Understanding Insulin Endocrinology in Decapod Crustacea: Molecular Modelling Characterization of an Insulin-Binding Protein and Insulin-Like Peptides in the Eastern Spiny Lobster, Sagmariasus verreauxi

 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
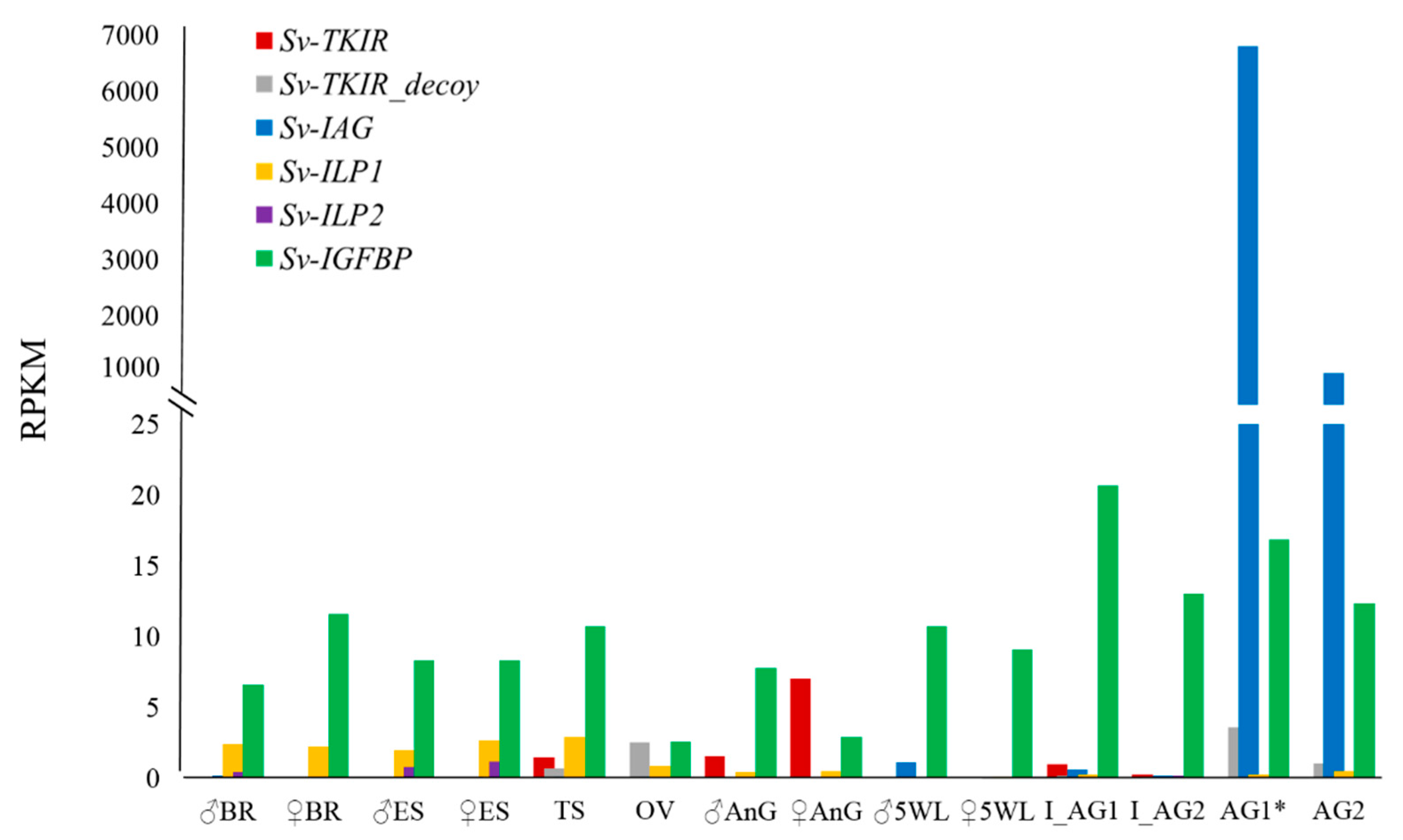
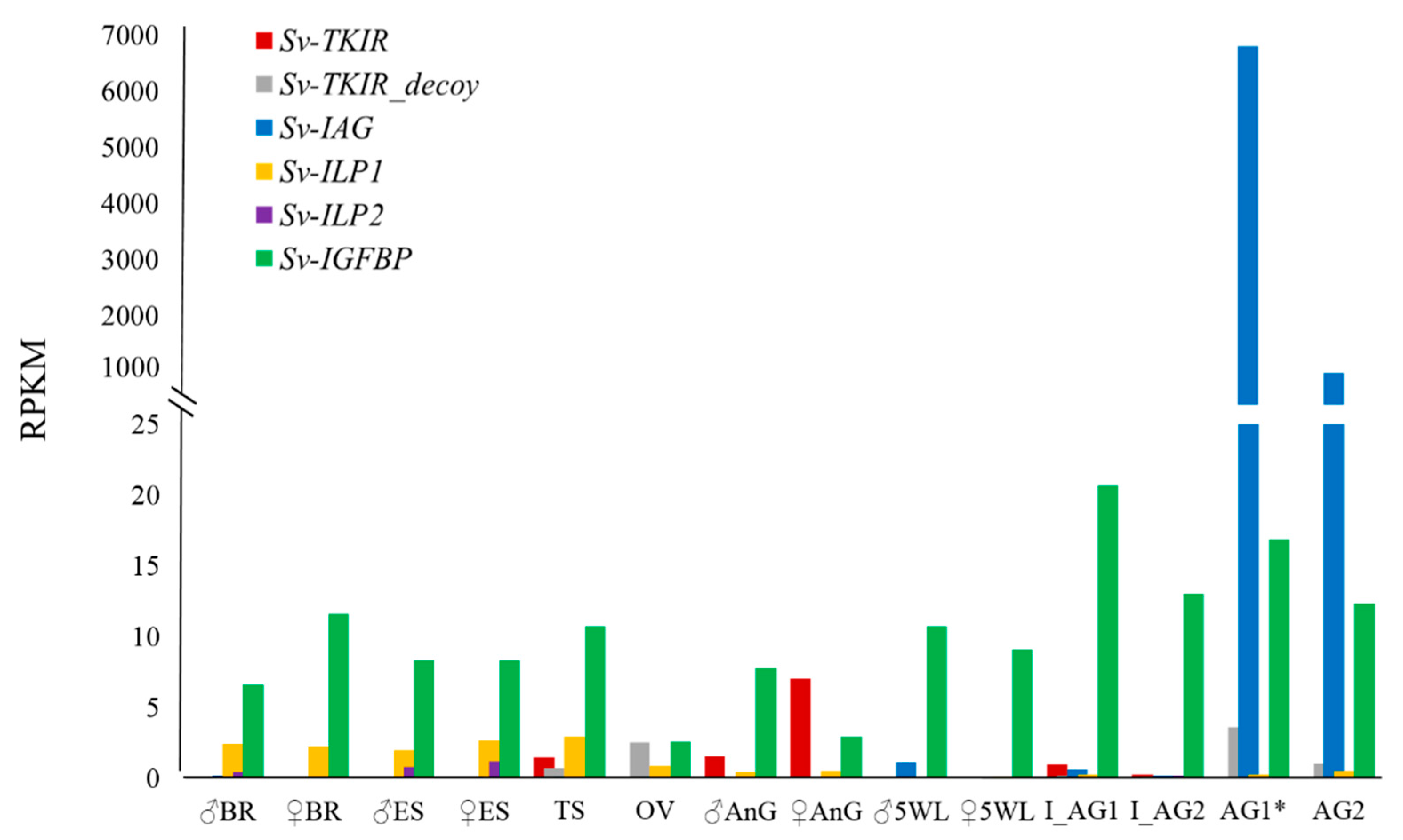
2. Results
