Human CD3+ T-Cells with the Anti-ERBB2 Chimeric Antigen Receptor Exhibit Efficient Targeting and Induce Apoptosis in ERBB2 Overexpressing Breast Cancer Cells

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
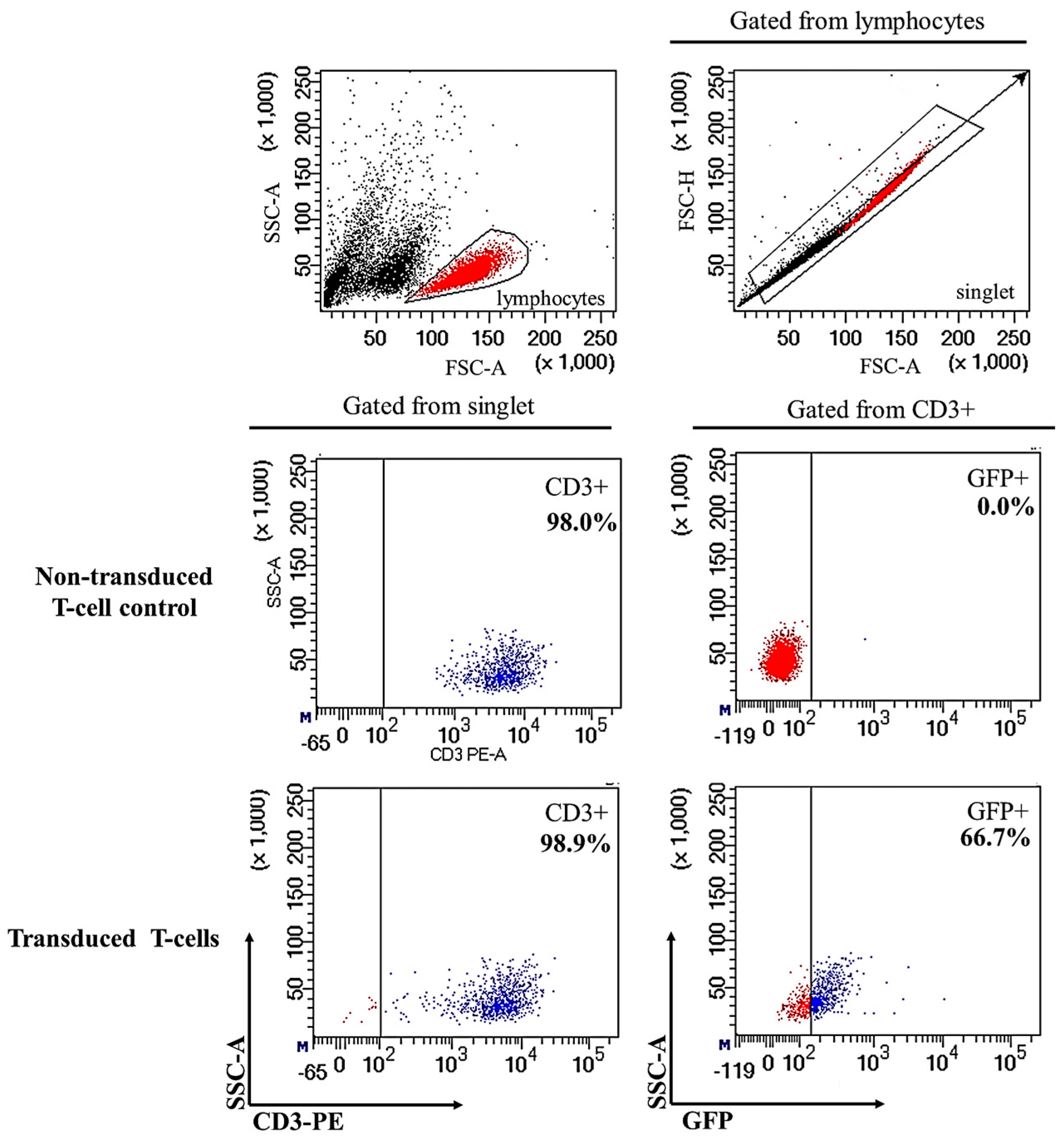
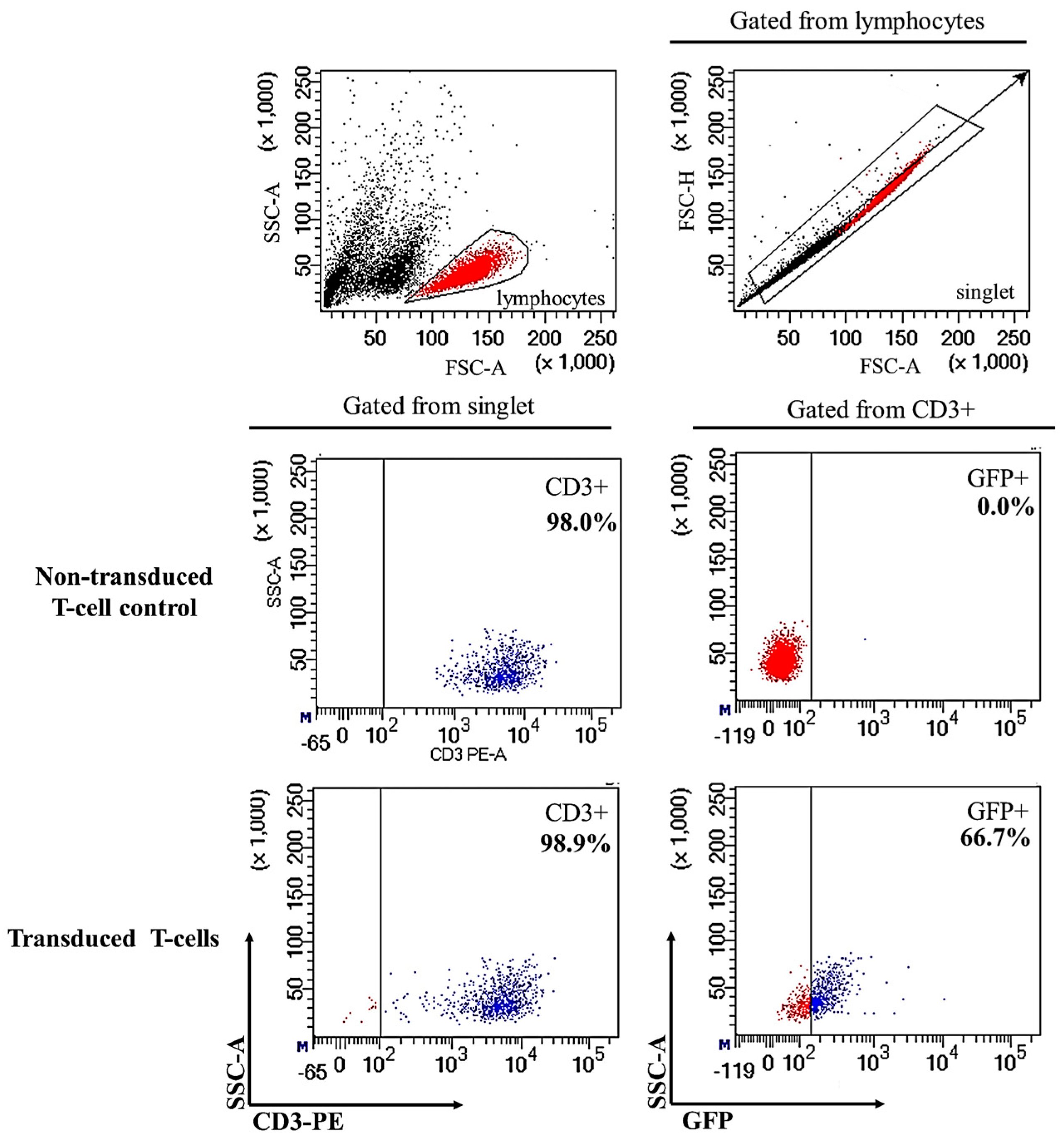
2.1. Successful Transduction of Lentiviral Particles Encoding Chimeric Antigen Receptor (CAR) into Human CD3+ T-Cells
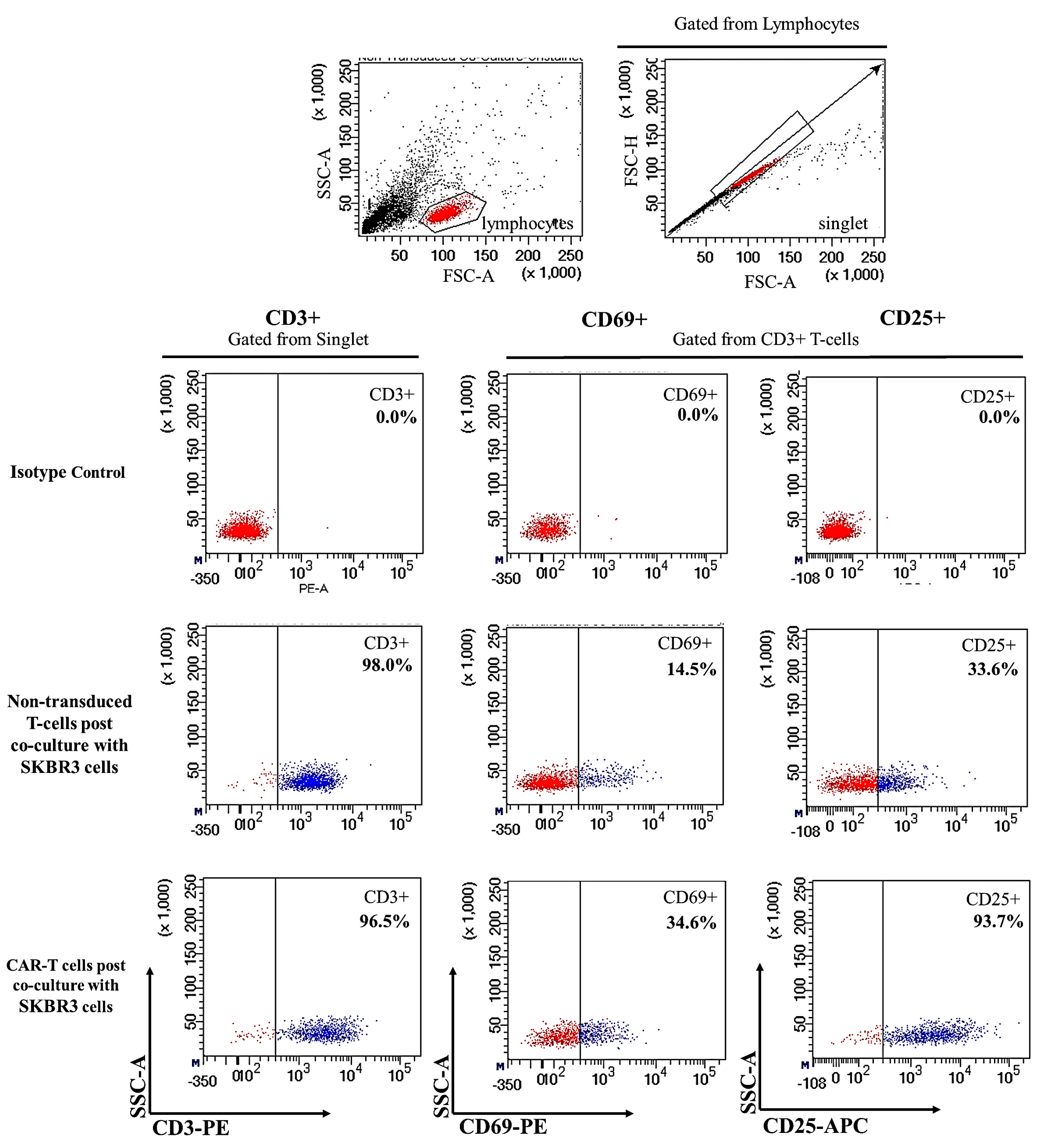
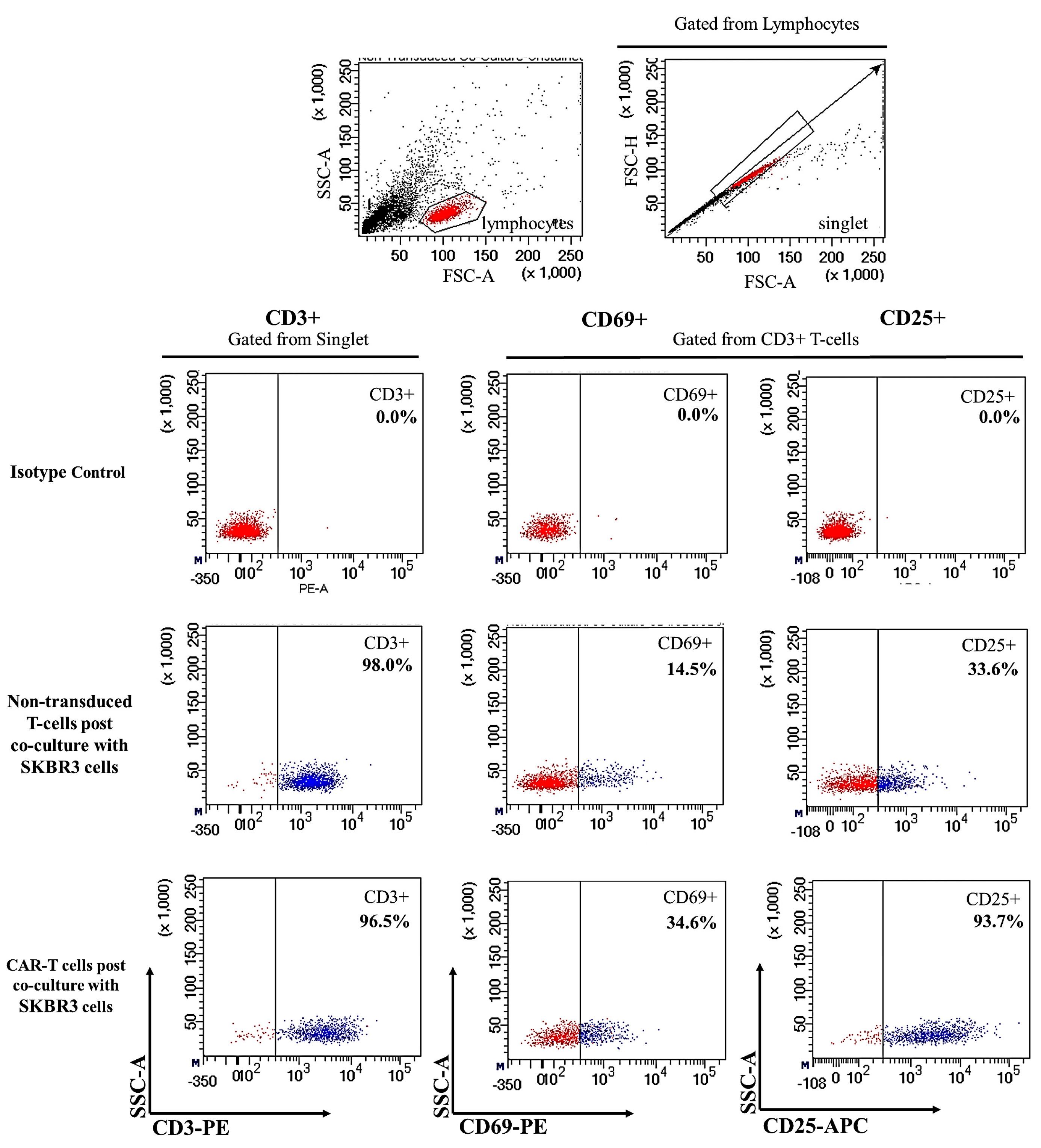
2.2. CAR-T Cells Showed Presence of Cell Surface Activation Markers upon Co-Culture with SKBR3 Cells
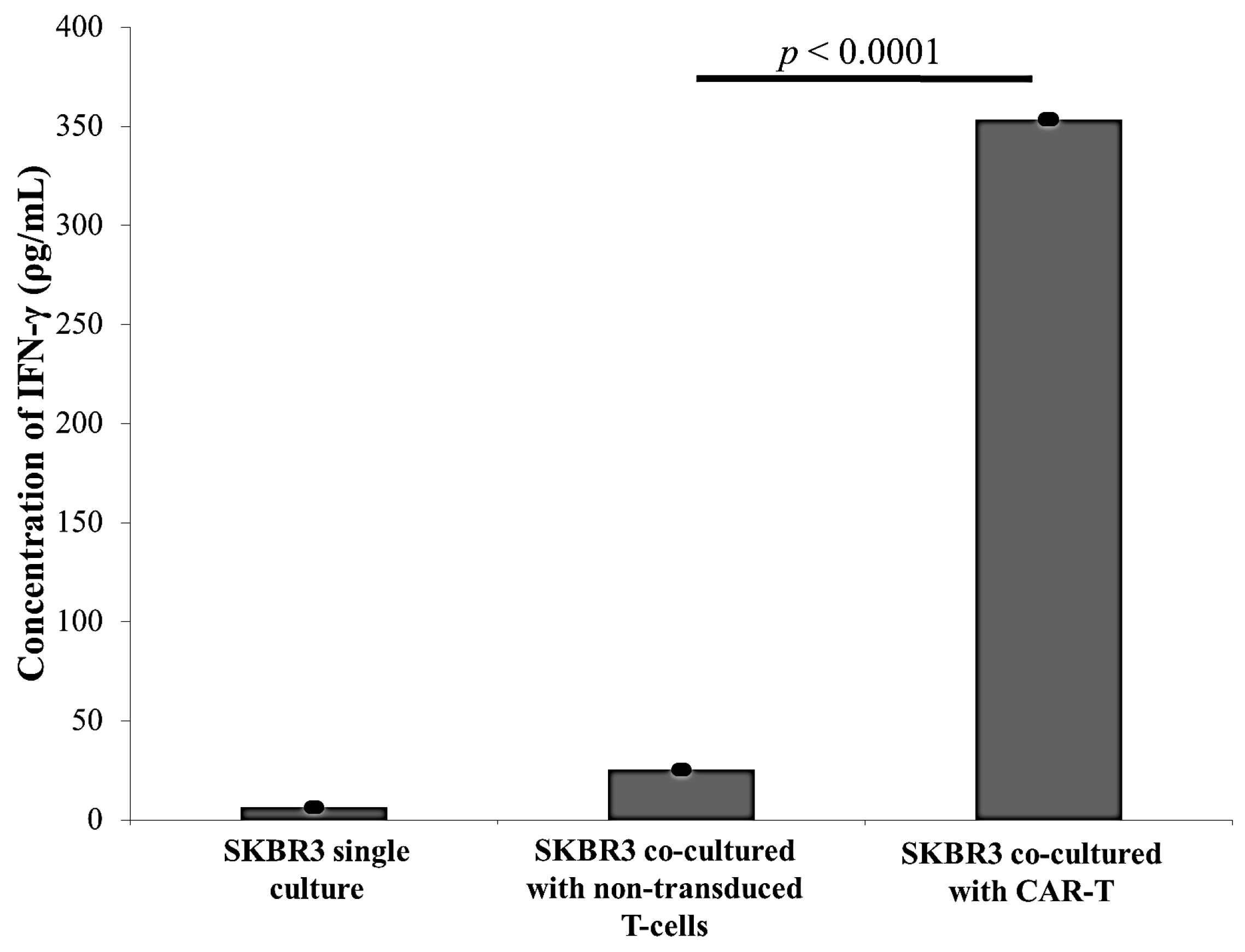
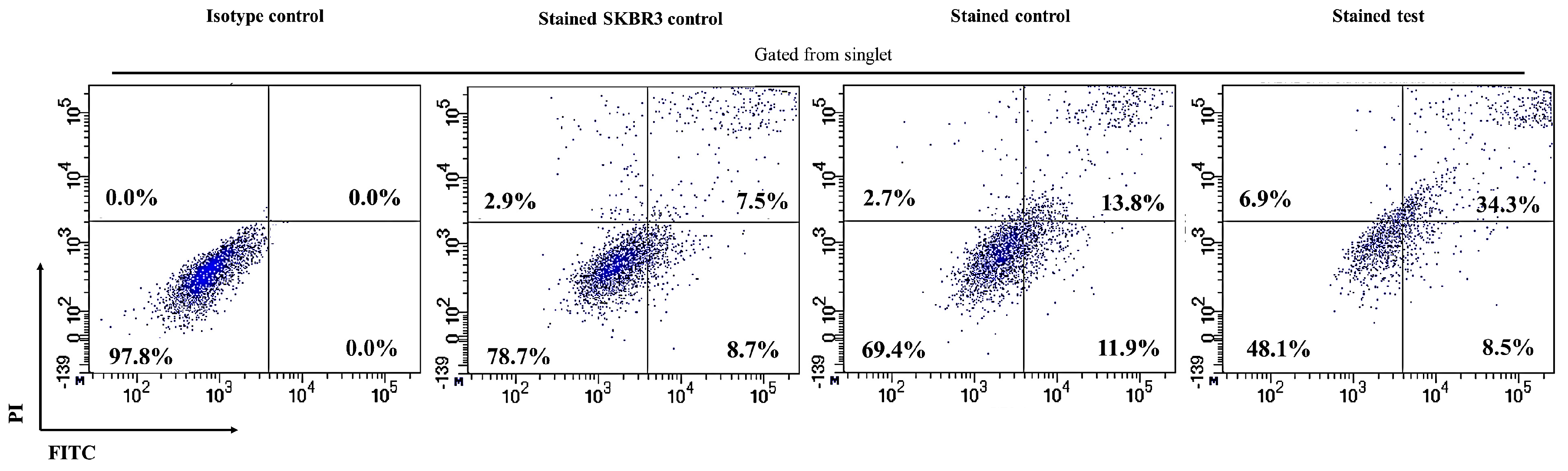
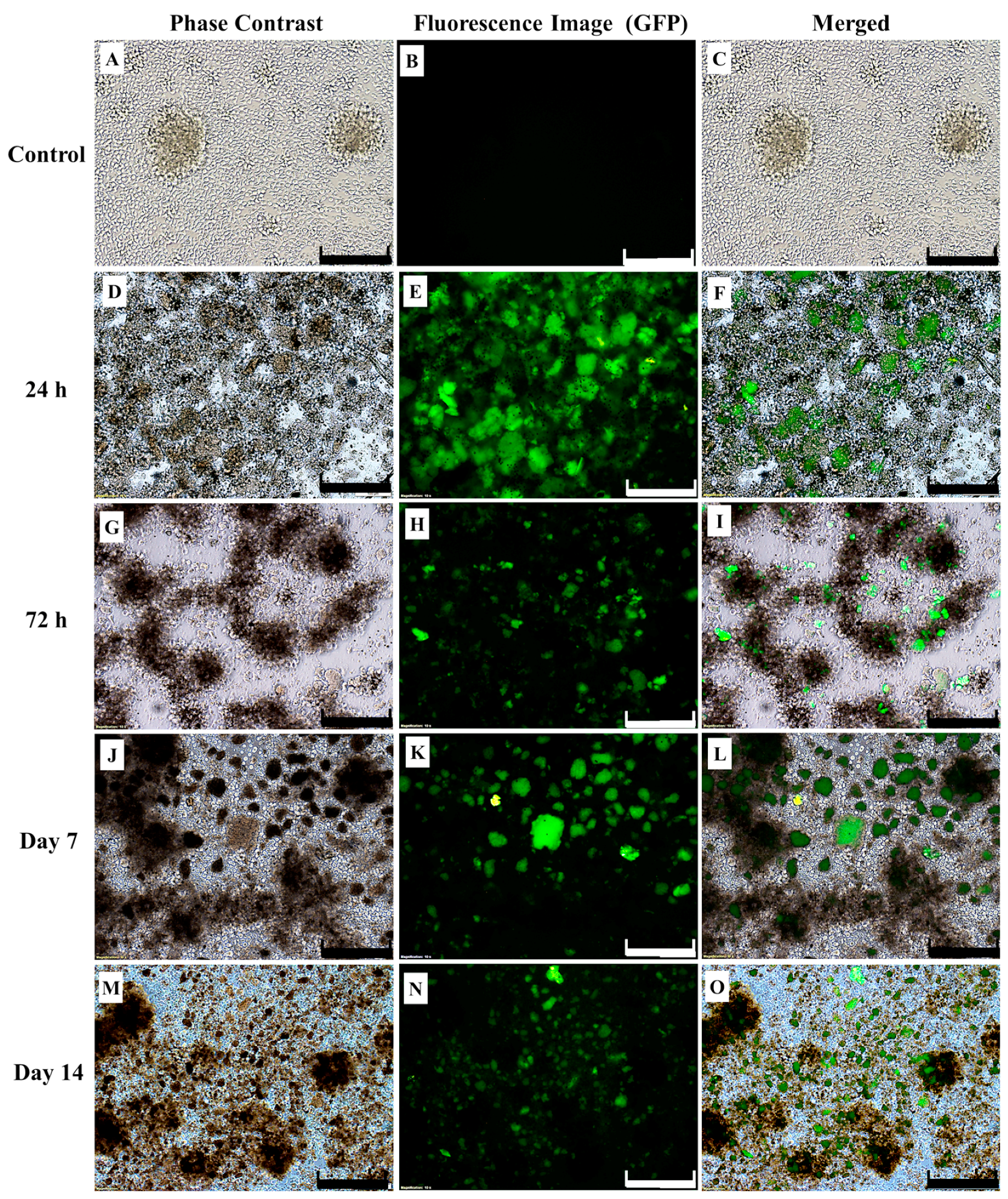
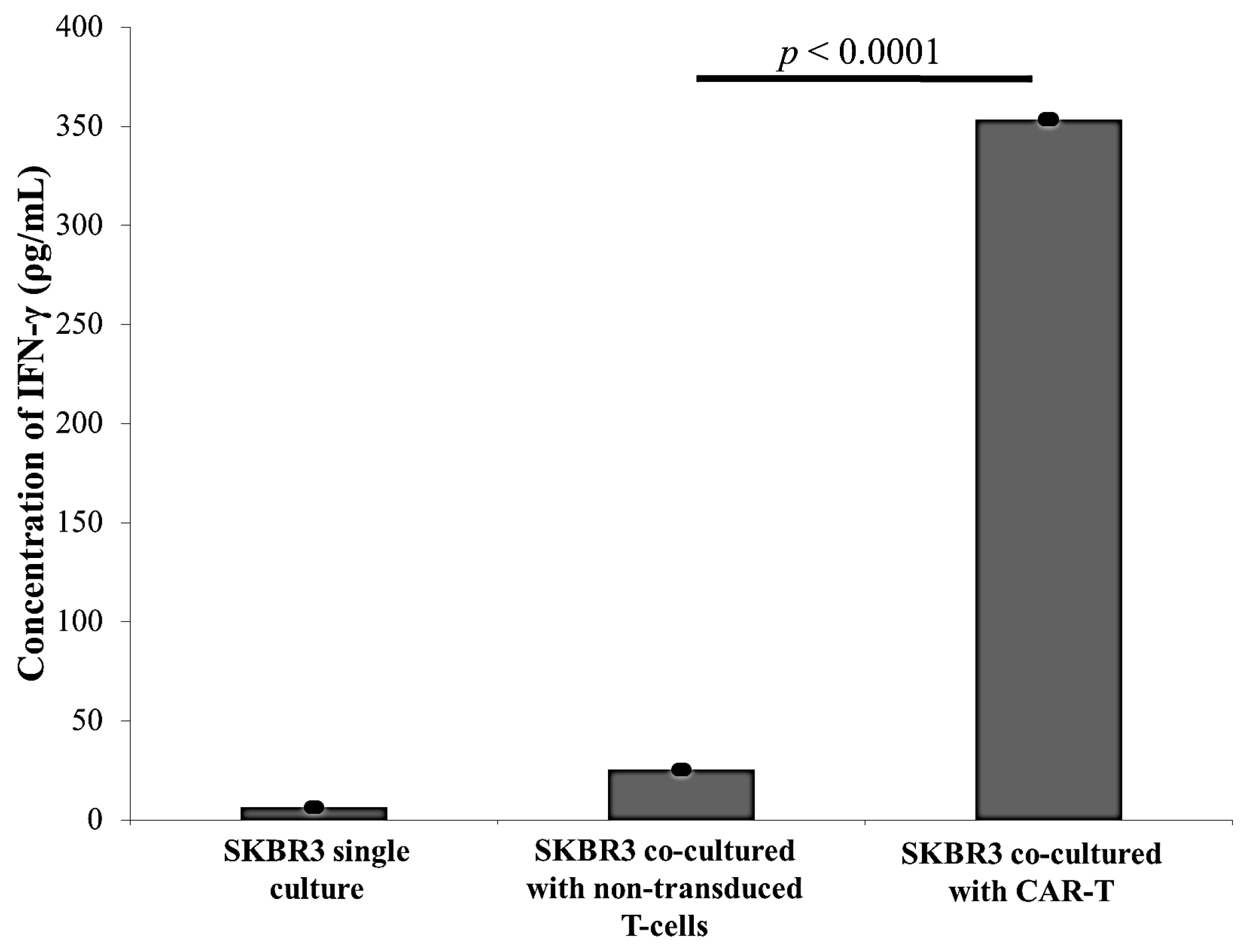
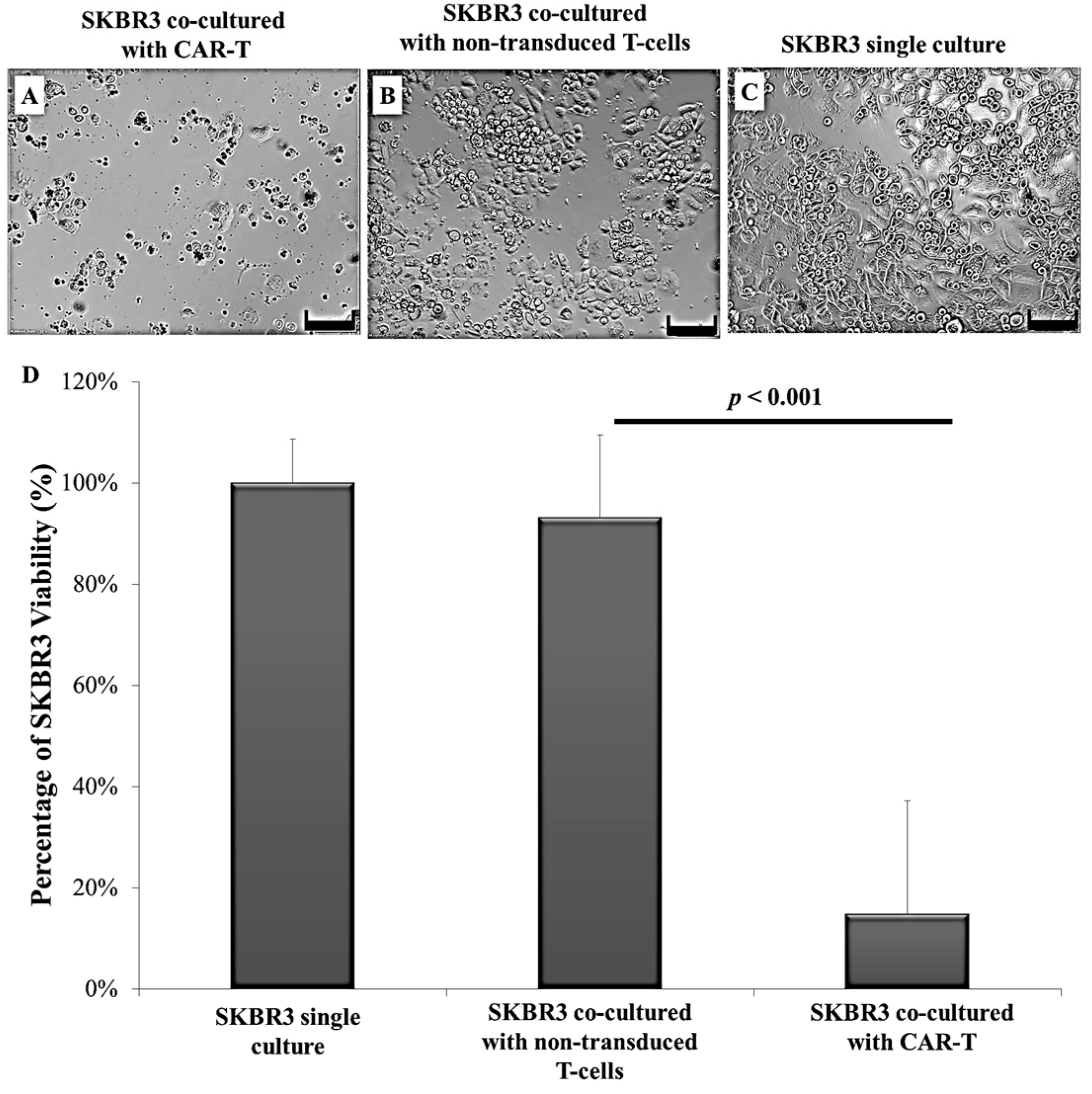
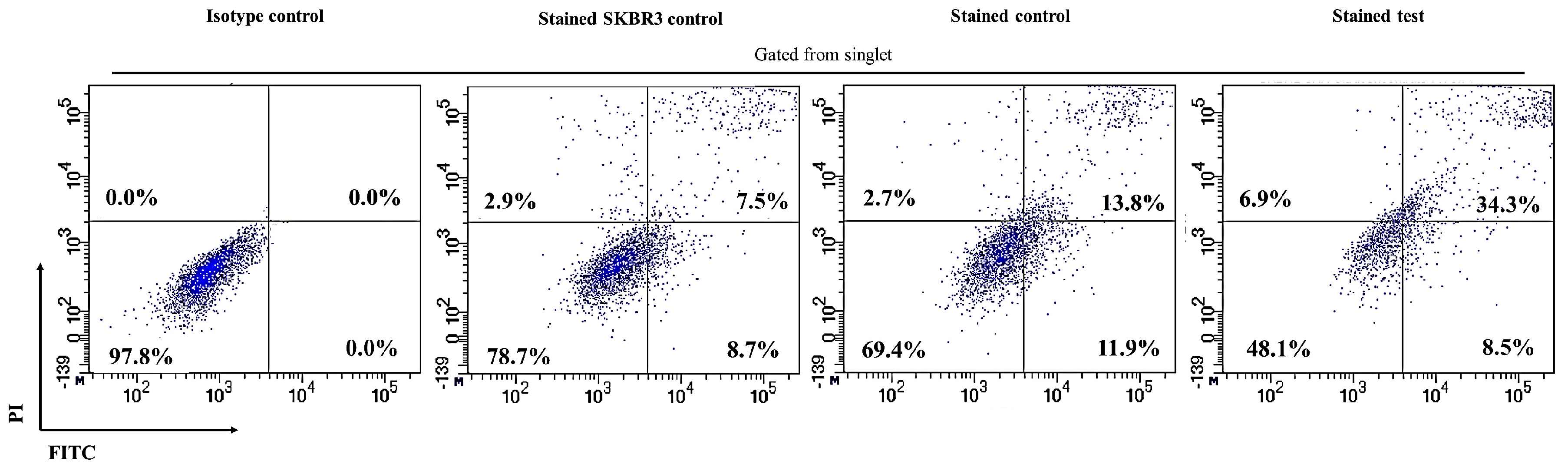
2.3. CAR-T Cells Effectively Secreted IFN-γ and Subsequently Lysed the SKBR3 Cells
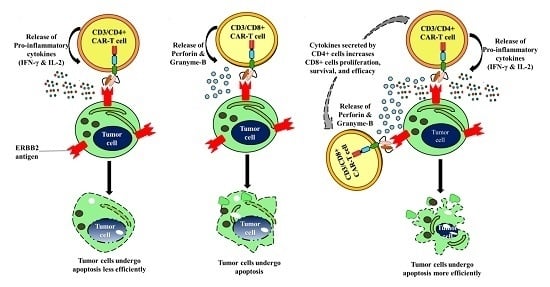
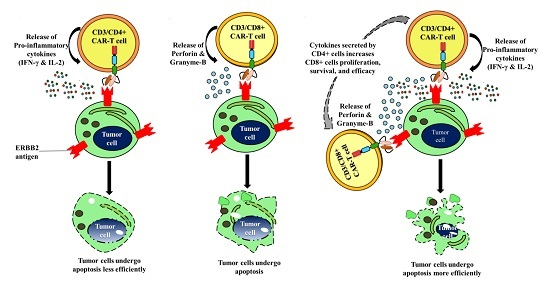
3. Discussion
4. Materials and Methods
4.1. Collection, Isolation, and Processing of Human T-Cells
4.2. Isolation of Human CD3+ T-Cells from Peripheral Blood Mononuclear Cells (PBMC)
4.3. Culture, Expansion, and Characterisation of Human T-Cells and the SKBR3 Cell Line
4.4. Immunophenotyping of Human T-Cells
4.5. Generation of Lentiviral Plasmid Coding for CAR
4.6. Lentiviral Production and Transduction to Produce CAR-Expressing Human T-Cells (CAR-T Cells)
