Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity

1
Department of Basic Medical Sciences, Laboratory of Histology & Embryology, School of Medicine, National and Kapodistrian University of Athens, 75 MikrasAsias Avenue, Goudi, Athens 11527, Greece
2
Department of Pharmacology, School of Medicine, National and Kapodistrian University of Athens, 75 MikrasAsias Avenue, Goudi, Athens 11527, Greece
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(8), 1821; https://doi.org/10.3390/ijms18081821
Submission received: 10 July 2017
/
Revised: 11 August 2017
/
Accepted: 14 August 2017
/
Published: 22 August 2017
(This article belongs to the Special Issue Mechanisms Leading to Genomic Instability)
Abstract
:Mitochondria have emerged as key players regarding cellular homeostasis not only due to their contribution regarding energy production through oxidative phosphorylation, but also due to their involvement in signaling, ion regulation, and programmed cell death. Indeed, current knowledge supports the notion that mitochondrial dysfunction is a hallmark in the pathogenesis of various diseases. Mitochondrial biogenesis and function require the coordinated action of two genomes: nuclear and mitochondrial. Unfortunately, both intrinsic and environmental genotoxic insults constantly threaten the integrity of nuclear as well as mitochondrial DNA. Despite the extensive research that has been made regarding nuclear genome instability, the importance of mitochondrial genome integrity has only recently begun to be elucidated. The specific architecture and repair mechanisms of mitochondrial DNA, as well as the dynamic behavior that mitochondria exert regarding fusion, fission, and autophagy participate in mitochondrial genome stability, and therefore, cell homeostasis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Owing to the symbiotic relationship that mitochondria and eukaryotic cells established about 1.5 billion years ago, mammalian mitochondrial biogenesis and function require the coordinated action of two genomes: nuclear and mitochondrial [1]. Both nuclear genes (between 1000 and 2000 genes) and thousands of copies of the maternally inherited mitochondrial DNA (mtDNA) genes participate in the assembly of these multifaceted organelles [2]. Nuclear genes code for the vast majority of mitochondrial proteins which are located within the organelle, as well as regulating factors regarding cellular bioenergetics. Human mtDNA genes code for 13 critical polypeptides that are involved in oxidative phosphorylation, plus the 22 transfer ribonucleic acids (tRNAs) and two ribosomal RNAs required for their expression. None of the polypeptides required for replication, transcription, and repair of mtDNA, or for processing and translation of the mitochondrial RNA (mtRNA) transcripts are encoded by the mtDNA [3]. Surprisingly, this bi-genomic organization has been retained through evolution, and it seems to serve bioenergetic benefits [4].
MtDNA is the cornerstone regarding energy production through oxidative phosphorylation since it encodes components of four out of five mitochondrial respiratory complexes [5]. Indeed, advances in molecular and cell biology, as well as in genetics and biochemistry gave us the opportunity to understand that mitochondria are much more than the powerhouses of the cells, exerting a role in crucial cellular functions, including gene expression, ion homeostasis, and cell death signaling [6,7]. Therefore, the integrity of mtDNA is crucial not only for optimal substrate utilization and energy production, but also for cell homeostasis in general.
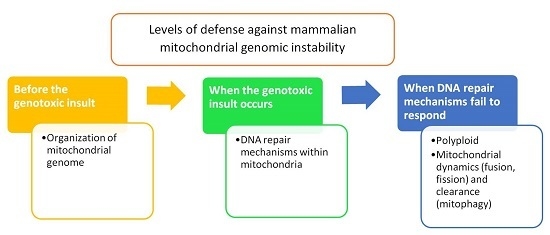
Due to their involvement in reactive oxygen species (ROS) production, mitochondria are both a major source and, at the same time, targets of genotoxic stress [8,9]. Instability regarding nuclear genome has been extensively studied; however, the importance of mitochondrial genome integrity has only recently begun to be elucidated [10,11]. Herein, we are summarizing basic principles of mechanisms regarding the integrity of the mitochondrial genome. Our approach has been stratified in three levels: (1) defensive mechanisms against mtDNA damage before the genotoxic insult, mostly referring to the specific organization of mitochondrial genome, and in particular mitochondrial nucleoid machinery and associated proteins, as well as other principal regulators of mitochondrial biogenesis;(2) when the DNA damage occurs, referring to DNA repair mechanisms that function efficiently within mitochondria; and (3) when mtDNA repair systems fail to deal with the DNA damage, referring to heteroplasmy and mitochondrial dynamic processes of fusion, fission, and mitochondrial autophagy.
2. Organization of Mitochondrial Genome
The mitochondrial genome was discovered about 60 years ago, after the pioneering studies of Nass and Schatz [12,13]. Human mtDNA comprises 16,569 bp of closed-circular double-stranded DNA [14]. The circularity of mtDNA does not refer to all eukaryotic organisms; accumulating evidence suggest that most of mtDNA in fungi and plants is linear in form, suggesting that the circular DNA found in animal mitochondria is an exception rather than the rule [15].
MtDNA contains a regulatory region, the displacement-loop (d-loop) region, which contains initiation sites for transcription and replication by the mitochondria-specific polymerase-γ (pol γ); pol γ is the only replicative DNA polymerase known to function within mitochondria [16,17]. Both replication and repair of mtDNA are dependent on this nuclear-encoded DNA polymerase [18]. Pol γ holoenzyme is actually a heterotrimer comprised of two accessory subunits (p55) and one pol γ catalytic subunit (p140); the accessory subunits are encoded by the POLG2 gene, whereas POLG encodes the catalytic subunit [18]. It is noteworthy that the isolated pol γ catalytic subunit exerts a remarkable fidelity of DNA replication, owing to its high nucleotide selectivity and exonucleolytic proofreading, but the inclusion of the p55 subunits reduces fidelity of replication by promoting extension of mismatched DNA termini [19]. Other major proteins involved in replication of mtDNA, together with the pol γ enzyme, are the mitochondrial DNA helicase, Twinkle, and the mitochondrial single stranded DNA binding protein (mtSSB) [20].
Over 200 mutations in POLG have been associated with numerous mitochondrial diseases [21,22,23]. The consequences of these mutations include alterations in the assembly of the replication apparatus, reductions of the enzymatic activities of the polymerase and helicase or the fidelity of DNA replication, as well as disruption of the polymerase holoenzyme [18]. Nevertheless, the frequency of these mutations is unusual. Surprisingly, it has been demonstrated, based on various mouse models with disrupted pol γ exonuclease activity, that mutations eliminating exonuclease function of pol γ are not embryonically lethal [24,25,26]. However, pol γ variants with decreased exonuclease activity targeted specifically to the heart proved to cause severe cardiomyopathy along with accelerated mtDNA mutagenesis [23]. Similarly, mice expressing defective mtDNA polymerase exhibited characteristics of premature aging, along with enlarged hearts [25,26].
Twinkle helicase is essential for mtDNA maintenance and a key regulator of mtDNA copy number; inhibition of Twinkle expression in cultured cells resulted in rapid mtDNA depletion, whereas overexpression of Twinkle in transgenic mice led to considerable increase in mtDNA copy number [27]. Dominant mutations of Twinkle are associated with multiple mtDNA deletions in post-mitotic tissues (namely, in the brain, the skeletal muscle, and the heart), whereas recessive mutations of Twinkle cause mtDNA depletion in the brain and liver; unfortunately, the molecular mechanistic basis of the tissue specificity regarding mtDNA maintenance disorders has not yet been elucidated [20,28,29,30,31,32,33]. Interestingly, Twinkle disorders mimic those of primary deoxynucleoside triphosphates (dNTP) pool defects [34]. According to Nikkanen et al, the mitochondrial replication machinery communicates with cytoplasmic dNTP pools and upregulation of glutathione synthesis through glucose-driven de novo serine biosynthesis contributes to the metabolic stress response; mtDNA depletion disorders show low and imbalanced dNTP pools, whereas multiple mtDNA deletion disorders show imbalanced and high dNTP pools [34].
