Electronegative Low-Density Lipoprotein L5 Impairs Viability and NGF-Induced Neuronal Differentiation of PC12 Cells via LOX-1


Abstract
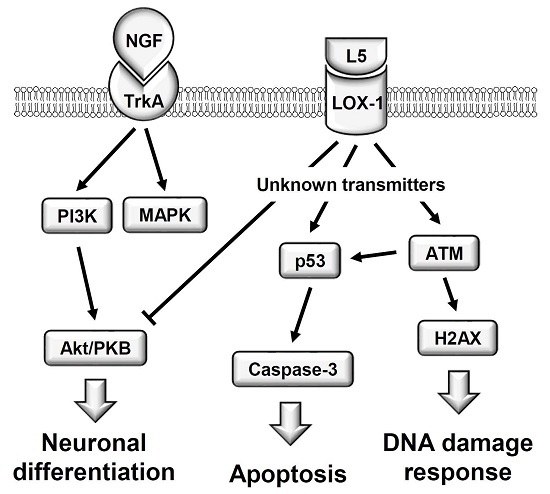
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
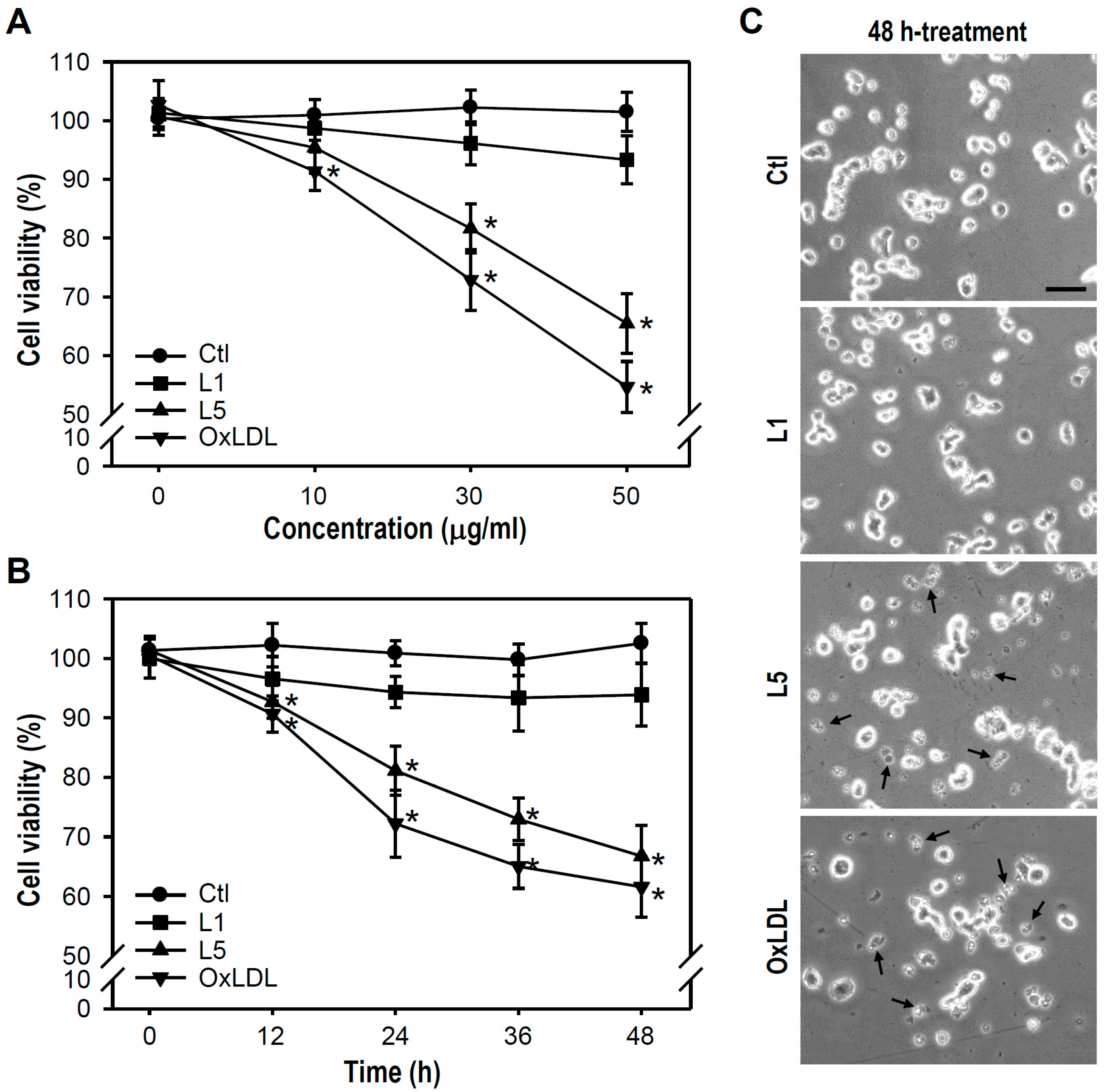
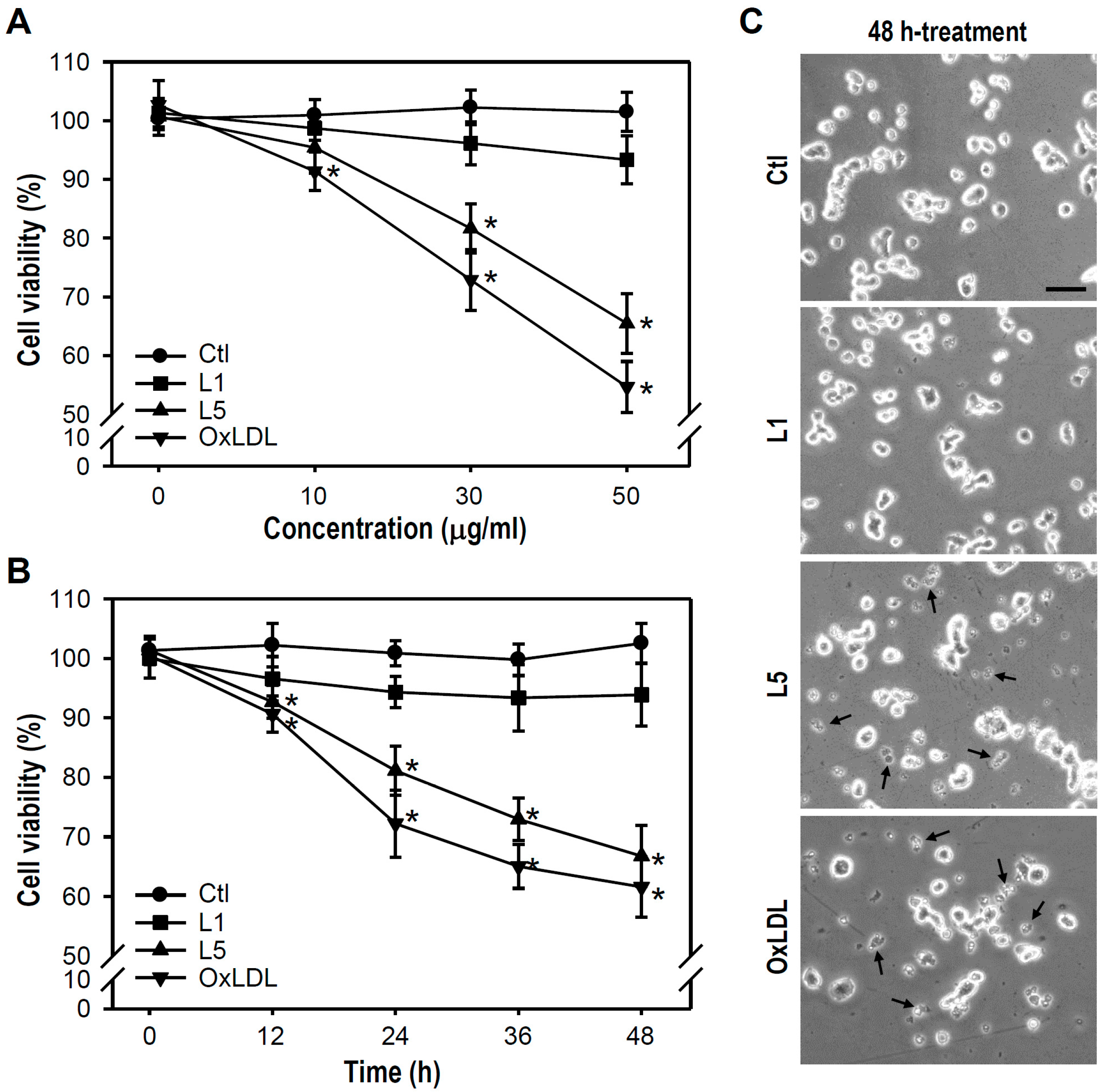
2.1. L5 Shows a Cytotoxic Property and Reduces the Viability of PC12 Cells
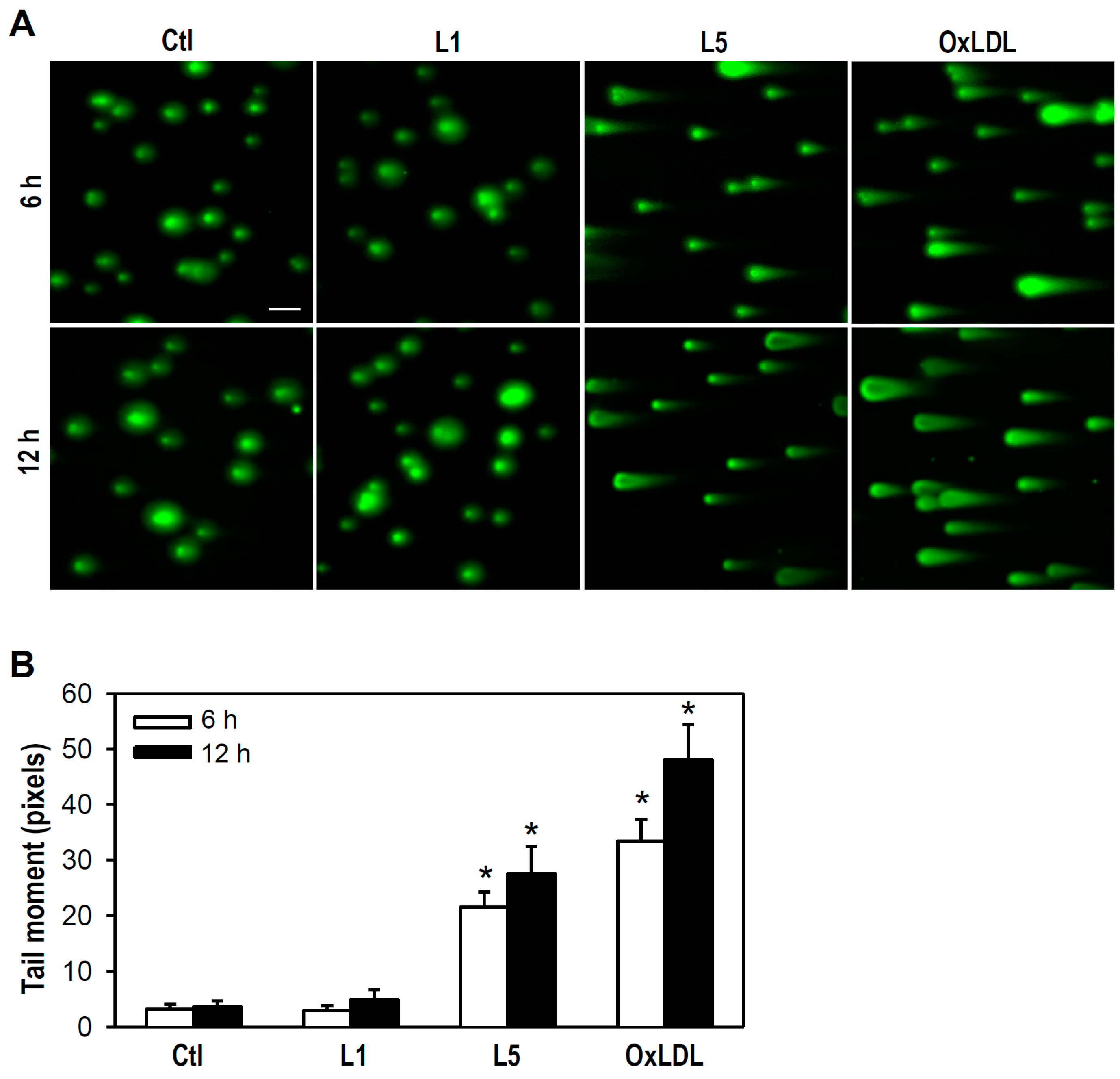
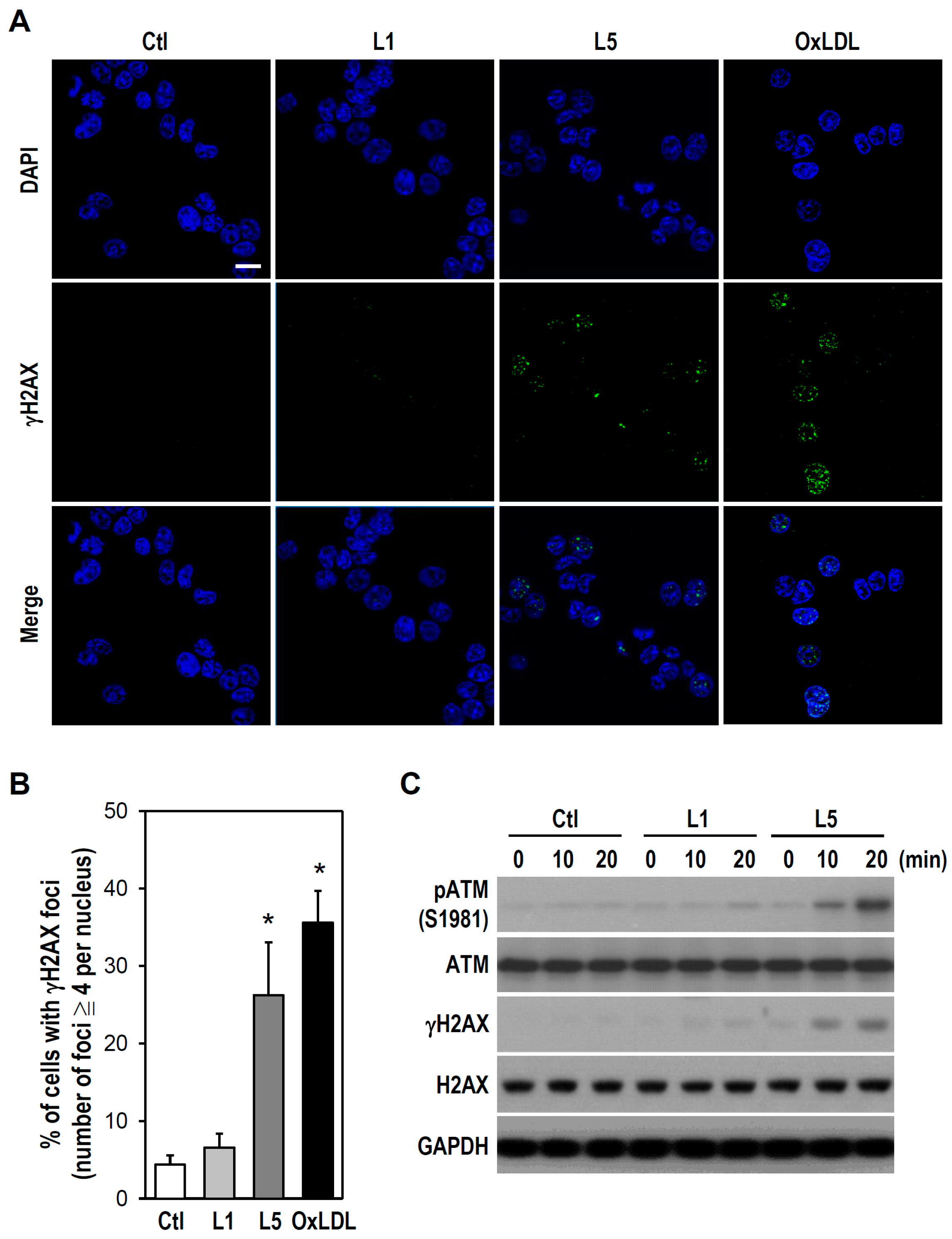
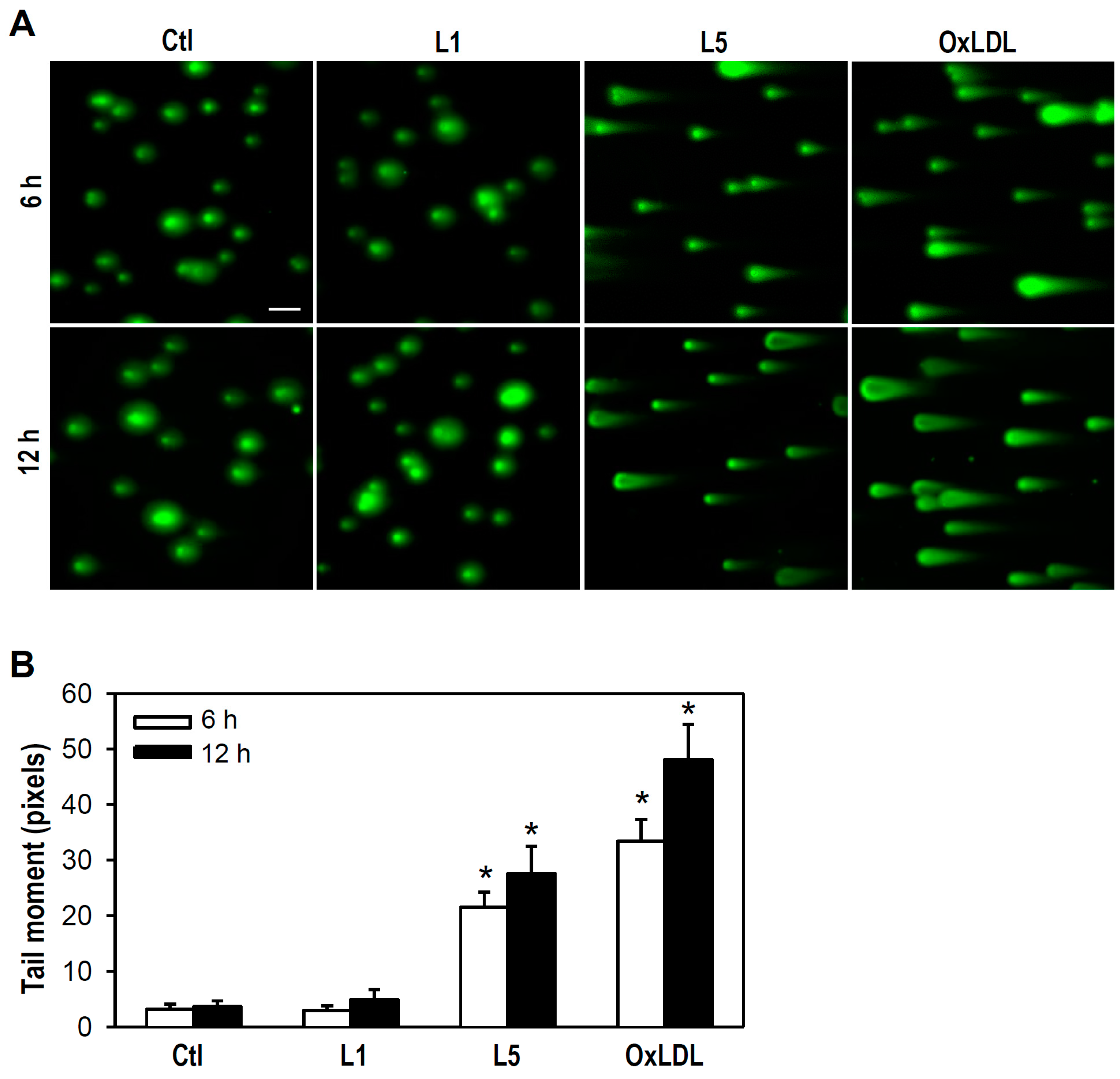
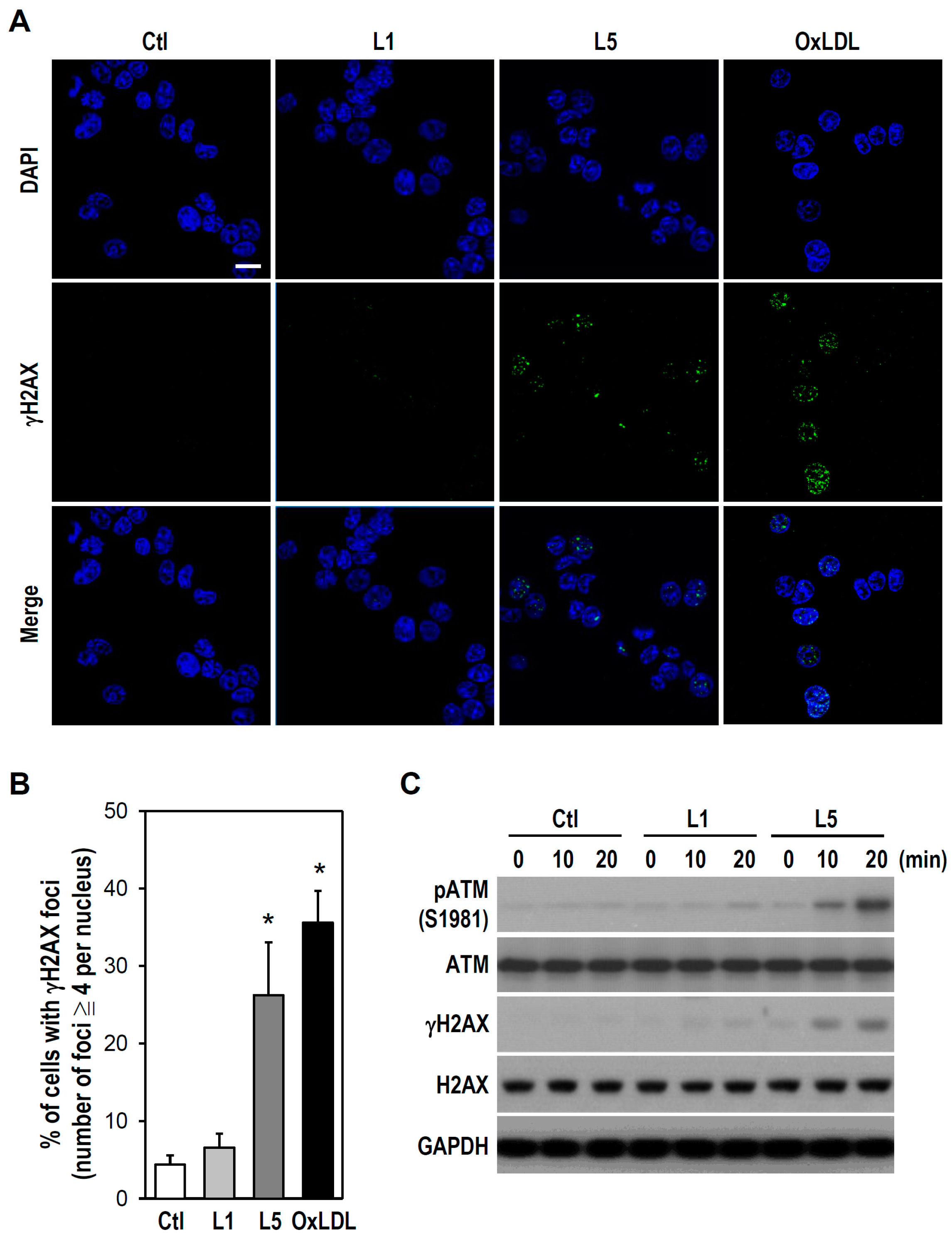
2.2. L5 Induces Genotoxicity via ATM/H2AX Activation in PC12 Cells
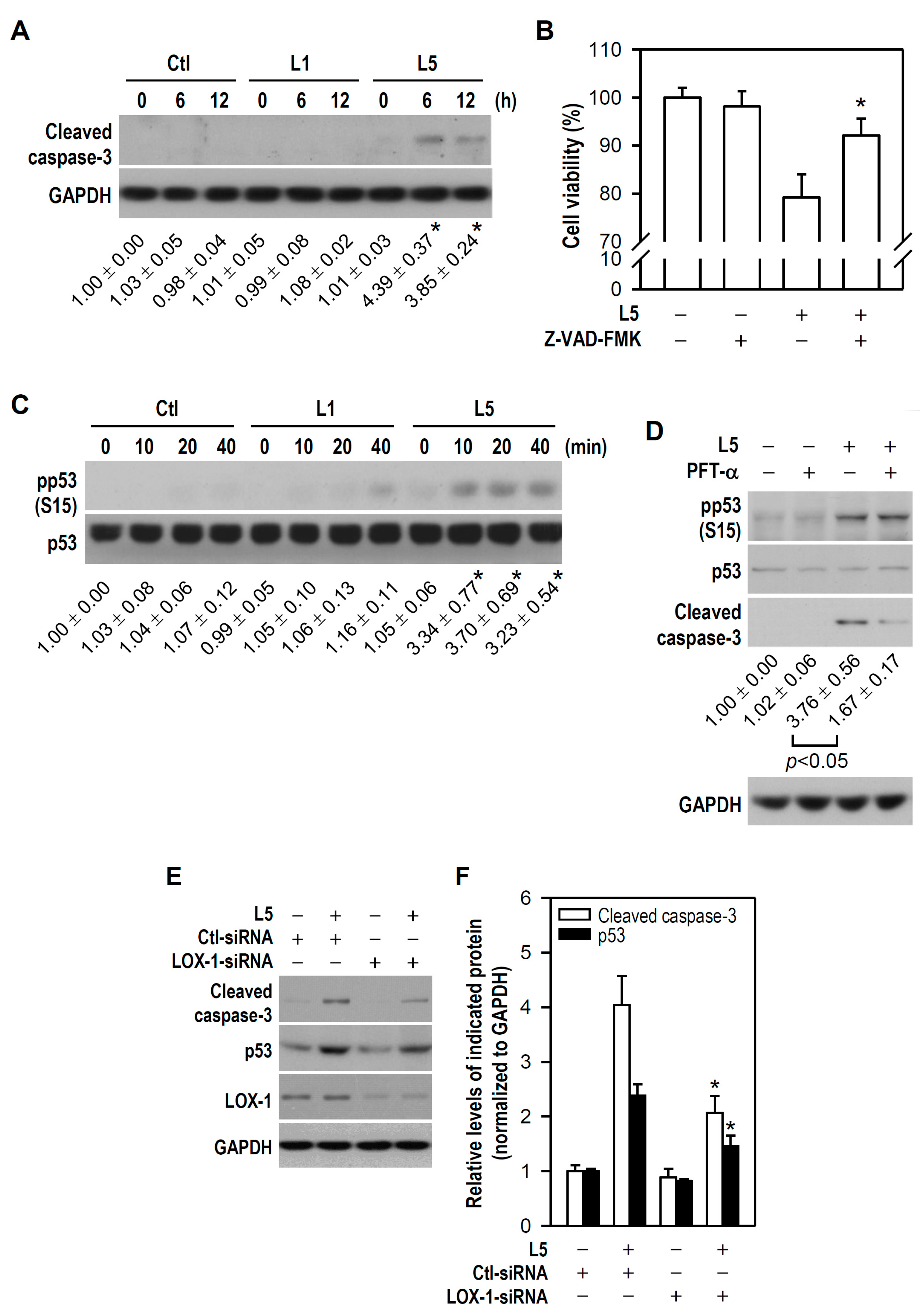
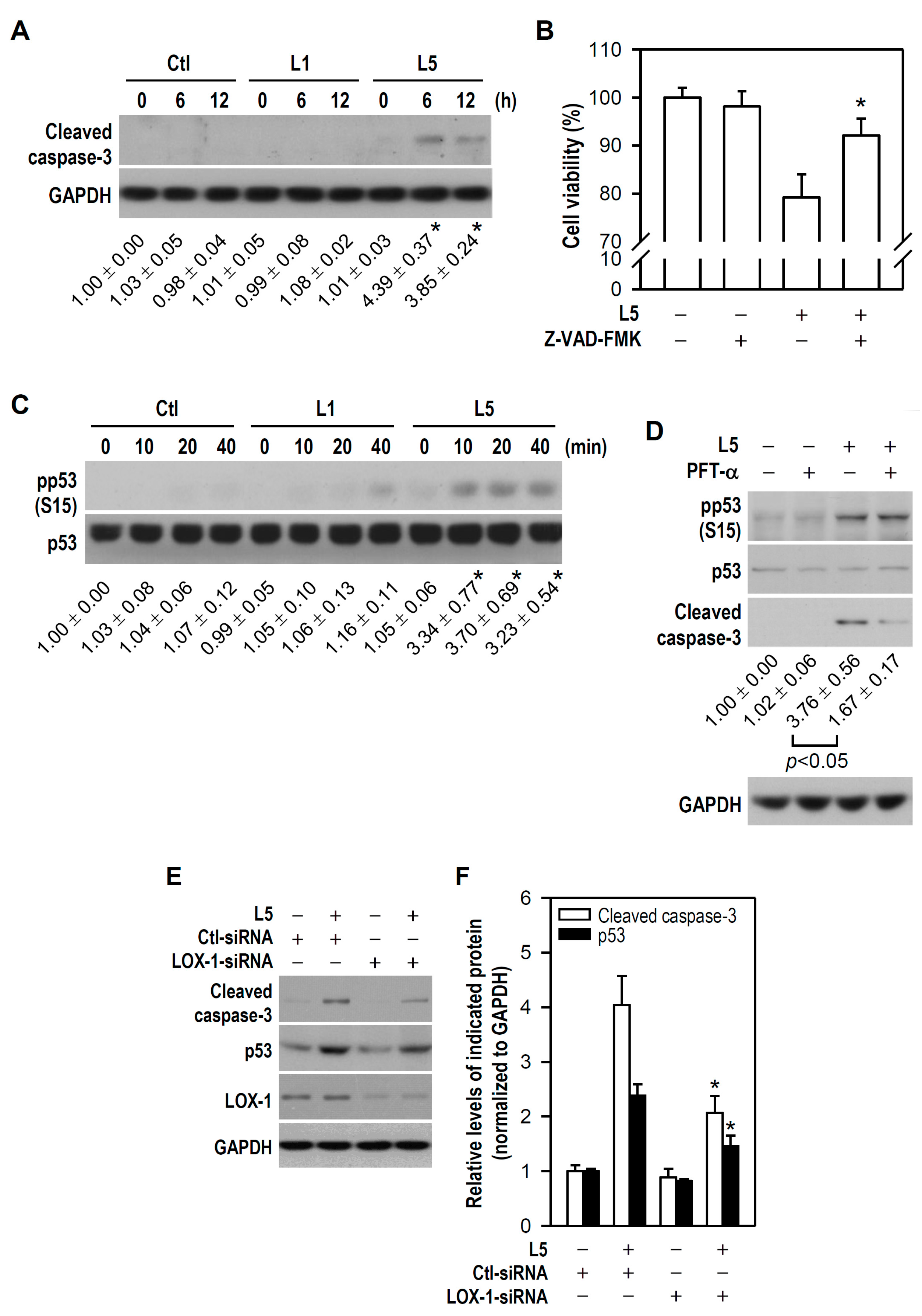
2.3. L5 Induces Apoptotic Death of PC12 Cells via the LOX-1/p53/Caspase-3 Pathway
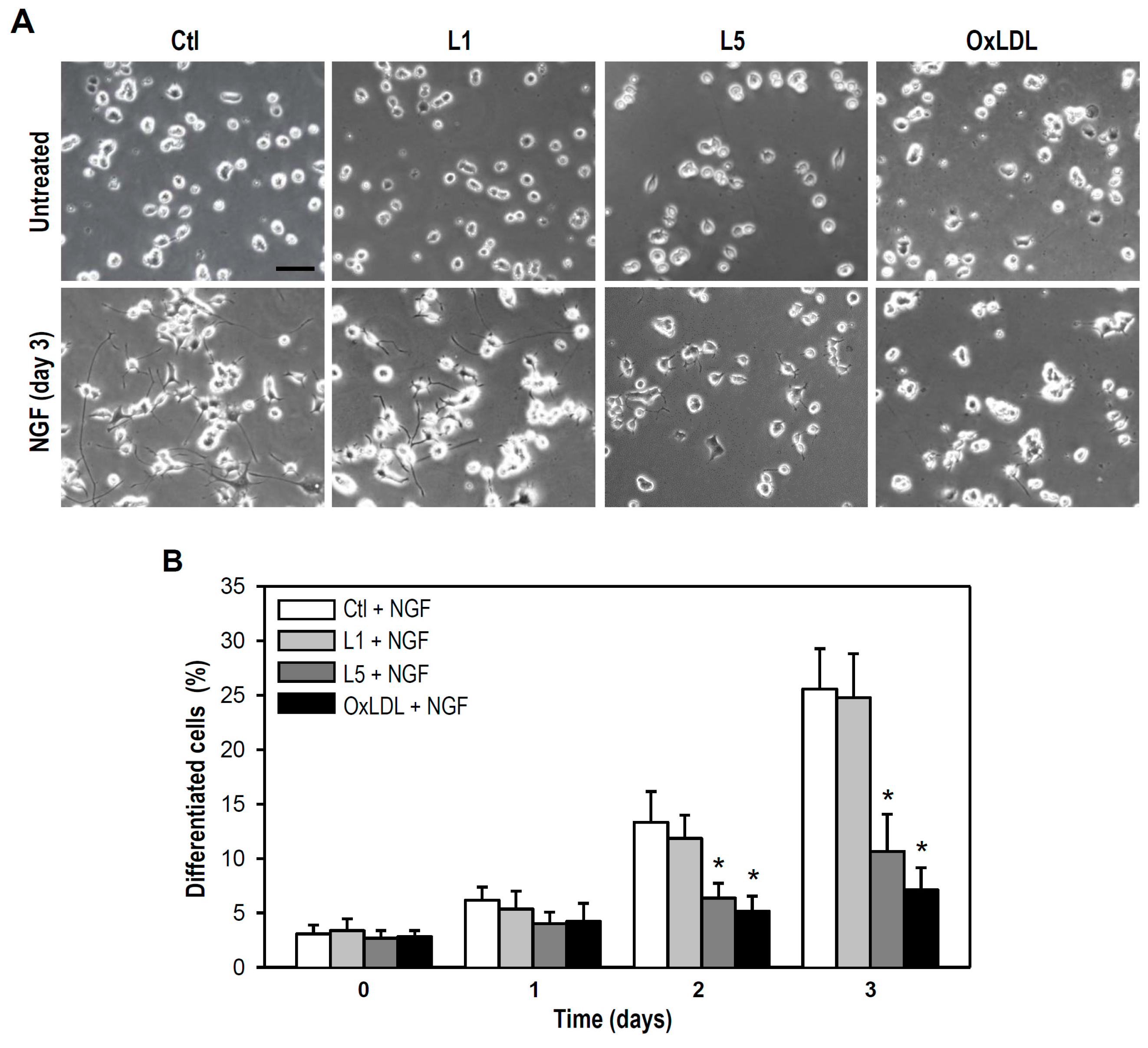
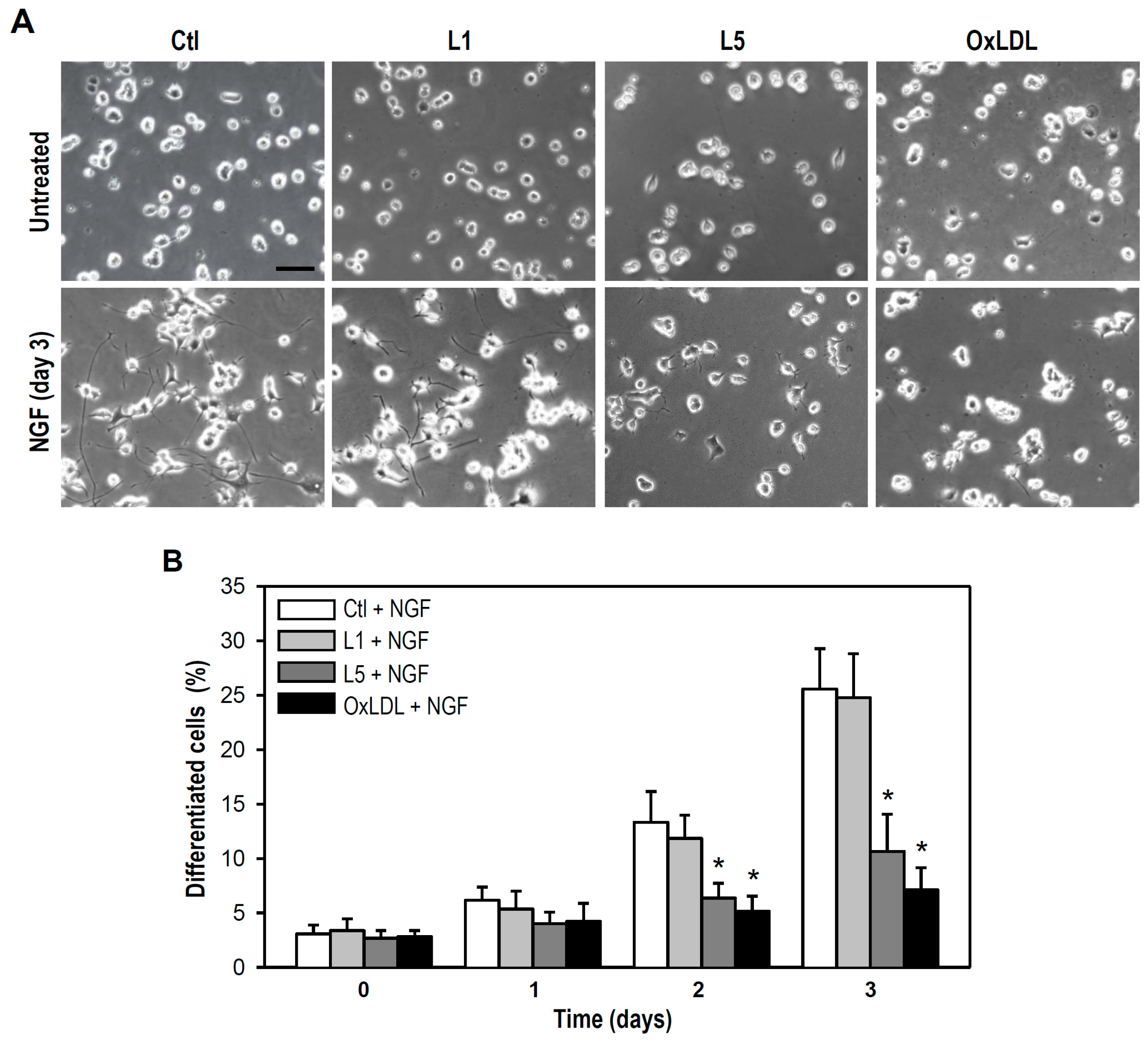
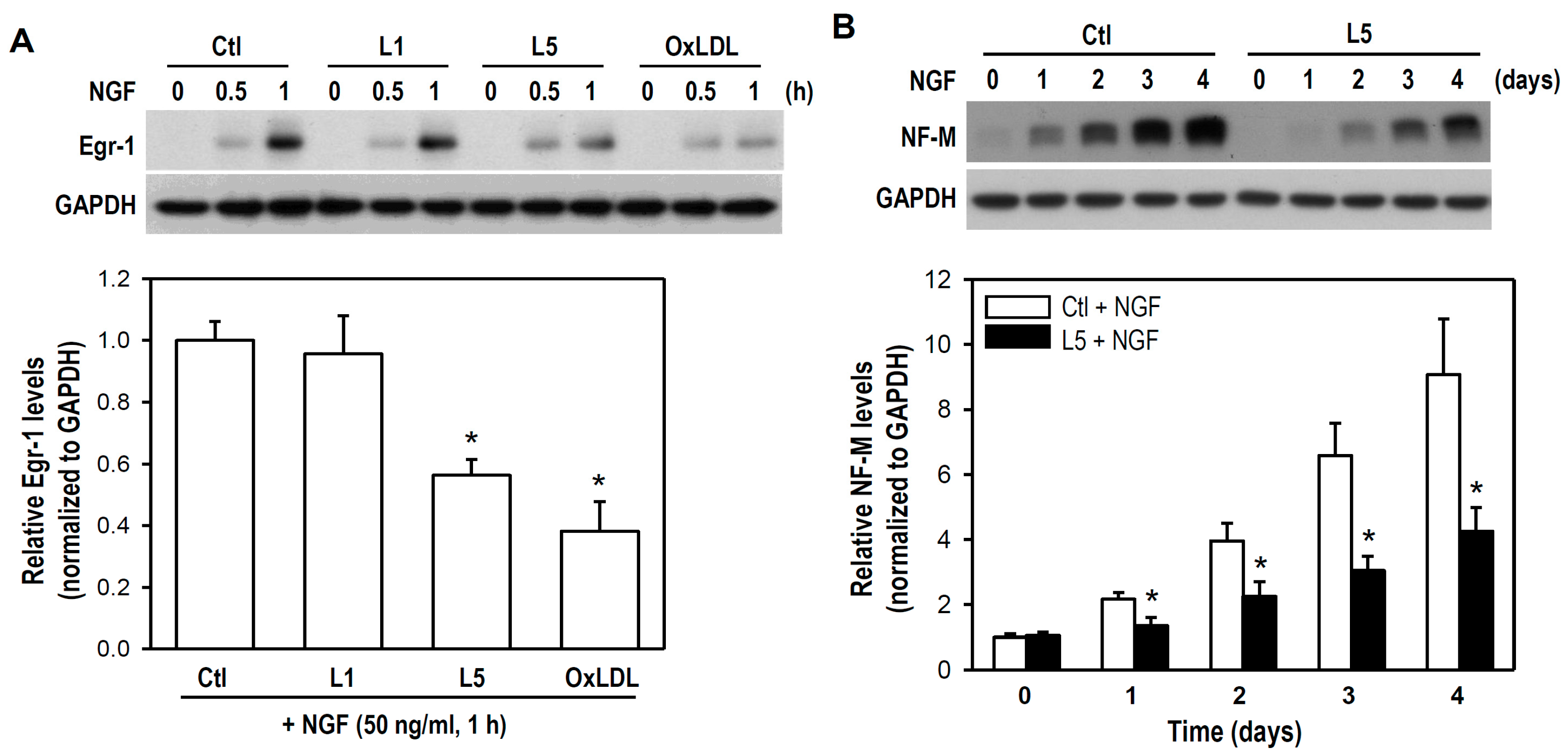
2.4. L5 Inhibits NGF-Induced Differentiation of PC12 Cells
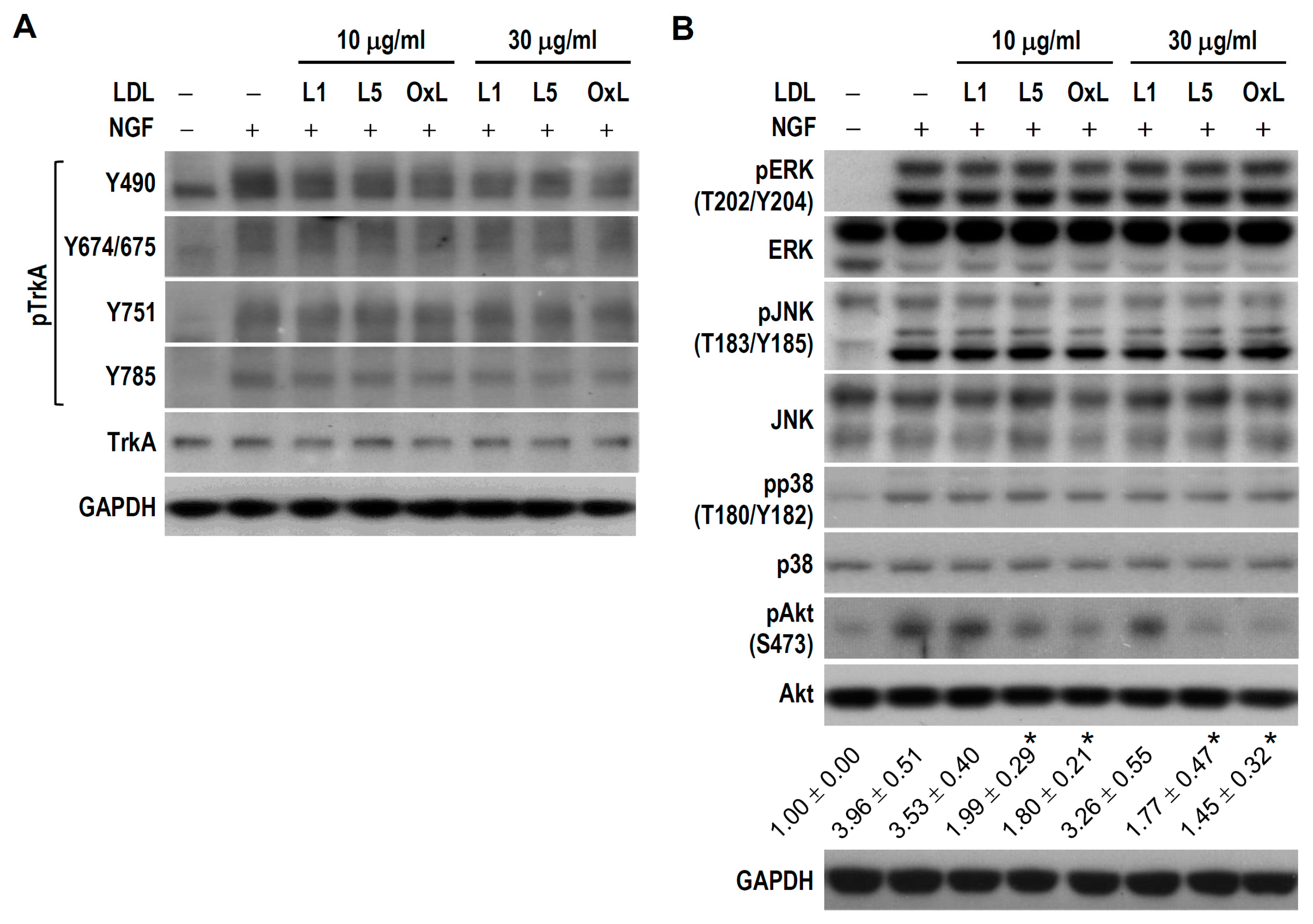
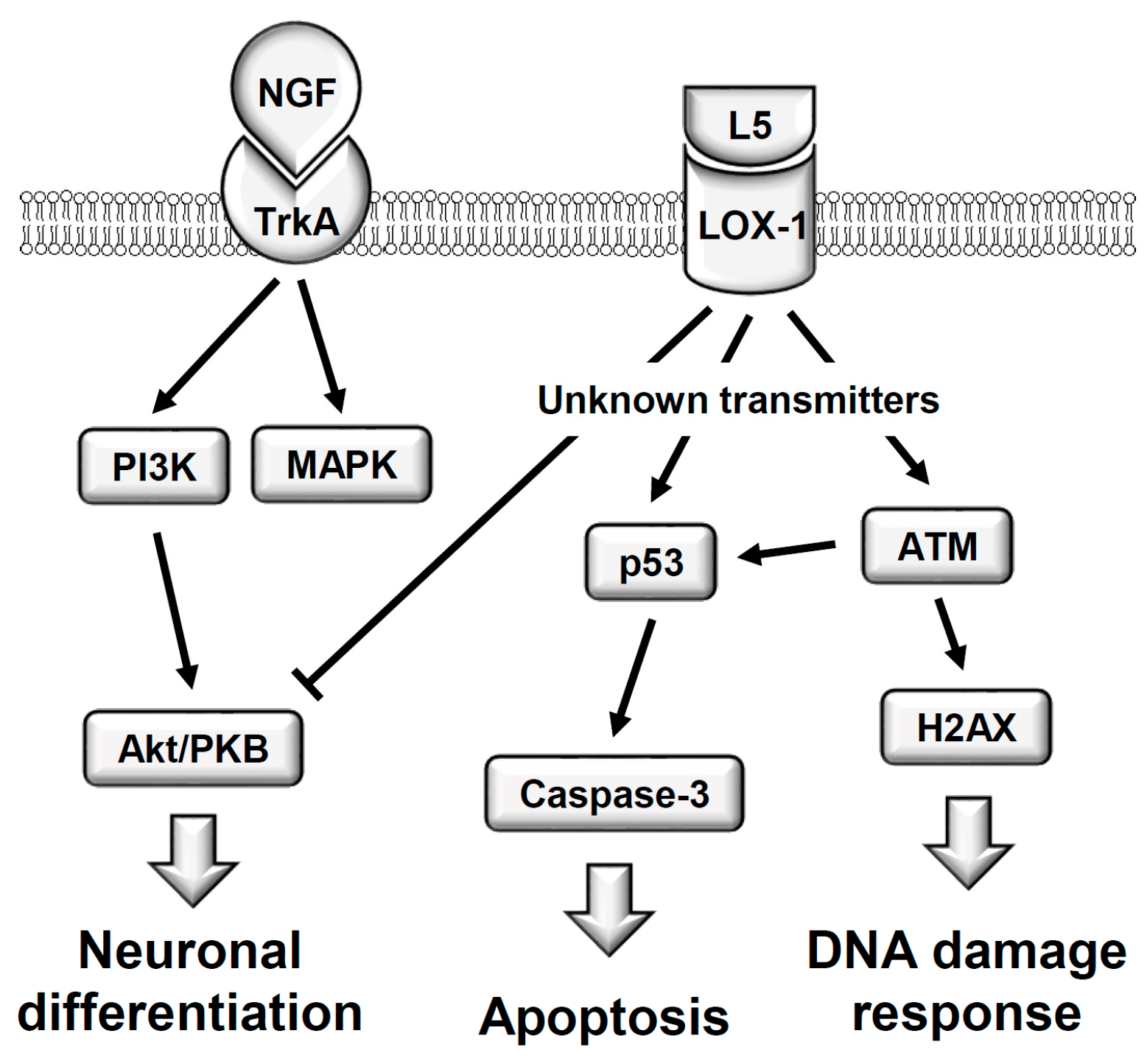
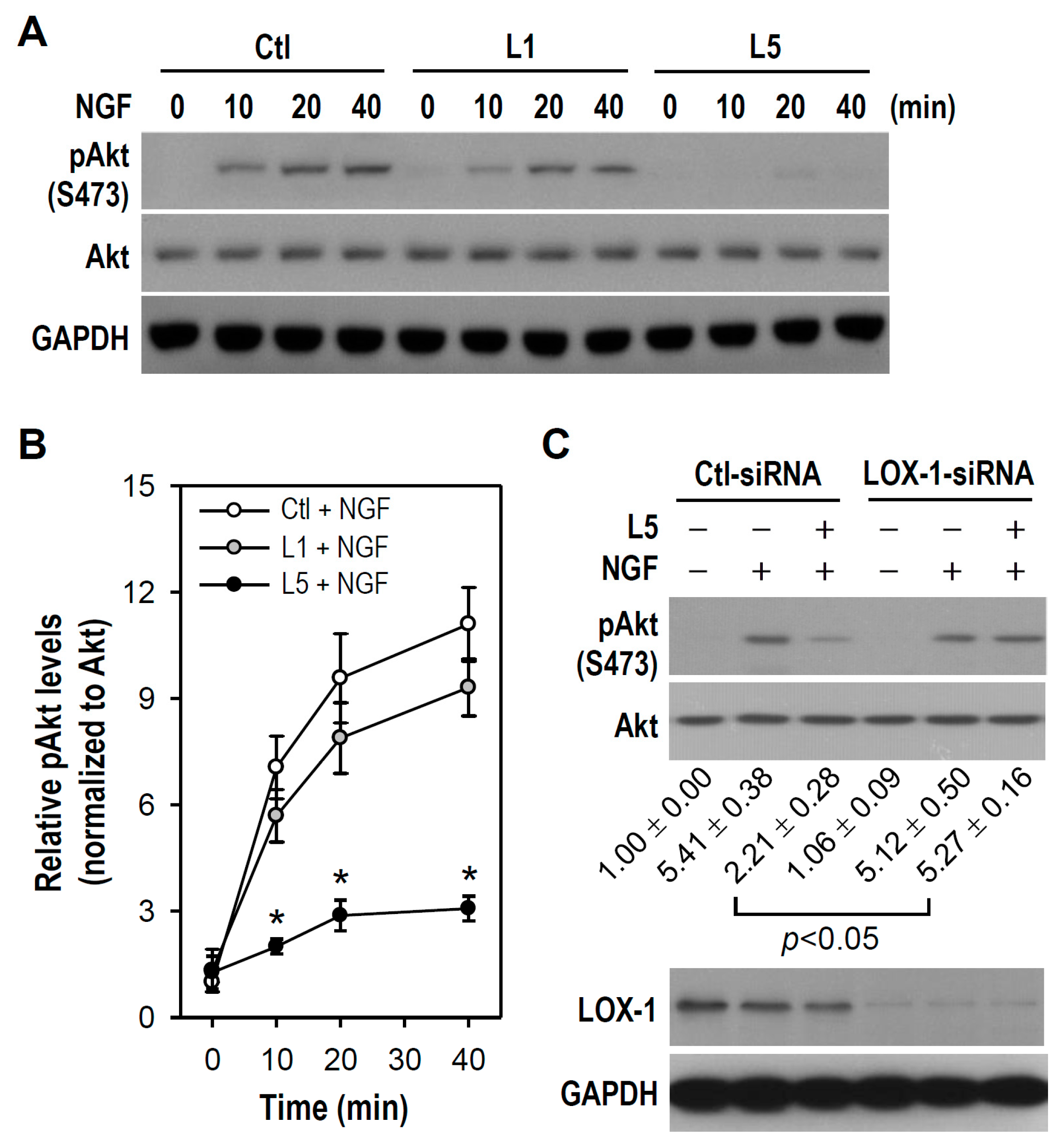
2.5. L5 Weakens NGF-Induced Akt but Not TrkA and MAPKs Activation
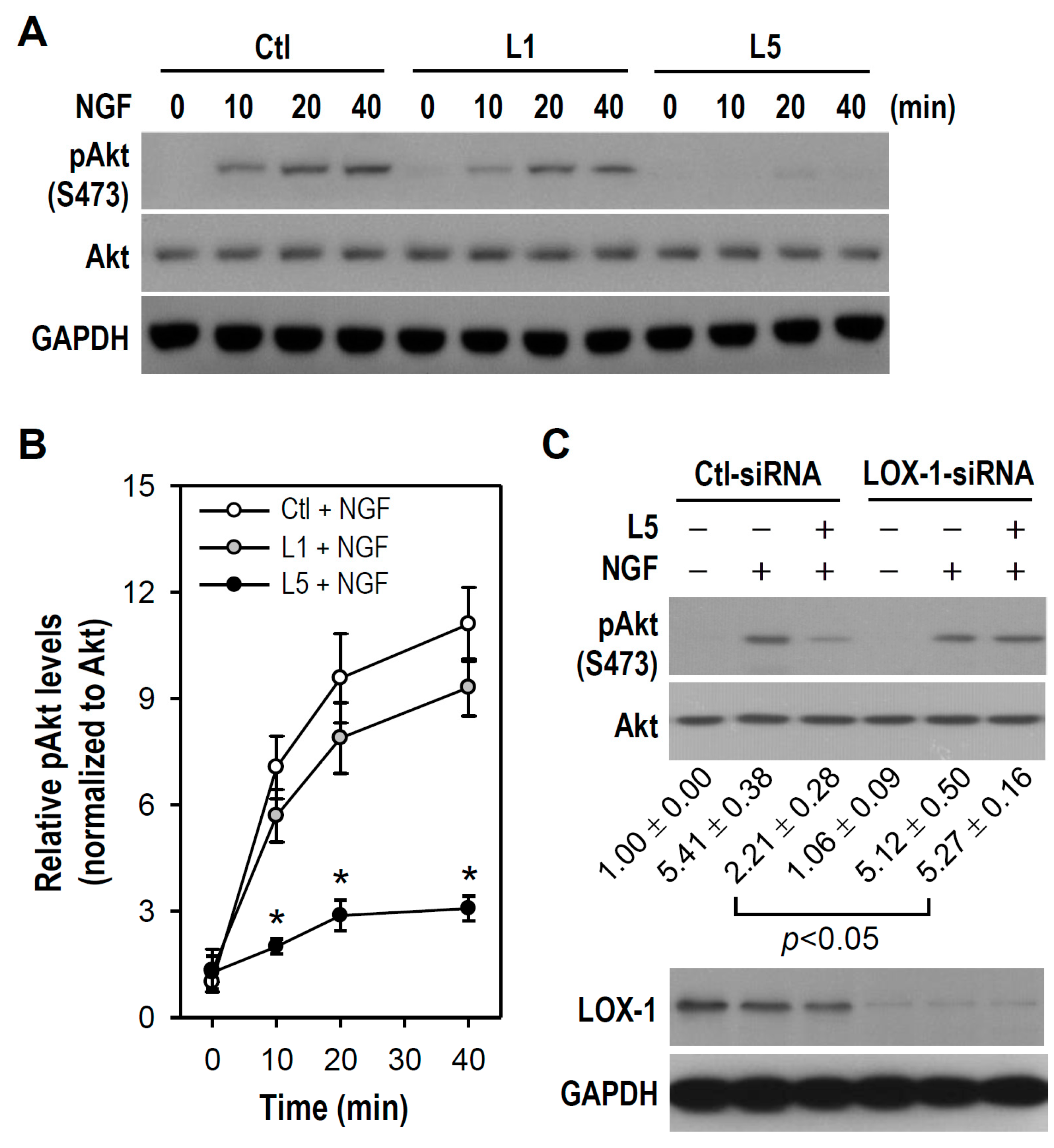
2.6. LOX-1 Is Required for L5 Suppression of NGF-Induced Akt Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Chemicals, Antibodies, Kits and Reagents
4.2. Obtainment of Human Plasma LDL
4.3. Cell Culture
4.4. MTT Reduction Assay
4.5. siRNA Transfection
4.6. Comet Assay
4.7. Immunofluorescence Detection of DNA DSBs
4.8. Western Blotting Analysis
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ApoB | Apolipoprotein B |
| ATM | Ataxia-telangiectasia mutated |
| BBB | Blood-brain barrier |
| CSF | Cerebrospinal fluid |
| CVD | Cardiovascular disease |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DSBs | Double strand breaks |