4.7. Co-Culture of CAR-T Cells with SKBR3 Cells and T-Cell Activation Marker Detection
4.8. Determination of IFN-γ Expression by Enzyme-Linked Immunosorbent Assay (ELISA)
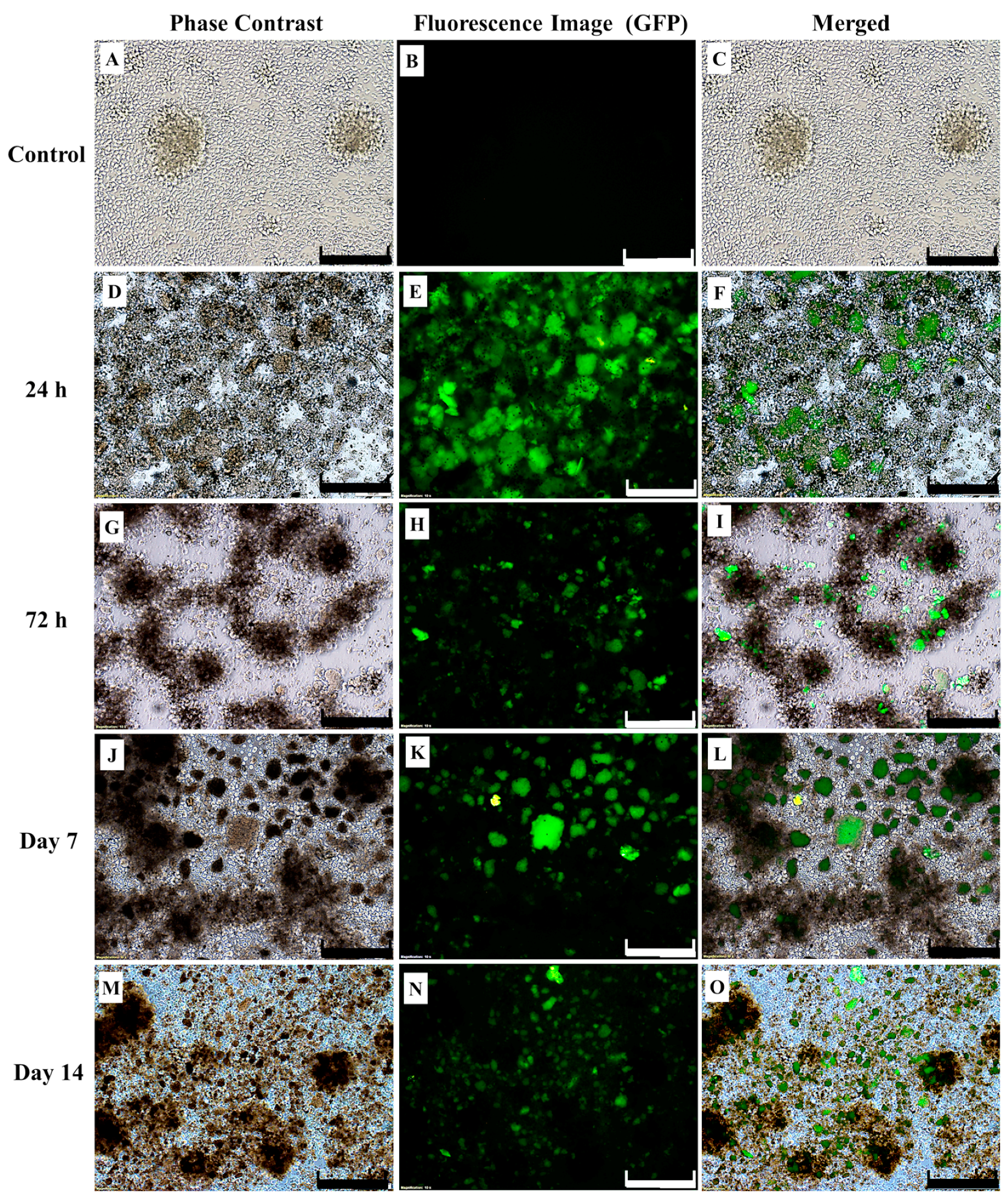
4.9. Effect of CAR-T Cells on Survivability of SKBR3 Cells In Vitro Following Co-Culture
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maher, J. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol. 2012, 27, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Irvin, W.; Muss, H.B.; Mayer, D.K. Symptom management in metastatic breast cancer. Oncologist 2011, 16, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Carels, N.; Spinassé, L.B.; Tilli, T.M.; Tuszynski, J.A. Toward precision medicine of breast cancer. Theor. Biol. Med. Model. 2016, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Saha Roy, S.; Vadlamudi, R.K. Role of estrogen receptor signaling in breast cancer metastasis. Int. J. Breast Cancer 2012, 65, 46–98. [Google Scholar] [CrossRef] [PubMed]
- Mahyari, H.M.; Khosravi, A.; Monfared, Z.E.; Khosravi, N. Overexpression of HER2/neu as a prognostic value in Iranian women with early stage breast cancer; a single institute study. Iran. Red Crescent Med. J. 2014, 16, e16005. [Google Scholar] [CrossRef]
- Tan, M.; Yu, D. Molecular mechanisms of ERBB2-mediated breast cancer chemoresistance. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2000–2013. [Google Scholar]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 85, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Heslop, H.E. Safer CARS. Mol. Ther. 2010, 18, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, M.; Olmez, O.F.; Kurt, E.; Cubukcu, E.; Evrensel, T.; Kanat, O.; Manavoglu, O. Prognostic significance of C-ERBB2 overexpression in patients with metastatic gastric cancer. Clin. Transl. Oncol. 2013, 15, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Koga, F.; Yoshida, S.; Tamura, T.; Fujii, Y.; Ito, E.; Kihara, K. Significance of ERBB2 overexpression in therapeutic resistance and cancer-specific survival in muscle-invasive bladder cancer patients treated with chemoradiation-based selective bladder-sparing approach. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Peters, S.; Lepage, B.; Cortot, A.B.; Barlesi, F.; Beau-Faller, M.; Besse, B.; Blons, H.; Mansuet-Lupo, A.; Urban, T.; et al. Lung cancer that harbors an HER2 mutation: Epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol. 2013, 31, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Fauvel, B.; Yasri, A. Antibodies directed against receptor tyrosine kinases: Current and future strategies to fight cancer. MAbs 2014, 6, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Claret, F. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Esteva, F.J. HER2 therapy: Molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.X.; Chen, H.P.; Yu, K.; Shen, L.-X.; Wang, C.-Y.; Su, S.-Z.; Sui, W.-J.; Shan, D.-M.; Li, H.-Z. Gene therapy of malignant solid tumors by targeting ERBB2 receptors and by activating T cells. Cancer Biother. Radiopharm. 