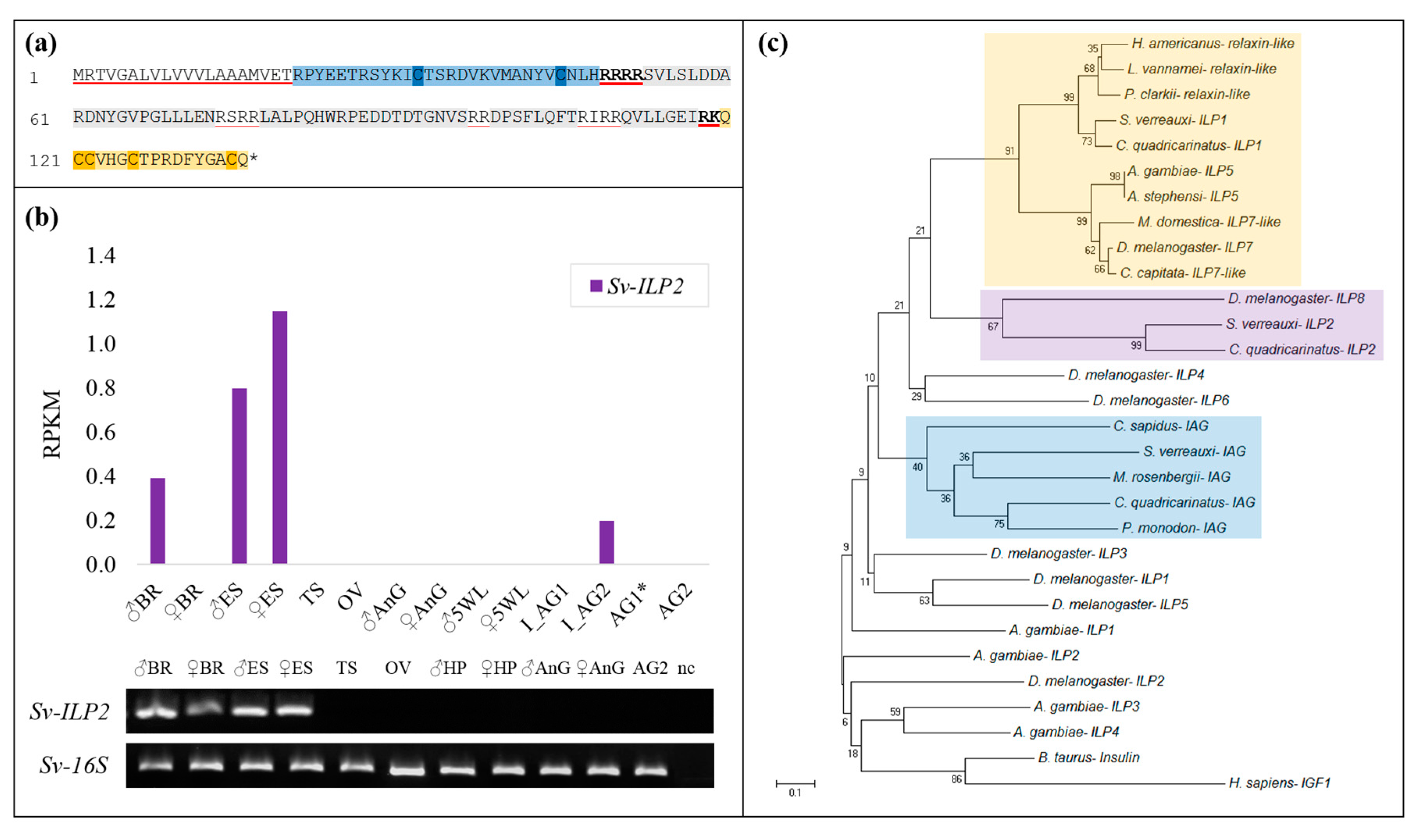
2.1. Identification of Sv-ILP2
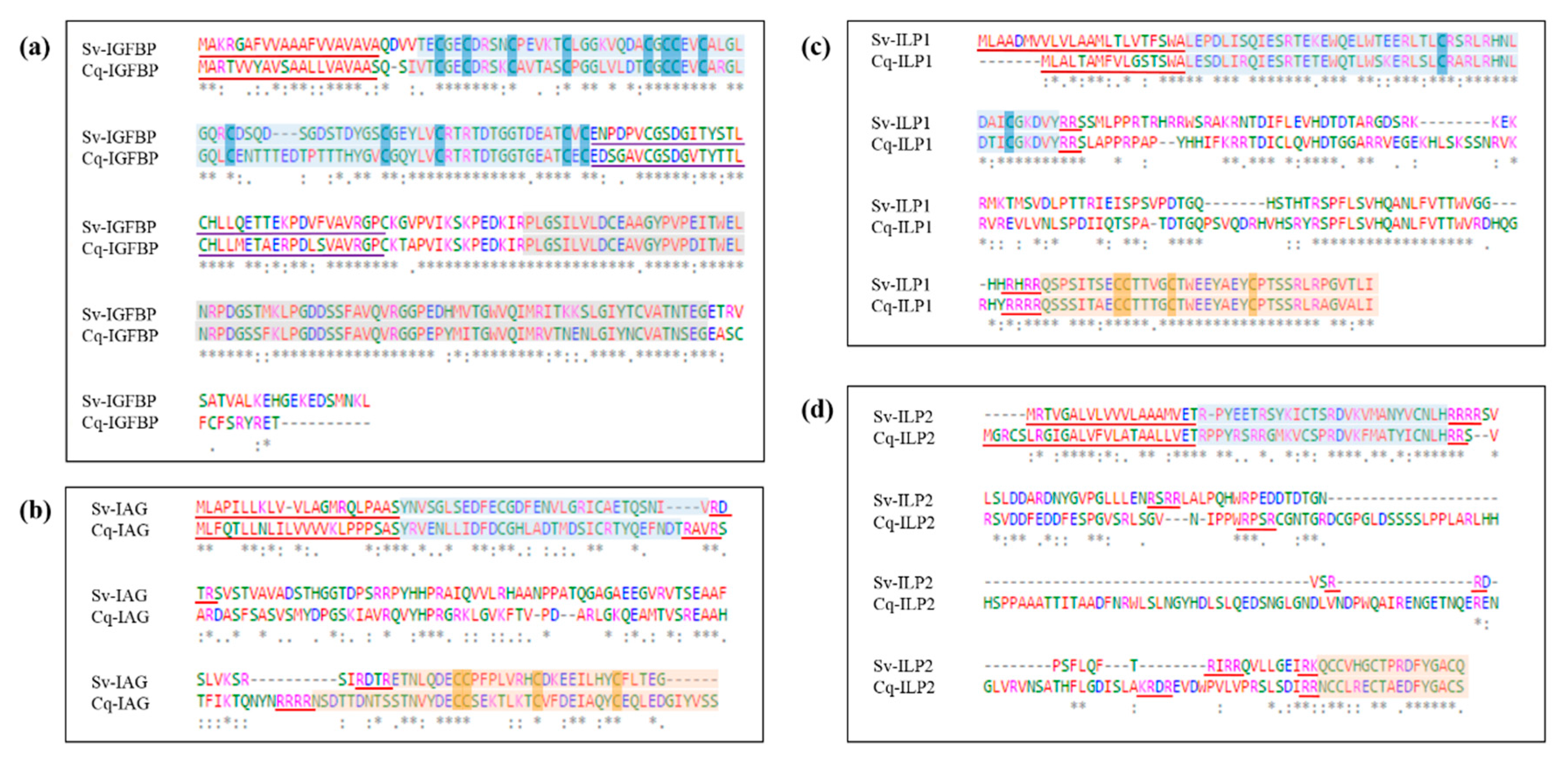