| EC | Endothelial cell |
| Egr-1 | Early growth response-1 |
| ERK | Extracellular signal-regulated kinase |
| JNK | c-JUN NH2-terminal protein kinase |
| LDL | Low-density lipoprotein |
| LDL(−) | Electronegative low-density lipoprotein |
| LDL-C | LDL-cholesterol |
| LDLR | LDL receptor |
| LOX-1 | Lectin-like oxidized low-density lipoprotein receptor-1 |
| MAPKs | Mitogen-activated protein kinases |
| MTT | 3-(4,5-Dimethylthianol-2-yl)-2,5 diphenyltetrazolium bromide |
| NGF | Nerve growth factor |
| NF-M | Neurofilament-medium |
| OxLDL | Oxidized LDL |
| PFT-α | Pifithrin-α |
References
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. Arteriosclerosis, apolipoprotein E, and Alzheimer’s disease. Lancet 1997, 349, 1174. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Mayeux, R. Cardiovascular risk factors and Alzheimer’s disease. Curr. Atheroscler. Rep. 2004, 6, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Emmerling, M.R.; Bisgaier, C.L.; Essenburg, A.D.; Lampert, H.C.; Drumm, D.; Roher, A.E. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1–42 levels. Biochem. Biophys. Res. Commun. 1998, 252, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Laakso, M.P.; Tuomilehto, J.; Nissinen, A.; Soininen, H. Hypertension and hypercholesterolaemia as risk factors for Alzheimer’s disease: Potential for pharmacological intervention. CNS Drugs 2002, 16, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Hsu, C.Y.; Liou, L.M.; Hsieh, H.Y.; Hsieh, Y.H.; Liu, C.K. Effect of cholesterol and CYP46 polymorphism on cognitive event-related potentials. Psychophysiology 2011, 48, 1572–1577. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.H.; MacQueen, G.M. Cognitive dysfunction associated with metabolic syndrome. Obes. Rev. 2007, 8, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Helzner, E.P.; Luchsinger, J.A.; Scarmeas, N.; Cosentino, S.; Brickman, A.M.; Glymour, M.M.; Stern, Y. Contribution of vascular risk factors to the progression in Alzheimer disease. Arch. Neurol. 2009, 66, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Stanyer, L.; Betteridge, D.J.; Cooper, M.B. Native and oxidized low-density lipoproteins modulate the vasoactive actions of soluble beta-amyloid peptides in rat aorta. Clin. Sci. 2007, 113, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.; Irundika, D.; Lip, G.; Spickett, C.; Polidori, C. Oxidised LDL lipids, statins and a blood-brain barrier. Free Radic. Biol. Med. 2014, 75, S15–S16. [Google Scholar] [CrossRef] [PubMed]
- Kuller, L.H. Statins and dementia. Curr. Atheroscler. Rep. 2007, 9, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Quesada, J.L.; Benitez, S.; Ordonez-Llanos, J. Electronegative low-density lipoprotein. Curr. Opin. Lipidol. 2004, 15, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Quesada, J.L.; Estruch, M.; Benitez, S.; Ordonez-Llanos, J. Electronegative LDL: A useful biomarker of cardiovascular risk? Clin. Lipidol. 2012, 7, 345–359. [Google Scholar] [CrossRef]
- Lu, J.; Yang, J.H.; Burns, A.R.; Chen, H.H.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.Y.; Sawamura, T.; Chen, C.H. Mediation of electronegative low-density lipoprotein signaling by LOX-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Hosken, B.D.; Huang, M.; Gaubatz, J.W.; Myers, C.L.; Macfarlane, R.D.; Pownall, H.J.; Yang, C.Y. Electronegative LDLs from familial hypercholesterolemic patients are physicochemically heterogeneous but uniformly proapoptotic. J. Lipid Res. 2007, 48, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Jiang, T.; Yang, J.H.; Jiang, W.; Lu, J.; Marathe, G.K.; Pownall, H.J.; Ballantyne, C.M.; McIntyre, T.M.; Henry, P.D.; et al. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation 2003, 107, 2102–2108. [Google Scholar] [CrossRef] [PubMed]