Transcriptional control of mitochondrial biogenesis involves a number of regulators such as the mitochondrial transcription factor A (TFAM), nuclear respiratory factors 1 and 2 (NRF 1/2), and peroxisome proliferator-activated receptor gamma co-activator (PGC1a) [35,36,37]. TFAM is a nuclear-encoded transcription factor that binds to a common upstream enhancer of the promoter sites of the two mitochondrial DNA strands [37]. Of note, cardiac-specific TFAM knockout mice displayed a progressive cardiomyopathy phenotype of cardiac hypertrophy, with decreased levels of mtDNA and an accompanying severe decline of respiratory chain enzyme activities along with a decreased mitochondrial ATP production rate [38]. NRF 1/2 control many, but not all of the mitochondrial genes [39]. They are both recognized by TFAM, thus mediating the coordinated activation of mitochondrial and nuclear genome during mitochondrial biogenesis [37]. The primary regulator of the coordinated transcription of mitochondrial and nuclear genomes is the PGC1a [37,40]. PGC-1a is enriched in tissue with high oxidative activity-like the heart- and it is rapidly induced under conditions of enhanced energy demand [41,42].
In contrast to the nuclear genome which is compacted and organized by histone octamers, mtDNA is devoid of nucleosomes and organized as protein-DNA complexes known as nucleoids (mitochondrial nucleoids/mt-nucleoids) [3,43]. The precise structure of these nucleoids in which mtDNA is organized is not fully elucidated. Initially, it was believed that each nucleoid carries six–10 copies of mtDNA, but later on, it has been established with super-resolution imaging techniques that each nucleoid may contain only one or two (on average, 1.4) molecules of mtDNA, or according to other findings, three mtDNA genomes [44,45]. This special molecular organizing unit of mitochondrial DNA serves as a mechanism of protection, maintenance and accurate propagation of the mitochondrial genome; disruption in any component of this assembly results inmtDNA deletionsand mutations [43].Measurements of the absolute mtDNA copy number per cell and per nucleoid using stimulated emission depletion (STED) and photo activating light microscopy (PALM) imaging studies revealed that mtDNA, compared with bacterial nucleoid or nuclear chromatin, exerts a greater DNA packaging density, in regard with nucleoid volume, nuclear volume, and genome size [44,46]. The clinical significance of the tight package of mtDNA is associated with the protection against mutagenesis [3].
Several proteins are involved in packaging mtDNA in the form of mt-nucleoids. The best-characterized and most abundant protein binding duplex mtDNA, is theTFAM [47]. TFAM has a dual role in the maintenance of mtDNA, through both transcription and nucleoid formation [48]. It enhances mtDNA transcription in a promoter-specific fashion in the presence of mtRNA polymerase and transcription factor B [49,50]. In addition, TFAM exerts DNA-binding properties; approximately 900 molecules of TFAM are bound to one molecule of mtDNA [51].Depletion of TFAM gene resulted in embryonic lethality while tissue-specific disruption of TFAM gene in heart and muscle led to a mosaic cardiac-specific progressive respiratory chain deficiency, dilated cardiomyopathy, and atrioventricular heart conduction blocks and proved lethal after two to four weeks of age [52,53].
Other components of nucleoids include the Twinkle helicase, the mitochondrial polymerase γ, as well as mtSSB protein, all of which have been identified as participants in mtDNA maintenance [54]. The array of proteins found associated with nucleoids further includes helicases and RNA binding proteins, chaperones, and quality control proteases, mitochondrial ribosomal proteins as well as lipid metabolic enzymes [3]. According to Bogenhagen, many of these elements may represent contaminants in nucleoid preparations during proteomic analysis, however one cannot exclude the possible functional role of these proteins regarding mitochondrial biogenesis [3]. Among these potential nucleoid-associated proteins are included prohibitin, ATPase family AAA domain-containing protein 3(ATAD3), and p53.Except for the former, the other two (ATAD3 and p53) have not been detected in nucleoid purifications [55].
Prohibitin (PHB) proteins (PHB1 and PHB2) belong to chaperones’ system and reside in the intermembrane space [56]. Prohibitins’ primary function is to stabilize newly synthesized polypeptides in mitochondria, as they serve as foldase-unfoldase molecular chaperones; importantly, prohibitins exert various intra-mitochondrial as well as extra-mitochondrial effects, both of which are implicated in mitochondrial DNA stability [57,58]. PHB1 maintains the organization of the mt-nucleoids through a TFAM-independent pathway or through an undefined nucleoid factor (Factor X); alternatively, PHB1 maintains the copy number of mtDNA through a TFAM-dependent pathway [59]. In addition, PHB1 contributes to the organization of the inner membrane via an interaction with Opa1 (optic atrophy protein 1), a dynamin-related GTPase involved in mitochondrial fusion, cristae organization, and control of apoptosis [60]. Moreover, PHB2 regulates the stability of mitochondrial proteins such as HS1-associated protein X-1 (Hax-1) and Opa1, and is involved in anti-apoptosis and regulation of mitochondrial morphology [61]. Of note, mutations (mis-sense point mutations) in Opa1 gene lead to genomic instability through mtDNA deletions [62,63,64]. Furthermore, prohibitins exert various extra-mitochondrial cellular functions including roles in apoptosis, cell cycle regulation, transmembrane signal transduction, and life span regulation [58,65].
ATAD3 belongs to the family of the AAA-ATPases (ATPases associated with diverse cellular activities). Human ATAD3 refers to two mitochondrial paralogs, ATAD3A and ATAD3B [66]. It is considered to be an inner membrane mitochondrial protein; however, C-terminus islocated in the mitochondrial matrix and could be associated with the nucleoid, possibly through an indirect manner [55,67]. In vitro, ATAD3 has been shown to bind directly to the D-loop of mtDNA [68]. The implication of ATAD3 in the mt-nucleoid organization may also be related to the exact localization of this protein at contact points where the outer and the inner membranes of mitochondria are juxtaposed; these contact points are in relation to contact areas between the endoplasmic reticulum and mitochondria and serve as transit zones for many molecules such as neo-synthesized proteins, calcium and lipids [67,69,70]. It has been demonstrated that ATAD3 deficiency destabilizes mt-nucleoid organization through the dissociation of mtDNA fragments which are held together by this protein [71]. Furthermore, ATAD3 plays a critical role in cellular cholesterol homeostasis; ATAD3 deficiency causes aberrant mtDNA organization and is associated with elevated free cholesterol and increased expression of genes involved in cholesterol metabolism [72]. Importantly, it seems that perturbed cholesterol metabolism affects both replication and segregation of mtDNA [72]. In addition, de novo mutations in ATAD3A were recently found to cause a neurological syndrome with developmental delay, hypotonia, spasticity, optic atrophy, axonal neuropathy, and hypertrophic cardiomyopathy [66].
P53 is a soluble predominantly nuclear protein; in the nucleus, p53 functions as a transcription factor through a specific DNA-binding domain, but also represses gene expression through an interaction with histone deacetylases [73]. A small fraction of the total cellular amount of nuclear transcription factor p53 seems to be located at and within mitochondria. Recent evidence suggest that after genetic stress or death signals, a fraction of p53 translocates into the mitochondria, where it acts in a “biphasic” manner, either inducing or attenuating apoptosis progression.P53 is localized to the inside face of the inner membrane i.e., in the matrix, in which mtDNA is also located [74,75]. Within the human mitochondrial genome a p53-recognition motif has been recognized [76]. Mitochondrial p53 also exerts physical interactions with mitochondria DNA and DNA polymerase γ and enhances error correction activities [77]. Furthermore, it has been demonstrated that death signals can induce its translocation to the mitochondria, whereas p53 was shown to physically and functionally interact with both, the mtDNA and pol γ in response to mtDNA damage induced by exogenous and endogenous insults [78].