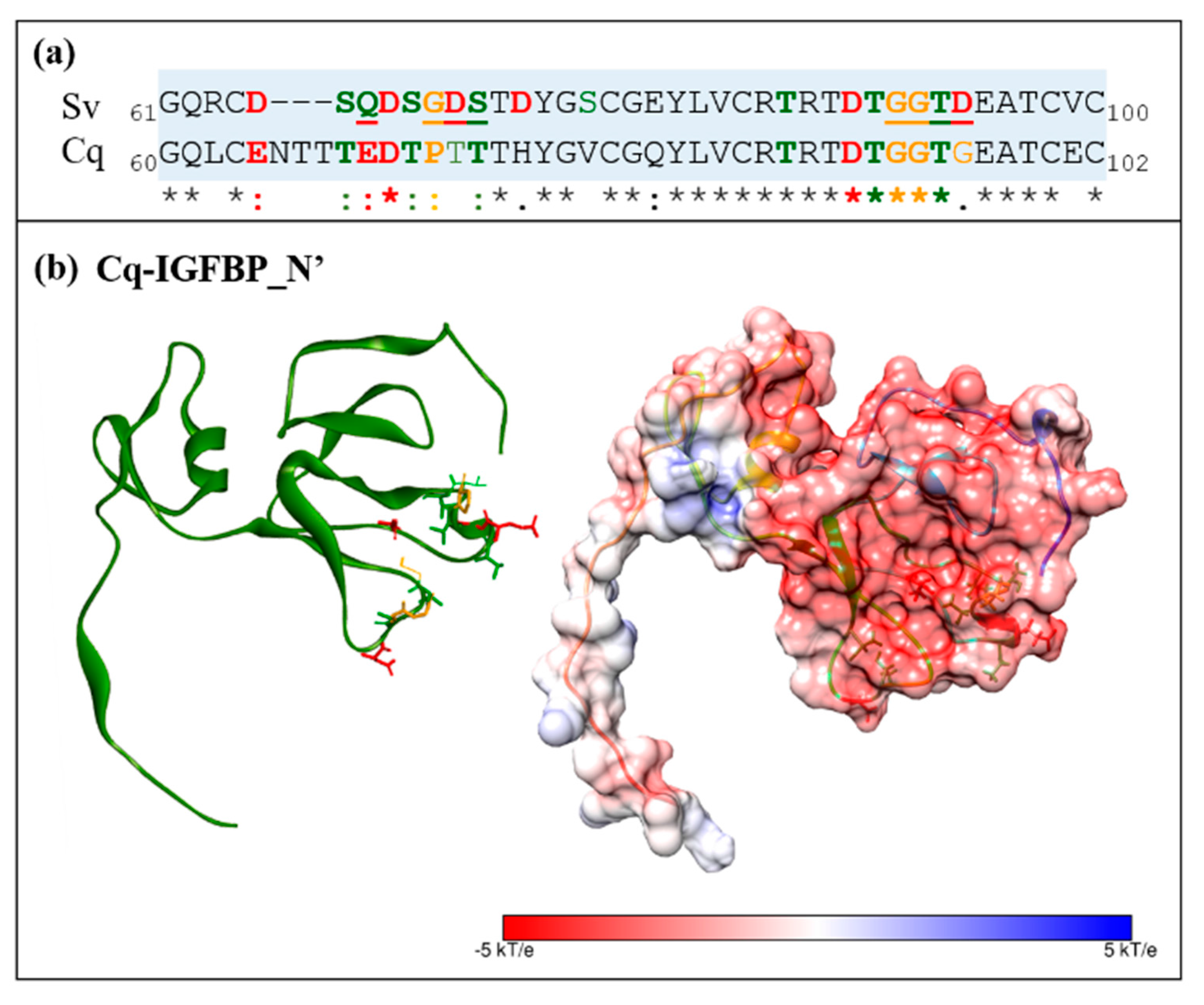
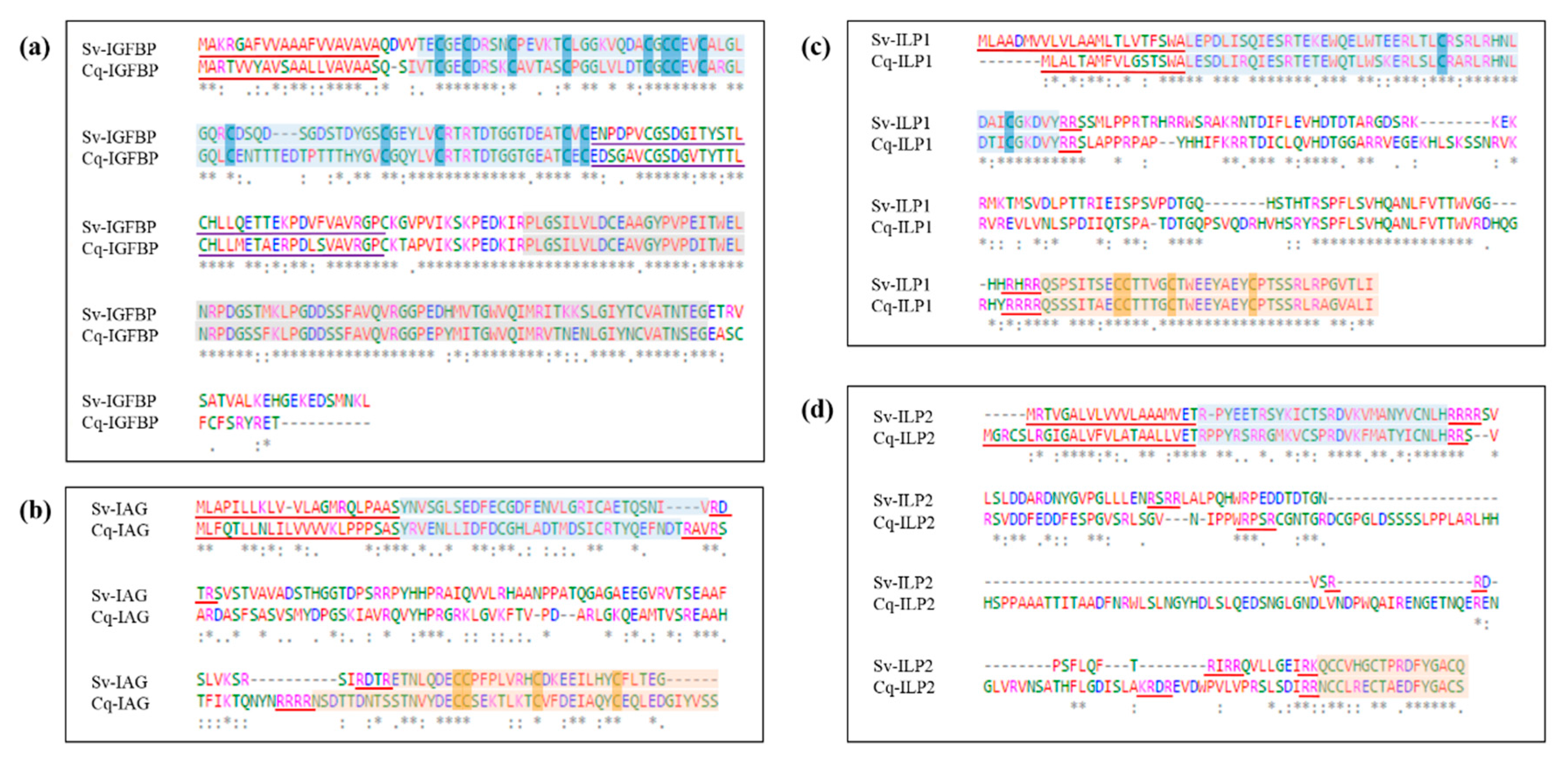
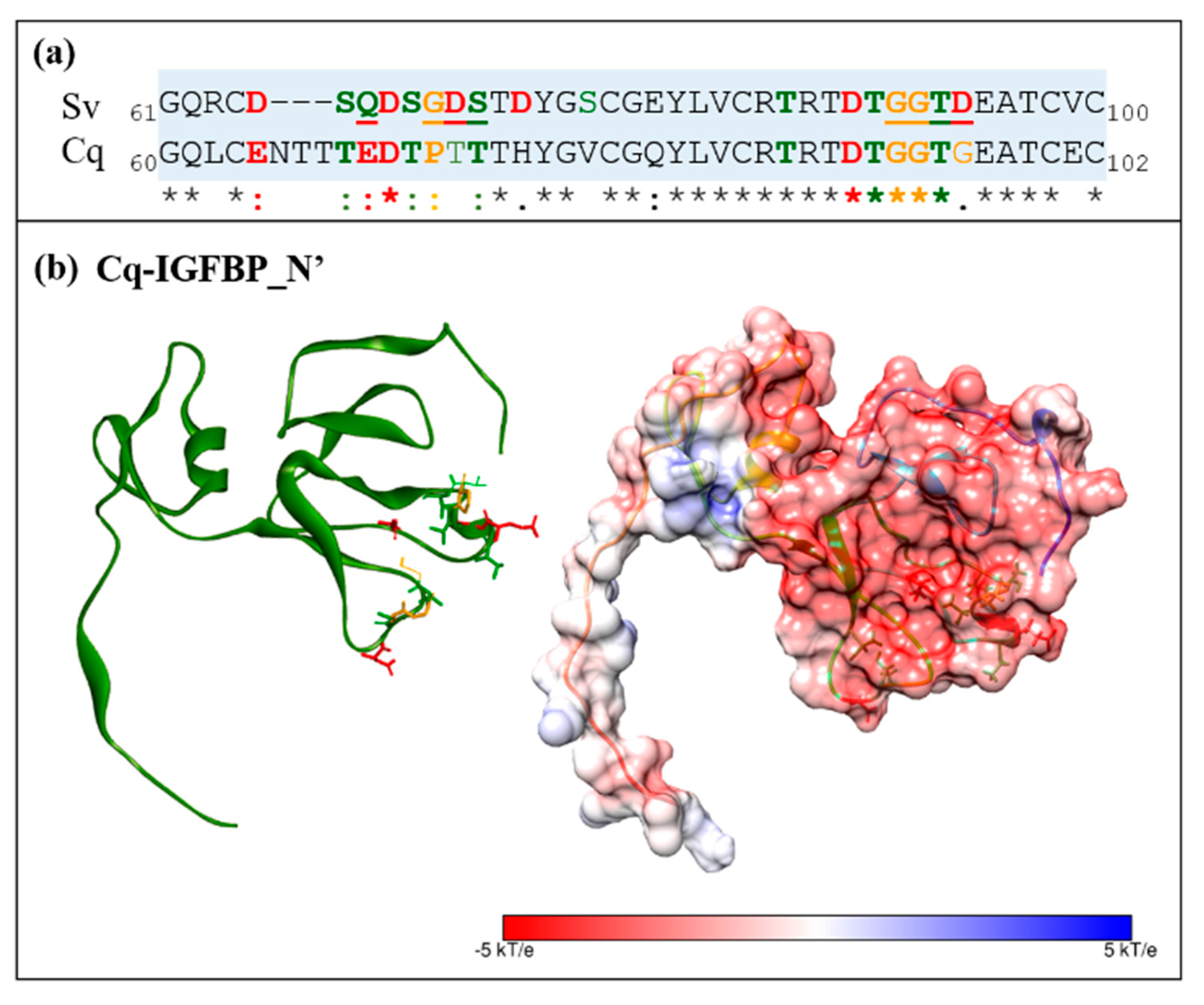