- Ke, L.Y.; Engler, D.A.; Lu, J.; Matsunami, R.K.; Chan, H.C.; Wang, G.J.; Yang, C.Y.; Chang, J.G.; Chen, C.H. Chemical composition-oriented receptor selectivity of L5, a naturally occurring atherogenic low-density lipoprotein. Pure Appl. Chem. 2011, 83, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Chau, L. Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2001, 280, C709–C718. [Google Scholar] [PubMed]
- Maziere, C.; Meignotte, A.; Dantin, F.; Conte, M.A.; Maziere, J.C. Oxidized LDL induces an oxidative stress and activates the tumor suppressor p53 in MRC5 human fibroblasts. Biochem. Biophys. Res. Commun. 2000, 276, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B. DNA damage-triggered apoptosis: Critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem. Pharmacol. 2003, 66, 1547–1554. [Google Scholar] [CrossRef]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Beckmann, A.M.; Wilce, P.A. Egr transcription factors in the nervous system. Neurochem. Int. 1997, 31, 477–510. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Miller, C.C. Neurofilaments and neurological disease. Bioessays 2003, 25, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Chen, F.Y.; Hsu, J.F.; Fu, R.H.; Chang, C.M.; Chang, C.T.; Liu, C.H.; Wu, J.R.; Lee, A.S.; Chan, H.C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Byzova, T.V. Prothrombotic lipoprotein patterns in stroke. Blood 2016, 127, 1221–1222. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.D.; Day, S.P.; Nicoll, J.A.; Packard, C.J.; Caslake, M.J. Remodelling of cerebrospinal fluid lipoproteins after subarachnoid hemorrhage. Atherosclerosis 2003, 170, 141–146. [Google Scholar] [CrossRef]