2012, 27, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Kempe, S.; Muller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 918471. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z. The T-body approach: Redirecting T cells with antibody specificity. Handb. Exp. Pharmacol. 2008, 181, 329–342. [Google Scholar]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor (CAR) design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Yang, J.; Kitano, M.; Dudley, M.; Laurencot, C.; Rosenberg, S. Case report of a serious adverse event following the administration of T Cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.; Haas, A.; Maus, M.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol. Res. 2013, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Levine, B.L. Smart CARS: Optimized development of a chimeric antigen receptor (CAR) T cell targeting epidermal growth factor receptor variant III (EGFRvIII) for glioblastoma. Ann. Transl. Med. 2016, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Haynes, N. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002, 100, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ERBB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Dotti, G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.; Voutsas, I.; Gritzapis, A.; Perez, S.; Papamichail, M. HER-2/Neu as a target for cancer vaccines. Immunotherapy 2010, 2, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Restifo, N.P. Reassessing target antigens for adoptive T-cell therapy. Nat. Biotechnol. 2013, 31, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.S.; Zhao, Y.; Schiro, P.G.; Ng, L.; Lim, D.S.; Shelby, J.P.; Chiu, D.T. Deformability considerations in filtration of biological cells. Lab Chip 2010, 10, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.; Grupp, S.; Porter, D.; June, C. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ERBB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.; Gogishvili, T.; Maloney, M.; Turtle, C.; Riddell, S. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2015, 30, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Tan Le, D.; van Pham, P. Synergistic effect of chimeric antigen receptors and cytokine induced killer cells: An innovative combination for cancer therapy. Biomed. Res. Ther. 2016, 3. [Google Scholar] [CrossRef]
- Gravano, D.; Hoyer, K. Promotion and prevention of autoimmune disease by CD8+ T cells. J. Autoimmun. 2013, 45, 68–79. [Google Scholar] [CrossRef] [PubMed]
- McGuire, H.; Walters, S.; Vogelzang, A.; Lee, C.M.Y.; Webster, K.; Sprent, J.; Christ, D.; Grey, S.; King, C. Interleukin-21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes 2011, 60, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Hopken, U. Lymphotoxin-keeps the gate open for T-cell infiltration in cHL. Blood 2014, 124, 2897–2898. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Brentjens, R.J. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov. Med. 2010, 9, 277–288. [Google Scholar] [PubMed]