2.2. Sequence Analyses of IGFBPs and ILP Ligands
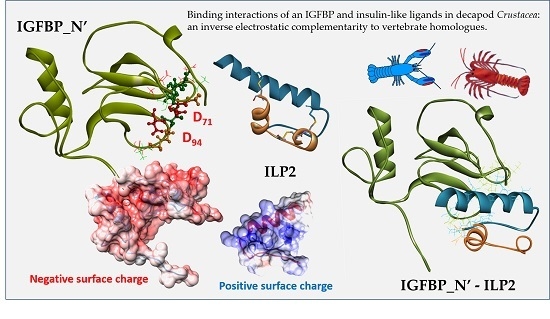
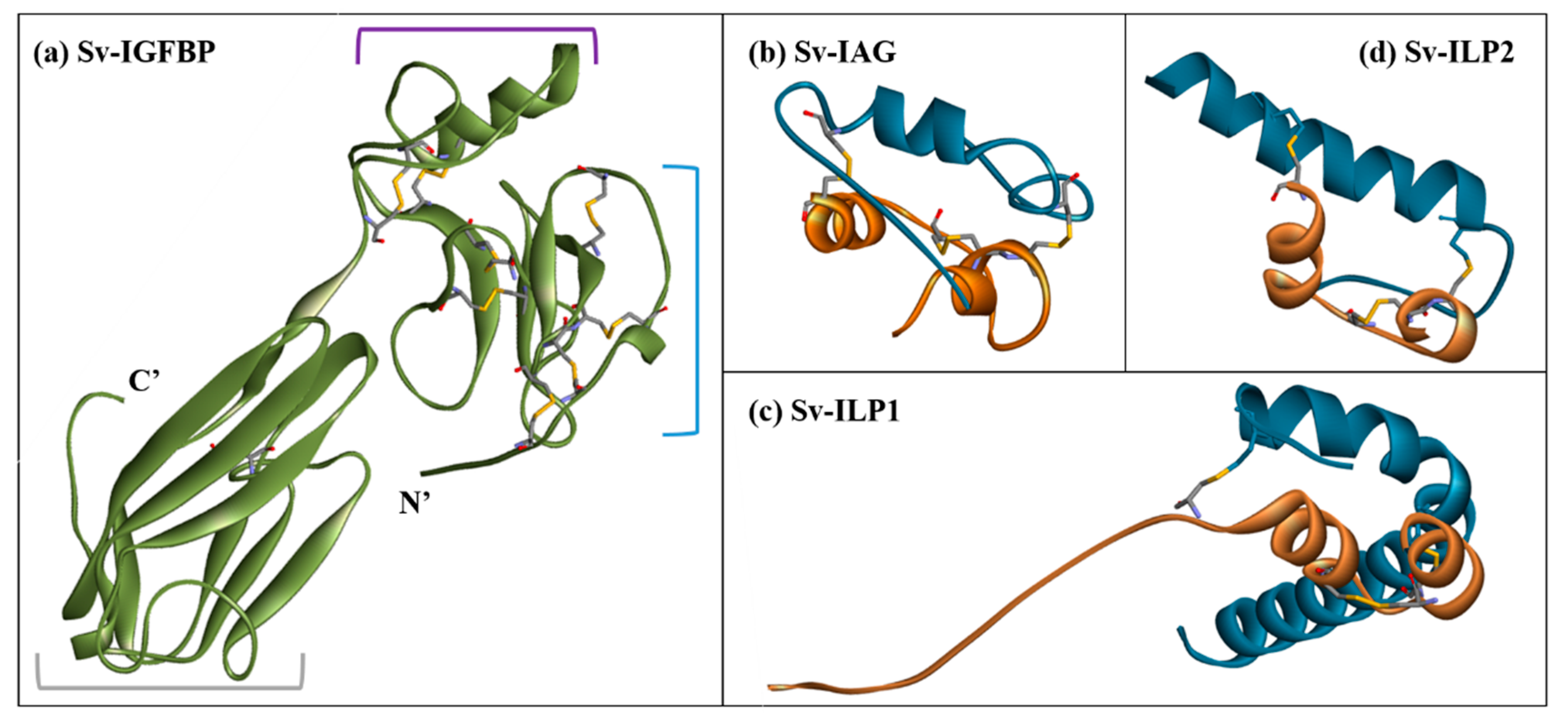