Given that contrary to what is happening regarding the nuclear genome, the mtDNA replication process is not restricted to the S phase of cell cycle, two models have been proposed to explain how mitochondrial nucleoids mediate mtDNA inheritance. The “faithful nucleoid model”, proposed by Jacobs, supports that individual nucleoids replicate only their own exact genetic content, thus mtDNA content within a nucleoid is generally static and nucleoids do not exchange mtDNAs between each other [79]. On the opposite site, D’ Aurelio proposed the “dynamic nucleoid model” according to which nucleoids are subject to remodeling and have a dynamic organization allowing nucleoids to exchange mtDNAs [80]. Current evidence supports that mitochondrial nucleoids tightly regulate their genetic content rather than freely exchanging mtDNAs, in agreement with the faithful nucleoid model [81].
3. Mitochondrial DNA Repair Machinery
Both environmental and intrinsic genotoxic insults constantly threaten the integrity of nuclear as well as mitochondrial DNA [82]. Exogenous factors include environmental insults such as ionizing radiation, ultraviolet (UV) radiation, and alkylating agents. Endogenous noxious agents include reactive oxygen species and products generated as a consequence (e.g., lipid peroxides), endogenous reactive chemicals (e.g., aldehydes and S-adenosylmethionine), and chemical DNA instability (e.g., deamination of bases, depurination or depyrimidination) [83]. Of note, the vast majority of mutations in human tissues are certainly of endogenous origin [74]. There are six main types of mtDNA damage: alkylation damage, hydrolytic damage, formation of adducts, mismatched bases, DNA strand breaks, and oxidative damage [84].
It is generally believed that mtDNA, compared with nuclear DNA, is more prone to damage; indeed, it has been estimated that mtDNA has 10–20 times higher mutation rate than the nuclear genome [85,86]. According to Yakes and Van Houten, several factors contribute to the increased susceptibility of mitochondrial genome to damage, such as the organization of mtDNA in nucleoids instead of chromatin, the limited DNA repair machinery of mtDNA, the presence of localized metal ions that may function as catalysts in the generation of ROS, or the propagation of mitochondrial damage through the generation of secondary ROS due to impairment of the electron transport chain and/or lipid peroxidation [87]. Importantly, recent studies, based on newer analytical techniques, support that specific oxidative lesions in specific tissues are less prevalent in mtDNA compared with nuclear DNA [88,89]. Experimental findings demonstrating that on some occasions, histones may enhance rather than reduce DNA damage, plus the possible protective role of mtDNA associated proteins, such as TFAM, which may serve as a shell for mtDNA, further challenge the belief about the vulnerability of mitochondrial genome [84].
Oxidative damage occurs basically from the generation of ROS within mitochondria; however, oxidative damage can also be caused by reactive nitrogen species, which are also present within the cell and their byproducts are able to oxidize or deaminate DNA bases and induce strand breaks [90,91]. Oxidative damage is the most common mechanism of damage as far as mitochondria are concerned. The first reaction against the potential genotoxic effect of ROS refers to antioxidant compounds and activities that are available within mitochondria, such as enzymatic antioxidants (i.e., catalase, glutathione peroxidase, thioredoxin, peroxiredoxin, and aldehyde dehydrogenase 2) and non-enzymatic antioxidants (i.e., glutathione, N-acetyl cysteine, ubiquinol, α-tocopherol, ascorbic acid, and lipoic acid) that have been shown to migrate into mitochondria due to oxidative stress [92,93]. During oxidative phosphorylation 1–2% of the oxygen that is consumed within the cell is released from mitochondria as ROS [94]. Under normal conditions, this amount of produced ROS results in low level of damage to mtDNA, which is rapidly repaired; however, during periods of increased and prolonged ROS exposure the generation of ROS leads to more extensive and persistent damage of mtDNA [87]. In any case, the proximity of mtDNA to the inner mitochondrial membrane where the electron transport chain constantly produces ROS as well as the mitochondrial membrane potential which facilitates the accumulation of lipophilic mutagenic cations, create a condition of susceptibility for mitochondrial DNA [95,96].
The maintenance of mitochondrial genome integrity is also supported by detection and repair mechanisms of DNA lesions [97]. Until recently, mitochondria were considered to exert a limited availability of DNA repair mechanisms, contrary to the several repair mechanisms that are definitely present in the nucleus; however, recent evidence suggest that some of the nuclear DNA repair pathways also function in mammalian mitochondria [9]. As mentioned above describing mt-nucleoid, a number of proteins have been reported to be localized in close proximity with the mtDNA assembly (such as RNA helicases, mitochondrial ribosomal proteins, chaperones, quality control proteases, and lipid metabolic enzymes) implicating their possible access to mtDNA following DNA damage [3,98]. Literally speaking, although mitochondria dispose of a competent and coherent base excision repair (BER) system, they lack effective mismatch repair (MMR) and completely lack nucleotide excision repair (NER) systems [20].
BER is the predominant DNA repair pathway in mitochondria; it is responsible for the removal of DNA bases altered by oxidation, alkylation, or deamination, and the repair of abasic sites and single-strand breaks resulting from spontaneous hydrolysis and oxidation, respectively [9,99]. ROS-induced oxidative lesions are preferentially eliminated by BER [84]. Of note, the repair of 8-hydroxyguanine (8-OHG or 8-oxoG), a major oxidative DNA base lesion which also serves as a biomarker of oxidative stress, is more efficient in the mitochondrial DNA than in the nuclear DNA [100,101]. It was initially believed that only short-patch BER (insertion of a single nucleotide) occurs in mitochondria, but recent evidence suggests that long-patch BER (insertion of a short sequence of two to six nucleotides) activity is present too [102,103,104,105,106]. Interestingly, the assembly of nuclear BER machinery needs a specific protein (XRCC1) which acts as a scaffold; this protein is absent from mitochondria [107]. However, given that BER within mitochondria takes place in the inner membrane where mtDNA is organized into nucleoids, it has been suggested that other nucleoid proteins, such as TFAM, regulate BER pathway within mitochondria as a result of its DNA-binding activity and protein interactions [108]. Of particular interest is the very recent detection of polymerase β (pol β), known as a key nuclear BER protein, in mitochondrial protein extracts from mammalian tissue and cells, contrary to what it has been believed about the “monopoly” of pol γ within mitochondria; indeed, human kidney cells with polβ knock-out exerted higher mtDNA damage [109].
The only repair pathway not yet identified in the mitochondria is the NER pathway which copes with the repair of bulky DNA adducts [82]. It involves, among others, the repair of bulky DNA adducts and cisplatin induced intrastrand crosslinks [9]. NER pathway involves the removal and resynthesis of a short fragment on the damaged strand [97]. To date, the only hint regarding NER pathway in the mitochondria is the localization of the transcription-coupled NER proteins CSA and CSB (Cockayne syndrome proteins) to mitochondria upon oxidative stress [110]. Current belief is that adducts requiring NER for their removal will accumulate into mitochondria resulting to mtDNA mutations and, ultimately, mtDNA degradation [9]. MMR activity and strand break repair (SBR) and double-strand, are also included in the DNA repair repertoire of mitochondria [84,111,112,113]. DNA mismatch repair corrects mis-incorporation and slippage errors introduced by DNA polymerase during replication and base mismatches caused either spontaneously or due to deamination, oxidation, and alkylation [84].
The lack of an efficient MMR and NER systems makes mitochondria vulnerable to the accumulation of miscoding lesions in mtDNA, which in turn can promote mtDNA mutations and block DNA replication [18,114]. Of great importance, pol γ can bypass some lesions by nucleotide incorporation opposite a template lesion and further extension of the DNA primer past the lesion [115]. This process of translesion synthesis is critical for the genetic integrity of mtDNA. Regarding translesion synthesis, PrimPol (primase-polymerase), an archaic enzyme with dual primase and polymerase activities has been identified in human cells; indeed, some PrimPol is present in the nuclear DNA compartment but a larger fraction is located inside mitochondria [116]. Regarding its role within mitochondria, PrimPol exerts synergy with polγ and facilitates replication fork progression by acting as a translesion DNA polymerase or as a specific DNA primase reinitiating downstream of lesions that block synthesis during mitochondrial DNA replication [116,117]. PrimPol has the unique feature ofde novoDNA synthesis solely with dNTPs, and the ability to tolerate lesions such as 8-oxoG and AP (apurinic/apyrimidinic or abasic) sites in DNA.