- Zhen, A.; Kamata, M.; Rezek, V.; Rick, J.; Levin, B.; Kasparian, S.; Chen, I.S.; Yang, O.O.; Zack, J.A.; Kitchen, S.G. HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol. Ther. 2015, 23, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.; Kershaw, M.H.; Cameron, R.; Westwood, J.A.; Trapani, J.A.; Smyth, M.J.; Darcy, P.K. Sustained antigen-specific antitumor recall response mediated by gene-modified CD4+ T helper-1 and CD8+ T cells. Cancer Res. 2007, 67, 11428–11437. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; King, J.; Xue, S.; Voisine, C.; Holler, A.; Wright, G. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef] [PubMed]
- Rivkina, A.; Holodnuka-Kholodnyuk, I.; Murovska, M.; Soloveichika, M.; Lejniece, S. Peripheral blood lymphocyte phenotype of ZAP-70+ and ZAP-70− patients with B-cell chronic lymphocytic leukaemia. Exp. Oncol. 2015, 37, 73–76. [Google Scholar] [PubMed]
- Schoenfeld, K.; Knopp, C.; Wels, W. Sequence 12 from Patent WO2012031744–Nucleotide. National Center for Biotechnology Information (NCBI), 2013. Available online: http://www.ncbi.nlm.nih.gov/nuccore/JB399738 (accessed on 12 Jan 2016).
- Ji, R.; Chasalow, S.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2011, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Köhler, H.; Rappl, G.; Abken, H. Human CD4+ T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J. Immunol. 2006, 177, 5668–5675. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.; Dudley, M.; Feldman, S.; Wilson, W.H.; Spaner, D.