- Salen, G.; Berginer, V.; Shore, V.; Horak, I.; Horak, E.; Tint, G.S.; Shefer, S. Increased concentrations of cholestanol and apolipoprotein B in the cerebrospinal fluid of patients with cerebrotendinous xanthomatosis. Effect of chenodeoxycholic acid. N. Engl. J. Med. 1987, 316, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Yang, Y.; Chen, J.; Cheng, K.; Li, Q.; Wei, Y.; Zhu, D.; Shao, W.; Zheng, P.; Xie, P. Elevated host lipid metabolism revealed by iTRAQ-based quantitative proteomic analysis of cerebrospinal fluid of tuberculous meningitis patients. Biochem. Biophys. Res. Commun. 2015, 466, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Takechi, R.; Galloway, S.; Pallebage-Gamarallage, M.; Wellington, C.; Johnsen, R.; Mamo, J.C. Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice. Histochem. Cell Biol. 2009, 131, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Takechi, R.; Galloway, S.; Pallebage-Gamarallage, M.M.; Wellington, C.L.; Johnsen, R.D.; Dhaliwal, S.S.; Mamo, J.C. Differential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-beta. Br. J. Nutr. 2010, 103, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska-Sadowska, E.; Cudnoch-Jedrzejewska, A.; Ufnal, M.; Zera, T. Brain and cardiovascular diseases: Common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J. Physiol. Pharmacol. 2010, 61, 509–521. [Google Scholar] [PubMed]
- Kerkar, S.; Williams, M.; Blocksom, J.M.; Wilson, R.F.; Tyburski, J.G.; Steffes, C.P. TNF-alpha and IL-1beta increase pericyte/endothelial cell co-culture permeability. J. Surg. Res. 2006, 132, 40–45. [Google Scholar] [CrossRef] [PubMed]
- de Vries, H.E.; Blom-Roosemalen, M.C.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Hedera, P. beta-Amyloid vasoactivity in Alzheimer’s disease. Lancet 1996, 347, 1492–1493. [Google Scholar] [PubMed]