Similarly, petite integration frequency 1 (Pif1) helicase is a major contributor regarding mtDNA replication. Pif1 localizes to both the nucleus and the mitochondrion; however, its mitochondrial localization in human cells is regulated by alternative splicing, which produces α and β isoforms. Isoform α localizes to the nucleus, whereas the majority of isoform β localizes to the mitochondrion, with some residual nuclear signal [118]. Pif1 is involved in mtDNA maintenance. It contributes to the reduction of DNA strand breaks in mtDNA as well as the repair of UV- and ethidium bromide-damaged mtDNA [119,120]. In addition, Pif1 helicase may assist Twinkleto overcome its inability to unwind G-quadruplex DNA, thus mediating the removal of the genotoxic G-quadruplexes from Twinkle [121]. Recently, it was demonstrated that Pif1 inactivation causes a phenotype of mitochondrial myopathy in mouse, due to deficiency to repair oxidative stress-induced mtDNA damage [122].
Furthermore, there is evidence that mammalian mitochondria may have the ability to directly repair simple alkylations, like methylated or ethylated derivatives [123,124,125]. The mitochondrial genome is known to retain a very low level of DNA methylation [126]. MtDNA can be methylated by machinery existing inside of the mitochondria, thus mediating the control of mitochondrial gene expression [127,128,129,130]. A recent study demonstrated that platelet mtDNA methylation is implicated in the etiology of cardiovascular diseases [126].
Moreover, it has been demonstrated that mammalian mitochondrial protein extracts possess both homologous recombination and non-homologous DNA end-joining activity [131,132]. Indeed, mitochondrial DNA end-binding (DEB) activity is present independently from nuclear DEB activity and is roughly equivalent in potency to that found in the nucleus [133]. The mitochondrial DEB factor shares functional and structural similarities with the nuclear Ku factor, a heterodimer composed of the Ku70 and Ku86 subunits, which is essential for efficient nuclear non-homologous DNA end joining [134]. A mitochondria-specific form of DNA ligase III is responsible for the mitochondrial end-joining activity and is encoded by the same gene that encodes nuclear DNA ligase III [135]. Interestingly, recent evidence suggest that mitochondria lack classical non-homologous DNA end-joining (NHEJ) but possess efficient microhomology-mediated end-joining (MMEJ) activity [136]. Mitochondrial MMEJ depends on ligase III, too. In addition, mitochondria-specific expression of poly (ADP-ribose) polymerase 1 (PARP-1), CtBP interacting protein 1 (CtIP), FEN1 (flap endonuclease 1), meiotic recombination 11 (MRE11), and RAD50 are also involved in mitochondrial MMEJ [136]. A minimum length of 5-nt microhomology was necessary for efficient MMEJ in mitochondria. Respectively, the efficiency of the MMEJ is improved when the length of the microhomology region is increased and when the microhomology region is more proximal to the DNA terminus of the double-strand break [136].
Deletion of mtDNA represents the most serious damage to the mitochondrial genome and may be produced during the repair of double-strand breaks [137,138,139,140]. Unfortunately, recombination errors could have important implications for mtDNA integrity in aged cells. It has been suggested that, in aged cells, mitochondrial dysfunction increases the production of ROS, thus activating a recombination mechanism of mtDNA, which in turn, if hyper-activated, increases the overall recombination errors, mtDNA deletions, ROS production and ultimately, damages mitochondria, through a vicious cycle model [141].
4. Heteroplasmy, Mitochondrial Dynamics, and Mitophagy
The third level of defense regarding mtDNA integrity, and therefore organelle homeostasis is the condition of heteroplasmy that characterizes mitochondria; mtDNA exists in numerous identical copies per cell in mammals (homoplasmy), but can also exist in multiple forms within the same tissue or cell (heteroplasmy) [142].Typically, approximately 1000 copies of mtDNA are present per cell, although the actual number may vary depending on the cell type [43]. Given that each cell has many mitochondria, it is concluded that a significant amount of mitochondrial genome is located within each cell, a fact of great importance regarding the accumulation of mutations in mtDNA [143]. Literally speaking, heteroplasmy does not offer protection against mtDNA instability per se, but, most importantly, for the organelle’s genomic stability as a whole. According to Wallace, there is a threshold regarding the number of accumulated mutations in mtDNA that define the transition to a disease state [143]. This threshold depicts the potential of the remaining wild-type mtDNAs within the organelle to compensate for the genetic defect of mutant molecules [3]. After reaching this threshold, mitochondria become dysfunctional, and at this point mechanisms regarding mitochondrial dynamics and clearance are employed.
Mitochondrial dynamics is a term used to describe two main macroscopic behaviors of these organelles: fusion (the joining of two organelles into one) and fission (the division of a single organelle into two), whereas mitochondrial clearance refers to mitophagy, a selective degradation of mitochondria via the autophagic pathway. The maintenance of the ideal balance between these two dynamic mitochondrial behaviors is of paramount importance [144]; derangement of the normal balance in either direction may result in the accumulation of damaged mitochondrial genome and dysfunctional organelles [145]. Multiple post-translational modifications are involved in the regulation of mitochondrial fusion and fission [146,147].
Mitochondrial fusion is primarily orchestrated by large dynamin-related GTPases termed mitofusins (MFN/ MFN1 and MFN2), plus Opa1. Fusion enables mitochondria to distribute mtDNA to all parts of the cell and to replenish damaged mitochondrial DNA [148,149,150]. Dilution of dysfunctional enzymes and mutated mtDNA is considered to be a repetitive process for dysfunctional mitochondria [122]. Fission is mediated by the mitochondrial fission proteins: dynamin-related protein 1 (Drp1); mitochondrial fission 1 protein (FIS1);mitochondrial fission factor(Mff);and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49/51) and is required for cell division and the removal of damaged mitochondria by mitophagy [149,150]. Mitochondrial fission is a major process of cell homeostasis: it distributes mitochondria along cytoskeletal tracks, contributes to the equal distribution of mitochondria into daughter cells during mitosis, and facilitates the removal of dysfunctional mitochondria by partitioning the damaged components to a daughter organelle that can be recovered by fusing with healthy mitochondria or targeted for degradation by mitophagy [146]. Interruption of either fusion, fission, or mitophagy due to loss-of-function mutations in the respective mitochondrial proteins disrupts mitochondrial quality control system, causes instability in mtDNA, and provokes compensatory mitochondrial proliferation, thus inducing neurodegenerative, cardiovascular, and other age-related diseases [149].