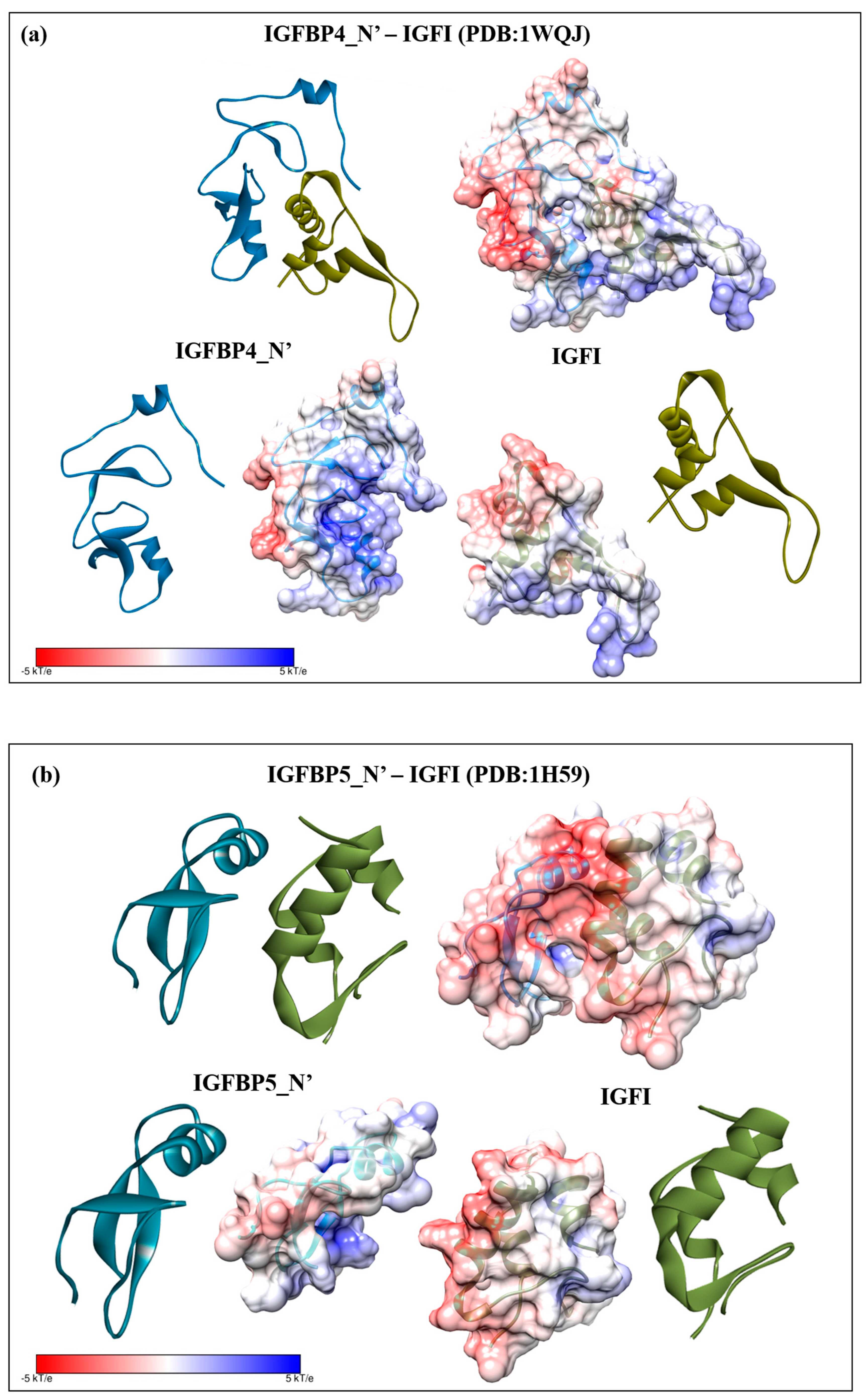
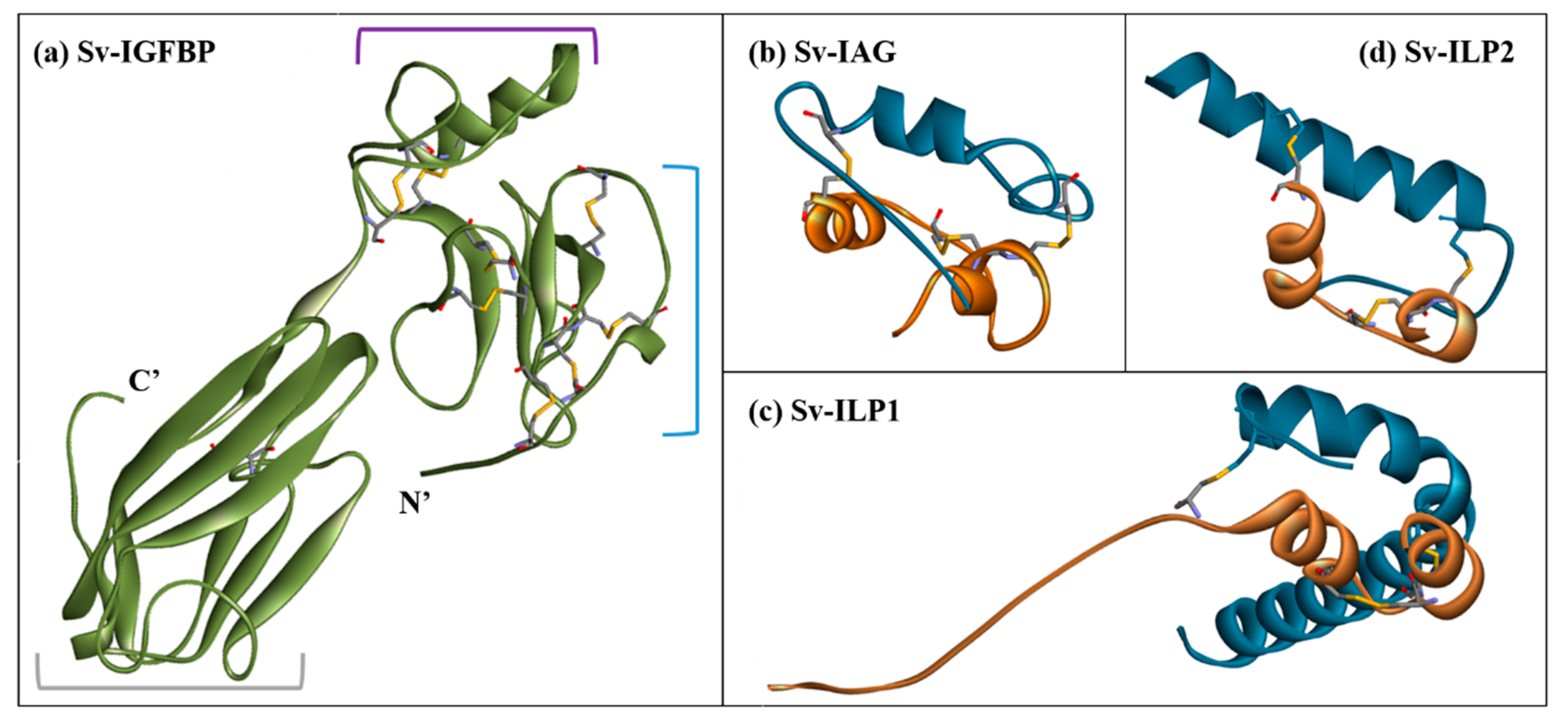
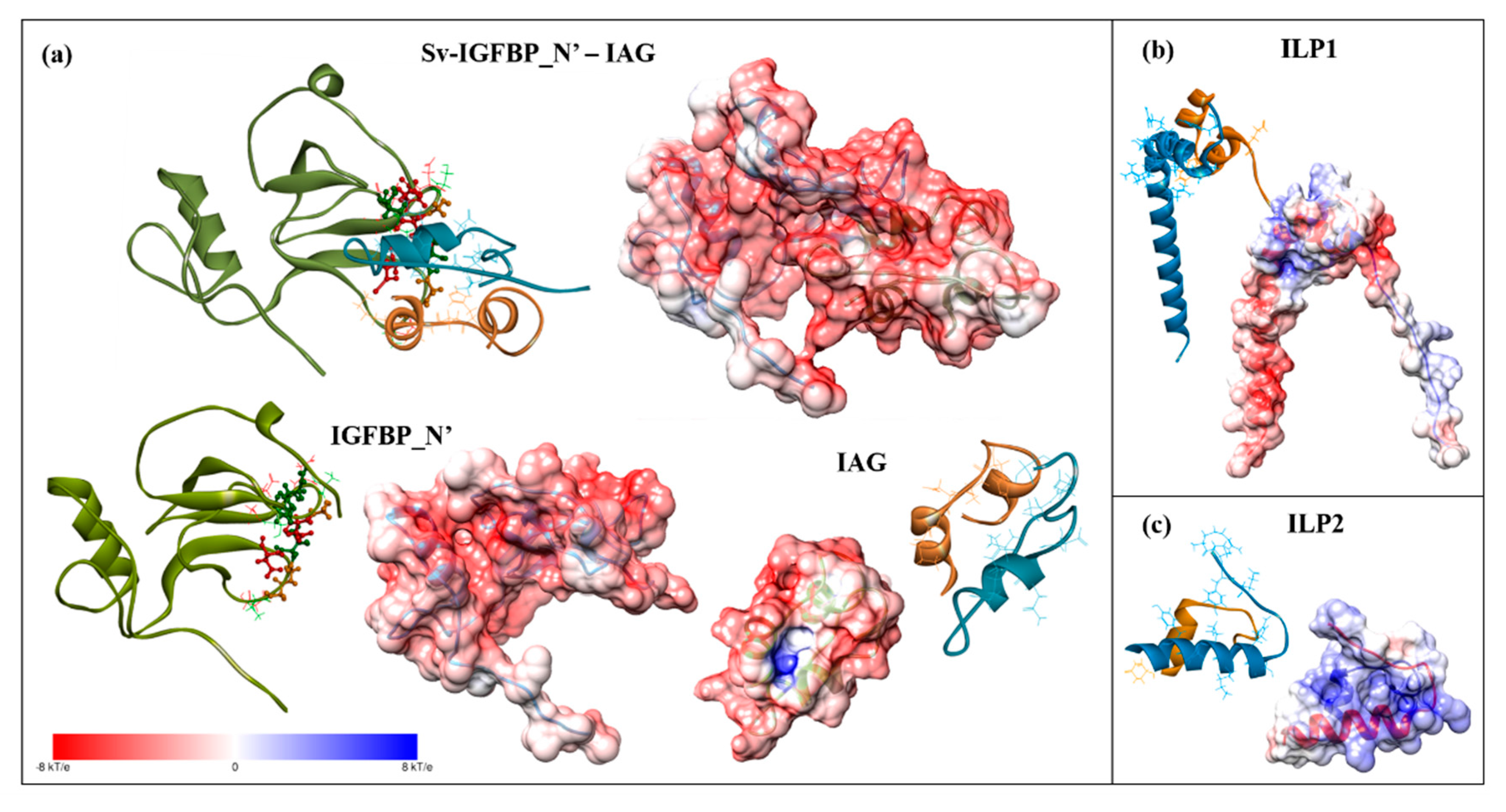