- Chu, C.S.; Wang, Y.C.; Lu, L.S.; Walton, B.; Yilmaz, H.R.; Huang, R.Y.; Sawamura, T.; Dixon, R.A.; Lai, W.T.; Chen, C.H.; et al. Electronegative low-density lipoprotein increases C-reactive protein expression in vascular endothelial cells through the LOX-1 receptor. PLoS ONE 2013, 8, e70533. [Google Scholar] [CrossRef] [PubMed]
- Estruch, M.; Bancells, C.; Beloki, L.; Sanchez-Quesada, J.L.; Ordonez-Llanos, J.; Benitez, S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis 2013, 229, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, P.; Bon, G.B.; Cazzolato, G. Presence of a modified low density lipoprotein in humans. Arteriosclerosis 1988, 8, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Benitez, S.; Villegas, V.; Bancells, C.; Jorba, O.; Gonzalez-Sastre, F.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Impaired binding affinity of electronegative low-density lipoprotein (LDL) to the LDL receptor is related to nonesterified fatty acids and lysophosphatidylcholine content. Biochemistry 2004, 43, 15863–15872. [Google Scholar] [CrossRef] [PubMed]
- Bancells, C.; Villegas, S.; Blanco, F.J.; Benitez, S.; Gallego, I.; Beloki, L.; Perez-Cuellar, M.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Aggregated electronegative low density lipoprotein in human plasma shows a high tendency toward phospholipolysis and particle fusion. J. Biol. Chem. 2010, 285, 32425–32435. [Google Scholar] [CrossRef] [PubMed]
- Bancells, C.; Benitez, S.; Ordonez-Llanos, J.; Oorni, K.; Kovanen, P.T.; Milne, R.W.; Sanchez-Quesada, J.L. Immunochemical analysis of the electronegative LDL subfraction shows that abnormal N-terminal apolipoprotein B conformation is involved in increased binding to proteoglycans. J. Biol. Chem. 