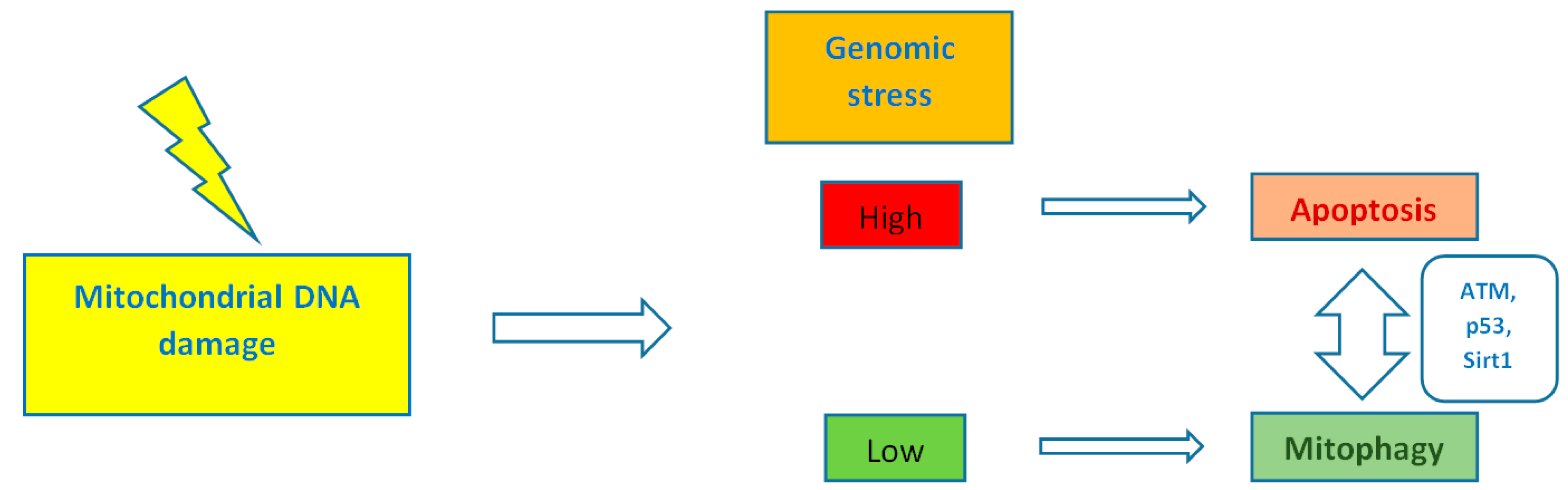
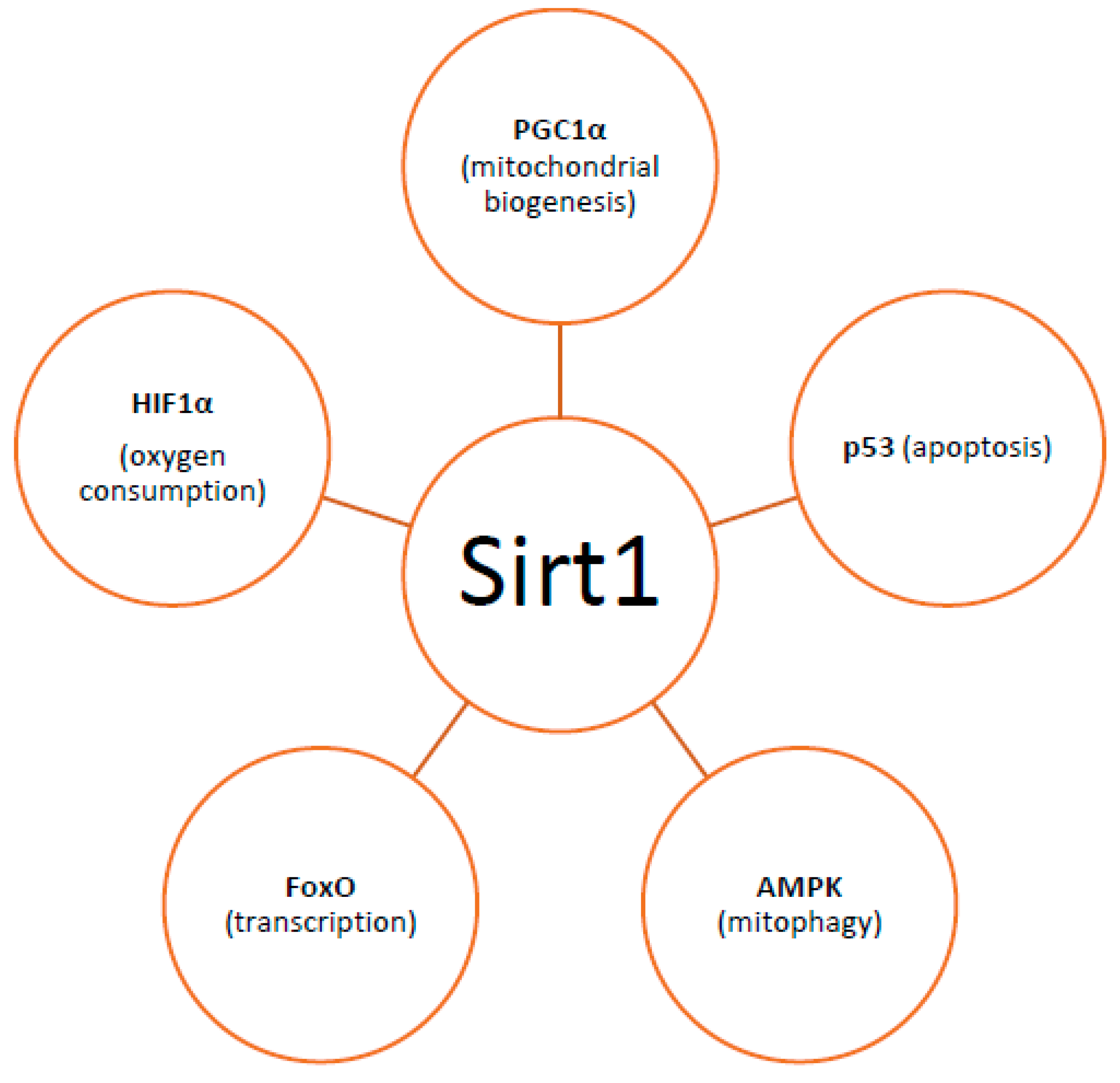
By controlling both apoptosis, which is mediated by Bcl2 (B-cell lymphoma 2) family mitochondrial death proteins, and necrosis, which is mediated by opening of the mitochondrial permeability transition pore, mitochondria regulate cell fate [151]. Importantly, whether DNA damage insults stimulate mitophagy or cell death, is a matter depending on the grade of DNA damage; low levels of DNA damage stress may induce mitophagy instead of apoptosis, whereas high levels of genomic stress stimulate apoptosis and inhibit mitophagy [152]. The crosstalk between mitophagy and apoptosis is mediated through three major nucleus-to-mitochondria signaling pathways linked to genomic stability and mitochondrial homeostasis [152]: (1) the protein kinase ATM (ATM serine/threonine kinase);(2) the tumor suppressor p53; and (3) sirtuin1 (Sirt1) (Figure 1). ATM is a major activator of the DNA damage response which at low levels of double-strand breaks it promotes cell survival, while in case of excessive double-strand break damage, it triggers apoptosis [153]. Similarly, under lethal genotoxic insult, p53 transcriptionally upregulates pro-apoptotic proteins, whereas under low levels of stress it promotes pro-autophagy proteins, thus inducing mitophagy [154]. Of note, ATM functions partly through p53 stabilization. ATM mediates the phosphorylation of MDM2 (murine double minute 2) protein (an E3 ubiquitin ligase); the phosphorylated MDM2 lacks the ability to poly-ubiquitinate p53, thus leading to its stabilization [155]. Except for the direct phosphorylation of MDM2, ATM mediates both the direct phosphorylation of N-terminal serine-15 residue of p53 and indirectly regulates serine-20 phosphorylation by controlling activation of the checkpoint kinase chk2, thus impeding the interaction of p53 with MDM2 and, therefore increasing the stability of the p53 protein and activating its transcriptional activity [156]. Sirt1 belongs to the family of mammalian class III histone deacetylases [157]. It is primarily a nuclear protein; however, Sirt1’s deacetylation of PGC1α has been extensively implicated in metabolic control and mitochondrial biogenesis. Sirt1 interacts with PGC1a, which is the master regulator of mitochondrial biogenesis; caloric restriction and energetic status that elevates Sirt1 levels or NAD+ (nicotinamide adenine dinucleotide) levels could promote the accumulation of PGC-1α in the nucleus, thus resulting in the transcription of genes that are necessary for mitochondrial function and biogenesis [157]. Furthermore, Sirt1 inhibits apoptosis after excessive toxic stress through inactivation of p53, and interacts with AMPK (5′ adenosine monophosphate-activated protein kinase), a key regulator of metabolism and energy homeostasis, in order to stimulate mitophagy [158,159]. Additionally, Sirt1 deacetylates members of the Forkhead box O (FoxO) transcription factor family, particularly in response to stress [160]. Moreover, Sirt1 deacetylates and represses the activity of hypoxia-inducible factor 1-α (HIF1α) which is known to down-regulate mitochondrial function and oxygen consumption by inducing pyruvate dehydrogenase kinase 1 (PDK1) [161,162] (Figure 2).