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.; Sherry, R.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2011, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.; Slaney, C.; Kershaw, M.; Gyorki, D.; Neeson, P.; Darcy, P. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Semin. Immunol. 2016, 28, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; DiGiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer. Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human epidermal growth factor receptor 2 (HER2)–Specific chimeric antigen receptor–modified T Cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.; Gerdemann, U.; Leen, A.M.; Shaffer, J.A.; Ku, S.; Tzou, B.; Horton, T.M.; Sheehan, A.; Copeland, A.; Younes, A.; et al. Improving T-cell therapy for relapsed EBV-negative Hodgkin lymphoma by targeting upregulated MAGE-A4. Clin. Cancer Res. 2011, 17, 7058–7066. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.; Chen, R.; Gong, C.; Yuan, L.; Jasper, J.; Ding, Y.; Markowitz, G.; Yang, P.; Xu, X.; McDonnell, D.; et al. Distinct receptor tyrosine kinase subsets mediate anti-HER2 drug resistance in breast cancer. J. Biol. Chem. 2016, 292, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.; Cogdill, A.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ERBB2 or EGFR chimeric antigen receptor T Cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [PubMed]
- Hillerdal, V.; Essand, M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs 2015, 29, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T Cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int. J. Cancer 2013, 133, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Kaufman, D.S. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front. Immunol. 2015, 6, 195. [Google Scholar] [CrossRef] [PubMed]
- Darcy, P.; Neeson, P.; Yong, C.; Kershaw, M. Manipulating immune cells for adoptive immunotherapy of cancer. Curr. Opin. Immunol. 2014, 27, 46–52. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munisvaradass, R.; Kumar, S.; Govindasamy, C.; Alnumair, K.S.; Mok, P.L. Human CD3+ T-Cells with the Anti-ERBB2 Chimeric Antigen Receptor Exhibit Efficient Targeting and Induce Apoptosis in ERBB2 Overexpressing Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1797. https://doi.org/10.3390/ijms18091797
Munisvaradass R, Kumar S, Govindasamy C, Alnumair KS, Mok PL. Human CD3+ T-Cells with the Anti-ERBB2 Chimeric Antigen Receptor Exhibit Efficient Targeting and Induce Apoptosis in ERBB2 Overexpressing Breast Cancer Cells. International Journal of Molecular Sciences. 2017; 18(9):1797. https://doi.org/10.3390/ijms18091797
Chicago/Turabian StyleMunisvaradass, Rusheni, Suresh Kumar, Chandramohan Govindasamy, Khalid S. Alnumair, and Pooi Ling Mok. 2017. "Human CD3+ T-Cells with the Anti-ERBB2 Chimeric Antigen Receptor Exhibit Efficient Targeting and Induce Apoptosis in ERBB2 Overexpressing Breast Cancer Cells" International Journal of Molecular Sciences 18, no. 9: 1797. https://doi.org/10.3390/ijms18091797