransmitters | CTRL | AC (500 mg/kg) | AlCl3 + d-gal | |||
|---|---|---|---|---|---|---|---|
| CTRL | AC (mg/kg) | ||||||
| 250 | 500 | 1000 | |||||
| Serum | Ach (μg/mL) | 115.4 ± 5.8 | 107.2 ± 6.6 | 87.2 ± 8.4 # | 98.3 ± 5.5 | 89.1 ± 3.1 | 108.8 ± 4.2 * |
| AchE (nmol/ L) | 9.7 ± 0.2 | 9.6 ± 0.1 | 11.2 ± 0.3 # | 10.7 ± 0.1 | 9.8 ± 0.2 * | 9.6 ± 0.1 * | |
| ChAT (pg/mL) | 181.6 ± 8.3 | 195.7 ± 23.2 | 163.4 ± 18.2 # | 182.7 ± 9.9 | 204.1 ± 13.7 * | 204.1 ± 15.2 * | |
| Tissue | Ach (μg/mgprot) | 2.1 ± 0.1 | 2.4 ± 0.2 | 1.3 ± 0.1 ## | 1.3 ± 0.2 | 1.2 ± 0.1 | 2.1 ± 0.2 * |
| AchE (nmol/gprot) | 0.28 ± 0.03 | 0.21 ± 0.02 | 0.47 ± 0.06 # | 0.48 ± 0.06 | 0.37 ± 0.02 * | 0.37 ± 0.05 * | |
| ChAT (pg/mgprot) | 6.9 ± 0.5 | 7.2 ± 0.3 | 5.0 ± 0.1 # | 5.1 ± 0.6 | 7.2 ± 0.3 * | 7.3 ± 0.4 * | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Chen, X.; Lu, W.; Zhang, S.; Guan, X.; Li, Z.; Wang, D. Anti-Oxidative Stress Activity Is Essential for Amanita caesarea Mediated Neuroprotection on Glutamate-Induced Apoptotic HT22 Cells and an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2017, 18, 1623. https://doi.org/10.3390/ijms18081623
Li Z, Chen X, Lu W, Zhang S, Guan X, Li Z, Wang D. Anti-Oxidative Stress Activity Is Essential for Amanita caesarea Mediated Neuroprotection on Glutamate-Induced Apoptotic HT22 Cells and an Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences. 2017; 18(8):1623. https://doi.org/10.3390/ijms18081623
Chicago/Turabian StyleLi, Zhiping, Xia Chen, Wenqian Lu, Shun Zhang, Xin Guan, Zeyu Li, and Di Wang. 2017. "Anti-Oxidative Stress Activity Is Essential for Amanita caesarea Mediated Neuroprotection on Glutamate-Induced Apoptotic HT22 Cells and an Alzheimer’s Disease Mouse Model" International Journal of Molecular Sciences 18, no. 8: 1623. https://doi.org/10.3390/ijms18081623