5. Conclusions
About 30 years ago, two reports suggested for the first time that deletions and point mutations of mitochondrial genome could be pathogenic [163,164]. Since then, a growing body of evidence has demonstrated the direct or indirect association between mitochondrial dysfunction and the pathogenesis of many neurodegenerative, cardiovascular, and metabolic diseases, as well as cancer and ageing. The hallmark which provokes the pathogenic cataract seems to be the failure of mitochondrial quality control systems, particularly the disruption of mitochondrial genome integrity. Mutations in mtDNA are generated due to spontaneous errors of DNA replication or through unrepaired damage to mtDNA that introduces mis-coding lesions. In addition, it has been demonstrated that except for mtDNA mutations, a reduction in the mtDNA copy number can be pathogenic too [165,166]. Indeed, mitochondrial dysfunction can arise not only from mutations and damage directly related to mitochondrial genes, but also from mutations in nuclear genes that encode proteins which translocate to the mitochondria and are necessary for the biogenesis and function of these multifaceted organelles, as well as from the impairment of nucleus-to-mitochondria signaling which refers to the ability of both the nuclear and mitochondrial genome to communicate and function synergistically [152,167]. More intriguingly, mtDNA repair itself may promote disease through the formation and accumulation of large deletions or recombination of mtDNA [168].From an opposite point of view, mitochondria can also promote genotoxic stress, thus affecting nuclear genome integrity, through the production of metabolic byproducts such as ROS or lipid peroxidation products [83].
Mitochondrial genome instability is associated not only with genetic defects in mtDNA or in nuclear genes encoding proteins that function in the mitochondria, but also with genes involved in supplying the mitochondrial nucleotide precursors needed for DNA replication [21,169]. The dNTPs represent the building blocks for DNA replication, and, as far as mitochondria are concerned, dNTPs pool arise either through active transport of cytosolic dNTPs or through the purine and pyrimidine salvage pathways by action of two mitochondrial deoxyribonucleoside kinases, thymidine kinase 2 (TK2), and deoxyguanosine kinase (DGUOK) [169]. Mutations in the genes that encode these enzymes involved in the salvage pathway cause several forms of mtDNA depletion syndromes [168]. In post-mitotic tissues, mtDNA replication is the major user of cytoplasmic dNTPs, thus forcing the burden of mitochondrial dNTP pool synthesis on the two mitochondrial deoxyribonucleoside kinases [170]. In contrast to the mitochondrial salvage pathway, nucleotide precursors required for DNA replication can be directly obtained by reduction of ribonucleoside diphosphates to deoxyribonucleoside diphosphates by ribonucleotide reductase, which is made up of two subunits, a large one (R1 subunit) and a smaller one (R2 subunit) [169]. Cells have two forms of the R2 subunit, a cell cycle regulated form that is maximally expressed in S-phase, and a p53-inducible form known as p53R2 [169]. The p53R2 form is required for a basal level of DNA repair and de novonucleotide synthesis in non-proliferating cells; it catalyzes the biosynthesis of deoxyribonucleotides by storing organic free radicals required for catalysis in the R2 subunit [170]. Mutations in the RRM2B (ribonucleoside-diphosphate reductase subunit M2 B) gene, encoding the p53-controlled ribonucleotide reductase subunit, has been implicated in the pathogenesis of mtDNA depletion syndrome [171].
The scope of this review is to highlight basic aspects of mitochondrial genome structure and DNA repair mechanisms present within mitochondria and how these features contribute to the maintenance of mtDNA integrity (Figure 3). Of great importance, a number of nuclear genome maintenance factors have been described as having also a mitochondrial location and are involved in mtDNA protection (Figure 4). In case of failure to restore or bypass troublemaking DNA damage, mitochondria exert an extra line of defense in order to retain cell homeostasis, which is basically relevant to mitochondrial dynamic behaviors. The identification of physiological regulators of mtDNA protection and repair mechanisms may open the way for the development of novel therapeutic strategies aiming to restore mitochondrial dysfunction which is an important component of various disease states. In this regard, thyroid hormone (TH) is shown to enhance mitochondrial function and reduce cellular oxidative stress in patients with mtDNA defects [172]. TH has been shown to stimulate mitochondrial biogenesis and increase mitochondrial mass, mitochondrial protein synthesis, and mtDNA content in both physiological and pathological conditions via induction of TFAM, PGC1a, and specific mitochondrial thyroid hormone receptors [173]. As mentioned above, especially TFAM, may also serve as a shell for mtDNA to enhance its resistance to genotoxic insults. From another point of view, it is interesting to note that TH may inhibit the genotoxic stress that is produced by mitochondria during myocardial ischemia and leads to apoptosis and heart failure [174,175]. Furthermore, TH was recently shown to induce selective mitophagy of damaged mitochondria blocking DNA damage and protecting hepatocytes from carcinogenesis [176].
Acknowledgments
In the memory of N. Sitaras.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gonzalez-Freire, M.; de Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the role of mitochondria in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Garesse, R.; Vallejo, C.G. Animal mitochondrial biogenesis and function: A regulatory cross-talk between two genomes. Gene 2001, 263, 1–16. [Google Scholar] [CrossRef]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Why do we still have maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.C.; Gomes, K.M.; Ferreira, J.C. Impact of exercise training on redox signaling in cardiovascular diseases. Food Chem. Toxicol. 2013, 62, 107–119. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Georgakilas, A.G.; Bonner, W.M. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 2010, 704, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 2012, 1819, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Gayarre, J.; Duran-Trío, L.; Criado Garcia, O.; Aguado, C.; Juana-López, L.; Crespo, I.; Knecht, E.; Bovolenta, P.; Rodríguez de Córdoba, S. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell. Biol. 2016, 18, 777–789. [Google Scholar] [Green Version]
- Nass, M.M.; Nass, S. Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J. Cell. Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Schatz, G.; Haslbrunner, E.; Tuppy, H. Deoxyribonucleic acid associated with yeast mitochondria. Biochem. Biophys. Res. Commun. 1964, 15, 127–132. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A. Sanger Fsequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.J. The size and form of chromosomes are constant in the nucleus, but highly variable in bacteria, mitochondria and chloroplasts. Bioessays 2007, 29, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, K.; Kunkel, T.A. Functions of DNA polymerases. Adv. Protein Chem. 2004, 69, 137–165. [Google Scholar] [PubMed]
- Ropp, P.A.; Copeland, W.C. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics 1996, 36, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair (Amst.) 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Nguyen, D.; Kunkel, T.A.; Copeland, W.C. The fidelity of human DNA polymerase γ with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 2001, 276, 38555–38562. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008, 59, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Graziewicz, M.A.; Bienstock, R.J.; Copeland, W.C. Consequences of mutations in human DNA polymerase γ. Gene 2005, 354, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, J.D.; Copeland, W.C. Mitochondrial DNA replication and disease: Insights from DNA polymerase γ mutations. Cell. Mol. Life Sci. 2011, 68, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Mott, J.L.; Chang, S.W.; Denniger, G.; Feng, Z.; Zassenhaus, H.P. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics 2000, 69, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Majander, A.; Haltia, M.; Somer, H.; Lönnqvist, J.; Savontaus, M.L.; Peltonen, L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Investig. 1992, 90, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the d-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lönnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.H.; Goffart, S.; Marjavaara, S.; Paetau, A.; Cooper, H.; Mattila, K.; Lampinen, M.; Sajantila, A.; Lönnqvist, T.; Spelbrink, J.N.; et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 2008, 17, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Lӧnnqvist, T.; Paetau, A.; Nikali, K.; von Boguslawski, K.; Pihko, H. Infantile onset spinocerebellar ataxia with sensory neuropathy (IOSCA): Neuropathological features. J. Neurol. Sci. 1998, 161, 57–65. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsstrӧm, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamäki, J.; Roivainen, A.; Marjamäki, P.; et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell. Metabol. 