2011, 286, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Fornage, M.; Yang, C.Y.; Bui-Thanh, N.A.; Wise, V.; Chen, H.H.; Rangaraj, G.; Ballantyne, C.M. L5, the most electronegative subfraction of plasma LDL, induces endothelial vascular cell adhesion molecule 1 and CXC chemokines, which mediate mononuclear leukocyte adhesion. Atherosclerosis 2007, 192, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Chen, F.Y.; Lee, A.S.; Ting, K.H.; Chang, C.M.; Hsu, J.F.; Lee, W.S.; Sheu, J.R.; Chen, C.H.; Shen, M.Y. Sesamol reduces the atherogenicity of electronegative L5 LDL in vivo and in vitro. J. Nat. Prod. 2015, 78, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Symonds, H.; Krall, L.; Remington, L.; Saenz-Robles, M.; Lowe, S.; Jacks, T.; van Dyke, T. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 1994, 78, 703–711. [Google Scholar] [CrossRef]
- Li, Y.; Duan, Z.; Gao, D.; Huang, S.; Yuan, H.; Niu, X. The new role of LOX-1 in hypertension induced neuronal apoptosis. Biochem. Biophys. Res. Commun. 2012, 425, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Zhu, X.Z. Reactive oxygen species-induced apoptosis in PC12 cells and protective effect of bilobalide. J. Pharmacol. Exp. Ther. 2000, 293, 982–988. [Google Scholar] [PubMed]
- Giuliano, P.; De Cristofaro, T.; Affaitati, A.; Pizzulo, G.M.; Feliciello, A.; Criscuolo, C.; De Michele, G.; Filla, A.; Avvedimento, E.V.; Varrone, S. DNA damage induced by polyglutamine-expanded proteins. Human Mol. Genet. 2003, 12, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Seger, R.; Suresh Babu, C.V.; Hwang, S.Y.; Yoo, Y.S. A positive role of the PI3-K/Akt signaling pathway in PC12 cell differentiation. Mol. Cells 2004, 18, 353–359. [Google Scholar] [PubMed]