2016, 23, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 2007, 115, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- François, R.; Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W. Mitochondrial DNA nucleoids determine mitochondrial genetics and dysfunction. Int. J. Biochem. Cell. Biol. 2009, 41, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobs, S.; Wurm, C.A. Super-resolution microscopy of mitochondria. Curr. Opin. Chem. Biol. 2014, 20, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.A.; Clayton, D.A. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 1991, 252, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor a in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Gaspari, M.; Rantanen, A.; Trifunovic, A.; Larsson, N.G.; Gustafsson, C.M. Mitochondrial transcription factors B1 and B2activate transcription of human mtDNA. Nat. Genet. 2002, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, V.; Seidel-Rogol, B.L.; Shadel, G.C. A human mitochondrial transcription factor is related to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol. Cell. Biol. 2002, 22, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Brüning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 1999, 21, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Haag, M.; Potting, C.; Rodenfels, J.; Dip, P.V.; Wieland, F.T.; Brügger, B.; Westermann, B.; Langer, T. The genetic interactome of prohibitins: Coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell. Biol. 2009, 184, 583–596. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.K.; Jupe, E.R.; Liu, X.T.; Dell’Orco, R.T. Prohibitin: Potential role in senescence, development, and tumor suppression. Exp. Gerontol. 1995, 30, 99–124. [Google Scholar] [CrossRef]
- Nijtmans, L.G.; Artal, S.M.; Grivell, L.A.; Coates, P.J. The mitochondrial PHB complex: Rolesin mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell. Mol. Life Sci. 2002, 59, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Sumitani, M.; Satoh, M.; Endo, H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp. Cell Res. 2008, 314, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Löwer, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 793, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, G.; Wang, S.; Chellappan, S. Differential regulation of Rb family proteins and prohibitin during camptothecin-induced apoptosis. Oncogene 2002, 21, 4539–4548. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.M.; Yang, Y.; Ylikalli, E.; Khairullin, R.; Woldegebriel, R.; Lin, K.L.; Euro, L.; Palin, E.; Wolf, A.; Trokovic, R.; et al. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 2017, 26, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Rousseau, D.J. ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J. Bioenerg. Biomembr. 2012, 44, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication of animal mitochondrial DNA. Cell 1982, 28, 693–705. [Google Scholar] [CrossRef]
- Gilquin, B.; Taillebourg, E.; Cherradi, N. The AAA+ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membrane. Mol. Cell. Biol. 2010, 30, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Chang, C.L.; Hsu, S.H.; Huang, C.Y.; Chiang, S.F.; Chiou, S.H.; Huang, C.H.; Hsiao, Y.T.; Lin, T.Y.; Chiang, I.P.; et al. ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci. 2010, 123, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Mao, C.C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Ahn, J.; Walker, K.K.; Hoffman, W.H.; Evans, R.M.; Levine, A.J.; George, D.L. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999, 13, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Achanta, G.; Sasaki, R.; Feng, L.; Carew, J.S.; Lu, W.; Pelicano, H.; Keating, M.J.; Huang, P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA pol γ. EMBO J. 2005, 24, 3482–3492. [Google Scholar] [CrossRef] [PubMed]
- Lilling, G.; Novitsky, E.; Sidi, Y.; Bakhanashvili, M. p53-associated 3′→5′ exonuclease activity in nuclear and cytoplasmic compartments of the cells. Oncogene 2003, 22, 233–245. [Google Scholar] [PubMed]
- Heyne, K.; Mannebach, S.; Wuertz, E.; Knaup, K.X.; Mahyar-Roemer, M.; Roemer, K. Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS Lett. 2004, 578, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M.; Grinberg, S.; Bondal, E.; Simon, A.J.; Moshitch-Moshkovitz, S.; Rahav, G. p53 in mitochondria enhances the accuracy of DNA synthesis. Cell Death Differ. 2008, 15, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, N.D.; Zaika, A.; Moll, U.M. Death signal-induced localization of p53 protein to mitochondria. J. Biol. Chem. 2000, 275, 16202–16212. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.T.; Lehtinen, S.K.; Spelbrink, J.N. No sex please, we’re mitochondria: A hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays 2000, 22, 564–572. [Google Scholar] [CrossRef]
- D’ Aurelio, M.; Gajewski, C.D.; Lin, M.T.; Mauck, W.M.; Shao, L.Z.; Lenaz, G.; Moraes, C.T.; Manfredi, G. Heterologous mitochondrial DNA recombination in human cells. Hum. Mol. Genet. 2004, 13, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Schon, E.A.; Hernandez, E.; Davidson, M.M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 2008, 181, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublié, S. Base excision repair in the mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity-critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Tatarenkov, A.; Avise, J.C. Rapid concerted evolution in animal mitochondrial DNA. Proc. Biol. Sci. 2007, 274, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear-DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Anson, R.M.; Hudson, E.; Bohr, V.A. Mitochondrial endogenous oxidative damage has been overestimated. FASEB J. 2000, 14, 355–360. [Google Scholar] [PubMed]
- Alexeyev, M.F. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009, 276, 5768–5787. [Google Scholar] [CrossRef] [PubMed]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 1999, 424, 37–49. [Google Scholar] [CrossRef]
- Burney, S.; Niles, J.C.; Dedon, P.C.; Tannenbaum, S.R. DNA damage in deoxynucleo-sides and oligonucleotides treated with peroxynitrite. Chem. Res. Toxicol. 1999, 12, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in cardiovascular therapy: Panacea or false hope? Front. Cardiovasc. Med. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hyperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- Singer, T.P.; Ramsay, R.R. Mechanism of the neurotoxicity of MPTP: An update. FEBS Lett. 1990, 274, 1–8. [Google Scholar] [PubMed]
- Maybury, B.D. Mitochondrial DNA damage is uncommon in cancer but can promote aggressive behavior. Anticancer Res. 2013, 33, 3543–3552. [Google Scholar] [PubMed]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2013, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.A.; Mayard, S.; Hashiguchi, K.; Souza-Pinto, N.C.; Bohr, V.A. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucl. Acids Res. 2005, 33, 3722–3732. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-Hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Sunesen, M.; Bohr, V.A.; Stevnsner, T. Repair of 8-oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair (Amst.) 2002, 1, 261–273. [Google Scholar] [CrossRef]
- Akbari, M.; Visnes, T.; Krokan, H.E.; Otterlei, M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair 2008, 7, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Boesch, P.; Ibrahim, N.; Dietrich, A.; Lightowlers, R.N. Membrane association of mitochondrial DNA facilitates base excision repair in mammalian mitochondria. Nucleic. Acids Res. 2010, 38, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Qian, L.; Sung, J.S.; de Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol. 2008, 28, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef] [PubMed]
- Stierum, R.H.; Dianov, G.L.; Bohr, V.A. Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic. Acids Res. 1999, 27, 3712–3719. [Google Scholar] [CrossRef] [PubMed]
- Hanssen-Bauer, A.; Solvang-Garten, K.; Sundheim, O.; Peña-Diaz, J.; Andersen, S.; Slupphaug, G.; Krokan, H.E.; Wilson, D.M.; Akbari, M.; Otterlei, M. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ. Mol. Mutagen. 2011, 52, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.C.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst.) 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A. DNA polymerase beta participates in mitochondrial DNA repair. Mol. Cell. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dollé, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Wilson, D.M.; Bohr, V.A. Repair of persistent strand breaks in the mitochondrial genome. Mech. Ageing Dev. 2012, 133, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic. Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst.) 2009, 8, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Kasiviswanathan, R.; Longley, M.J. Analysis of translesion DNA synthesis by the mitochondrial DNA polymerase γ. Methods Mol. Biol. 2016, 1351, 19–26. [Google Scholar] [PubMed]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA polymerase γ in mitochondrial DNA replication and repair. Chem. Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.J.; Doherty, A.J. Mitochondrial DNA replication: A PrimPol perspective. Biochem. Soc. Trans. 2017, 45, 513–529. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Shimamoto, A.; Furuichi, Y. Mitochondrial and nuclear localization of human PIF1 helicase. Biol. Pharm. Bull. 2007, 30, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dunaway, S.; Ivessa, A.S. The role of Pif1p, a DNA helicase in Saccharomyces cerevisiae, in maintaining mitochondrial DNA. Mitochondrion 2007, 7, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Kolodynski, J. PIF mutation blocks recombination between mitochondrial ρ+ and ρ− genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1983, 80, 5345–5349. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.L.; Brosh, R.M., Jr. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Berg-Alonso, L.; Augé, G.; Fragaki, K.; Kolesar, J.E.; Lespinasse, F.; Lacas-Gervais, S.; Burel-Vandenbos, F.; Villa, E.; Belmonte, F.; et al. Inactivation of Pif1 helicase causes a mitochondrial myopathy in mice. Mitochondrion 2016, 30, 126–137. [Google Scholar] [CrossRef] [PubMed]