- Read, D.E.; Gorman, A.M. Involvement of Akt in neurite outgrowth. Cell Mol. Life Sci. 2009, 66, 2975–2984. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Wang, G.J.; Chan, H.C.; Chen, F.Y.; Chang, C.M.; Yang, C.Y.; Lee, Y.T.; Chang, K.C.; Chen, C.H. Electronegative low-density lipoprotein induces cardiomyocyte apoptosis indirectly through endothelial cell-released chemokines. Apoptosis 2012, 17, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Lu, J.; Walterscheid, J.P.; Chen, H.H.; Engler, D.A.; Sawamura, T.; Chang, P.Y.; Safi, H.J.; Yang, C.Y.; Chen, C.H. Electronegative LDL circulating in smokers impairs endothelial progenitor cell differentiation by inhibiting Akt phosphorylation via LOX-1. J. Lipid Res. 2008, 49, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.E.; Lai, C.L.; Lee, C.T.; Wang, J.Y. Highly electronegative low-density lipoprotein L5 evokes microglial activation and creates a neuroinflammatory stress via Toll-like receptor 4 signaling. J. Neurochem. 2017, 142, 231–245. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.-Y.; Lai, C.-L.; Lee, C.-T.; Lin, C.-Y. Electronegative Low-Density Lipoprotein L5 Impairs Viability and NGF-Induced Neuronal Differentiation of PC12 Cells via LOX-1. Int. J. Mol. Sci. 2017, 18, 1744. https://doi.org/10.3390/ijms18081744
Wang J-Y, Lai C-L, Lee C-T, Lin C-Y. Electronegative Low-Density Lipoprotein L5 Impairs Viability and NGF-Induced Neuronal Differentiation of PC12 Cells via LOX-1. International Journal of Molecular Sciences. 2017; 18(8):1744. https://doi.org/10.3390/ijms18081744
Chicago/Turabian StyleWang, Jiz-Yuh, Chiou-Lian Lai, Ching-Tien Lee, and Chen-Yen Lin. 2017. "Electronegative Low-Density Lipoprotein L5 Impairs Viability and NGF-Induced Neuronal Differentiation of PC12 Cells via LOX-1" International Journal of Molecular Sciences 18, no. 8: 1744. https://doi.org/10.3390/ijms18081744