- LeDoux, S.P.; Wilson, G.L.; Beecham, E.J.; Stevnsner, T.; Wassermann, K.; Bohr, V.A. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 1992, 13, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Saffhill, R.; O’Connor, P.J. Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis 1988, 9, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Huh, N.; Rajewsky, M.F.; Kuroki, T. Enzymatic removal of O6-ethylguanine from mitochondrial DNA in rat tissues exposed to N-ethyl-N-nitrosoureain vivo. J. Biol. Chem. 1988, 263, 6854–6856. [Google Scholar] [PubMed]
- Baccarelli, A.A.; Byun, H.M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenetics 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.M.; Panni, T.; Motta, V.; Hou, L.; Nordio, F.; Apostoli, P.; Bertazzi, P.A.; Baccarelli, A.A. Effects of airborne pollutants on mitochondrial DNA methylation. Part. Fibre Toxicol. 2013, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, H.M.; Baccarelli, A.A. Environmental exposure and mitochondrial epigenetics: Study design and analytical challenges. Hum. Genet. 2014, 133, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Castegna, A.; Iacobazzi, F.; Spera, I.; Scala, I.; Andria, G.; Iacobazzi, V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol. Genet. Metab. 2011, 102, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Xiong, L.; Ji, Z.; Cheng, W.; Yang, H. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol. Med. Rep. 2012, 6, 125–130. [Google Scholar] [PubMed]
- Lakshmipathy, U.; Campbell, C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999, 27, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Padua, R.A.; Campbell, C. Mammalian mitochondria possess homologous DNA recombination activity. J. Biol. Chem. 1996, 271, 27536–27543. [Google Scholar]
- Coffey, G.; Lakshmipathy, U.; Campbell, C. Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic Acids Res. 1999, 27, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, U.; Campbell, C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 1999, 19, 3869–3876. [Google Scholar] [CrossRef] [PubMed]
- Tadi, K.S.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell. 2016, 27, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013, 77, 476–496. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Kalkhoran, S.B.; Hernández-Reséndiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-shaping proteins in cardiac health and disease-the long and the short of it! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015, 7, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell. Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM proteinkinase: Regulating the cellularresponse to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Turenne, G.A.; Paul, P.; Laflair, L.; Price, B.D. Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53. Oncogene 2001, 20, 5100–5110. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [PubMed]
- Yoshii, S.R.; Mizushima, N. Autophagy machinery in the context of mammalian mitophagy. Biochim. Biophys. Acta 2015, 1853, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negativecontrol of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively down regulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef]
- Rotig, A.; Poulton, J. Genetic causes of mitochondrial DNA depletion in humans. Biochim. Biophys. Acta 2009, 1792, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.T.; McAllister, K.; Worth, L. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009, 37, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, B.; Area, E.; Flanigan, K.M.; Ganesh, J.; Jayakar, P.; Swoboda, K.J.; Coku, J.; Naini, A.; Shanske, S.; Tanji, K.; et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul. Disord. 2008, 18, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Robinson, B.H.; Hood, D.A. Effect of thyroid hormone on mitochondrial properties and oxidative stress in cells from patients with mtDNA defects. Am. J. Physiol. Cell Physiol. 2009, 296, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312–6336. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I. Translating thyroid hormone effects into clinical practice: The relevance of thyroid hormone receptor α1 in cardiac repair. Heart Fail. Rev. 2015, 20, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I.; Cokkinos, D.V. Thyroid hormone and cardiac repair/regeneration: From Prometheus myth to reality? Can. J. Physiol. Pharmacol. 2012, 90, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnoveral. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The level of mitochondrial DNA damage determines whether mitophagy or cell death will be stimulated. ATM (ATM serine/threonine kinase), p53 and Sirt1 (sirtuin 1) mediate the signaling between the nucleus and the mitochondria.
Figure 1.
The level of mitochondrial DNA damage determines whether mitophagy or cell death will be stimulated. ATM (ATM serine/threonine kinase), p53 and Sirt1 (sirtuin 1) mediate the signaling between the nucleus and the mitochondria.

Figure 2.
Factors with which Sirt1 interacts regarding mitochondrial homeostasis. [PGC1a: peroxisome proliferator-activated receptor gamma co-activator (PGC1a), HIF1a: hypoxia-inducible factor 1-alpha, AMPK: 5′ adenosine monophosphate-activated protein kinase].
Figure 2.
Factors with which Sirt1 interacts regarding mitochondrial homeostasis. [PGC1a: peroxisome proliferator-activated receptor gamma co-activator (PGC1a), HIF1a: hypoxia-inducible factor 1-alpha, AMPK: 5′ adenosine monophosphate-activated protein kinase].

Figure 3.
Summary of factors mentioned in the text that are associated with instability of mammalian mitochondrial DNA. The list presented herein is not exhaustive. (Polγ: polymerase γ, mtSSB: mitochondrial single stranded DNA binding protein, TFAM: mitochondrial transcription factor A, NRF1/2: nuclear respiratory factors 1 and 2, PrimPol: primase-polymerase; Pif1: petite integration frequency 1, BER: base excision repair, Mfn: mitofusin, Opa1: optic atrophy protein 1,Drp1: dynamin related protein 1,hFIS1: fission 1 protein, Mff: mitochondrial fission factor, MiD49/51: mitochondrial dynamics proteins of 49 and 51 kDa, RRM2B: ribonucleoside-diphosphate reductase subunit M2 B).
Figure 3.
Summary of factors mentioned in the text that are associated with instability of mammalian mitochondrial DNA. The list presented herein is not exhaustive. (Polγ: polymerase γ, mtSSB: mitochondrial single stranded DNA binding protein, TFAM: mitochondrial transcription factor A, NRF1/2: nuclear respiratory factors 1 and 2, PrimPol: primase-polymerase; Pif1: petite integration frequency 1, BER: base excision repair, Mfn: mitofusin, Opa1: optic atrophy protein 1,Drp1: dynamin related protein 1,hFIS1: fission 1 protein, Mff: mitochondrial fission factor, MiD49/51: mitochondrial dynamics proteins of 49 and 51 kDa, RRM2B: ribonucleoside-diphosphate reductase subunit M2 B).

Figure 4.
Proteins involved in the maintenance of mitochondrial DNA integrity, in addition to their role in ensuring nuclear genomic homeostasis. (BRCA1: breast cancer type 1 protein, CtIP: CtBP-interacting protein, FEN1: flap endonuclease 1, MRE11: meiotic recombination 11, PARP: poly (ADP-ribose) polymerase).
Figure 4.
Proteins involved in the maintenance of mitochondrial DNA integrity, in addition to their role in ensuring nuclear genomic homeostasis. (BRCA1: breast cancer type 1 protein, CtIP: CtBP-interacting protein, FEN1: flap endonuclease 1, MRE11: meiotic recombination 11, PARP: poly (ADP-ribose) polymerase).

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vasileiou, P.V.S.; Mourouzis, I.; Pantos, C. Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. Int. J. Mol. Sci. 2017, 18, 1821. https://doi.org/10.3390/ijms18081821
AMA Style
Vasileiou PVS, Mourouzis I, Pantos C. Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. International Journal of Molecular Sciences. 2017; 18(8):1821. https://doi.org/10.3390/ijms18081821
Chicago/Turabian StyleVasileiou, Panagiotis V. S., Iordanis Mourouzis, and Constantinos Pantos. 2017. "Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity" International Journal of Molecular Sciences 18, no. 8: 1821. https://doi.org/10.3390/ijms18081821
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.