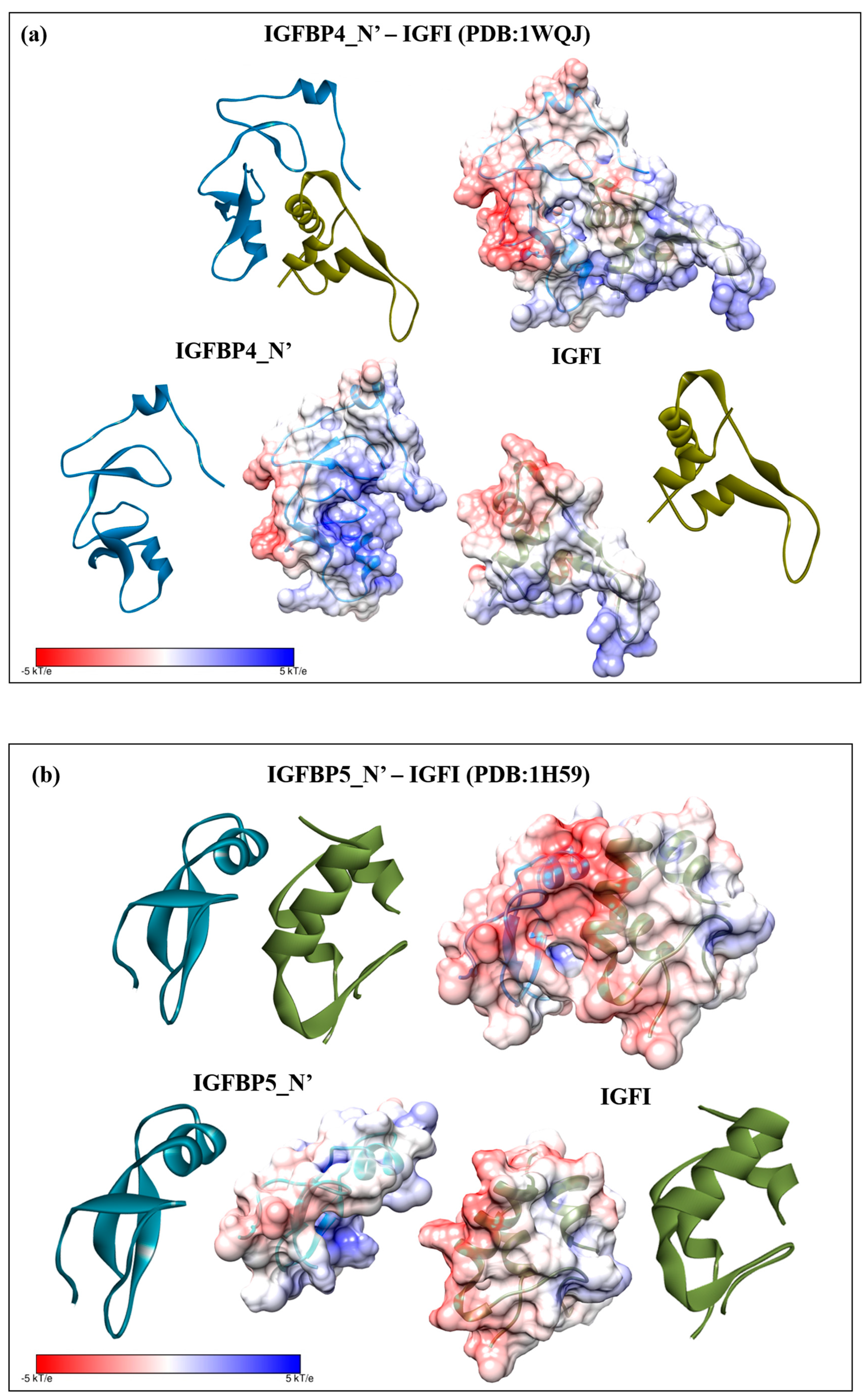
2.3. Structural Modelling
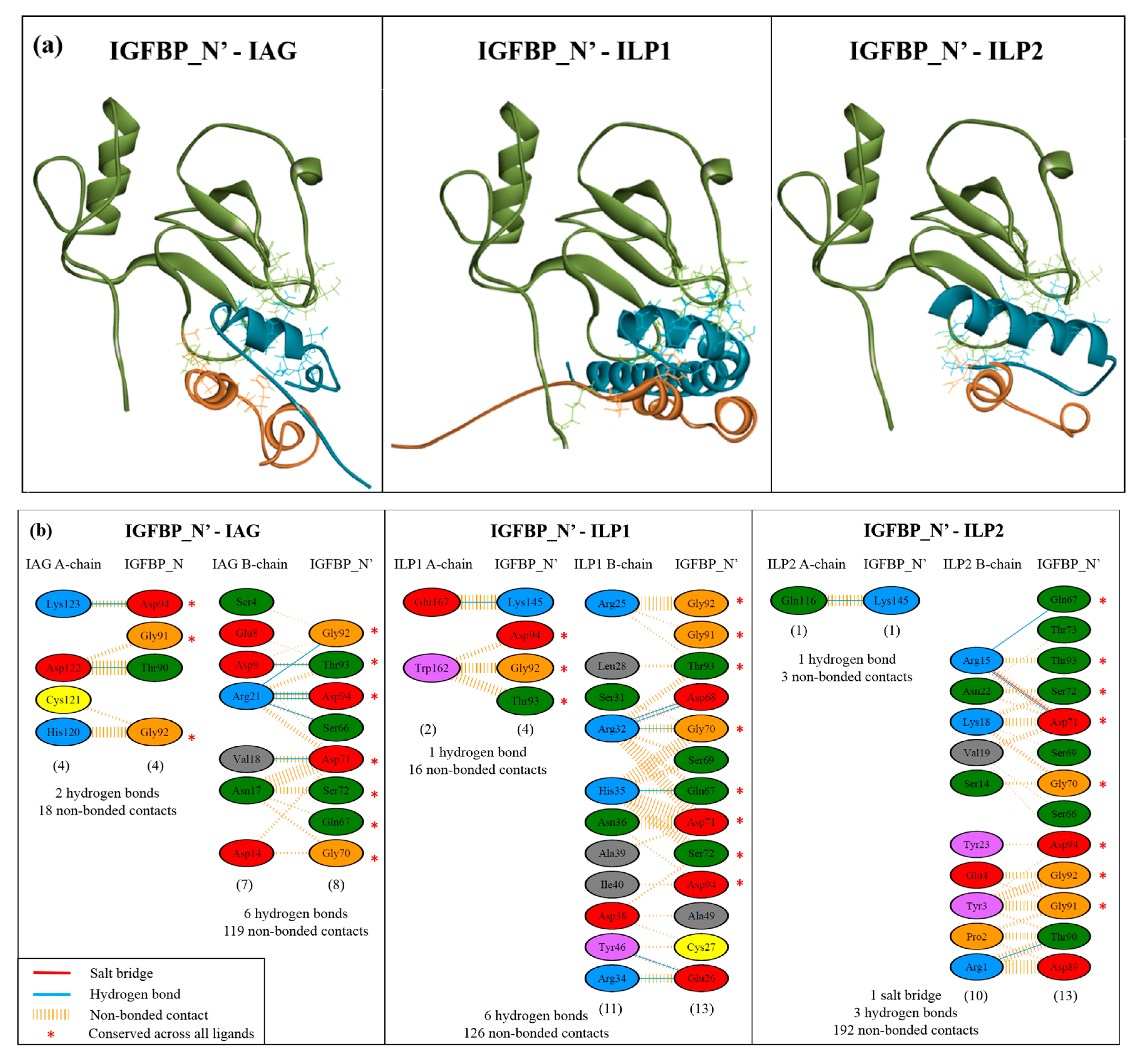
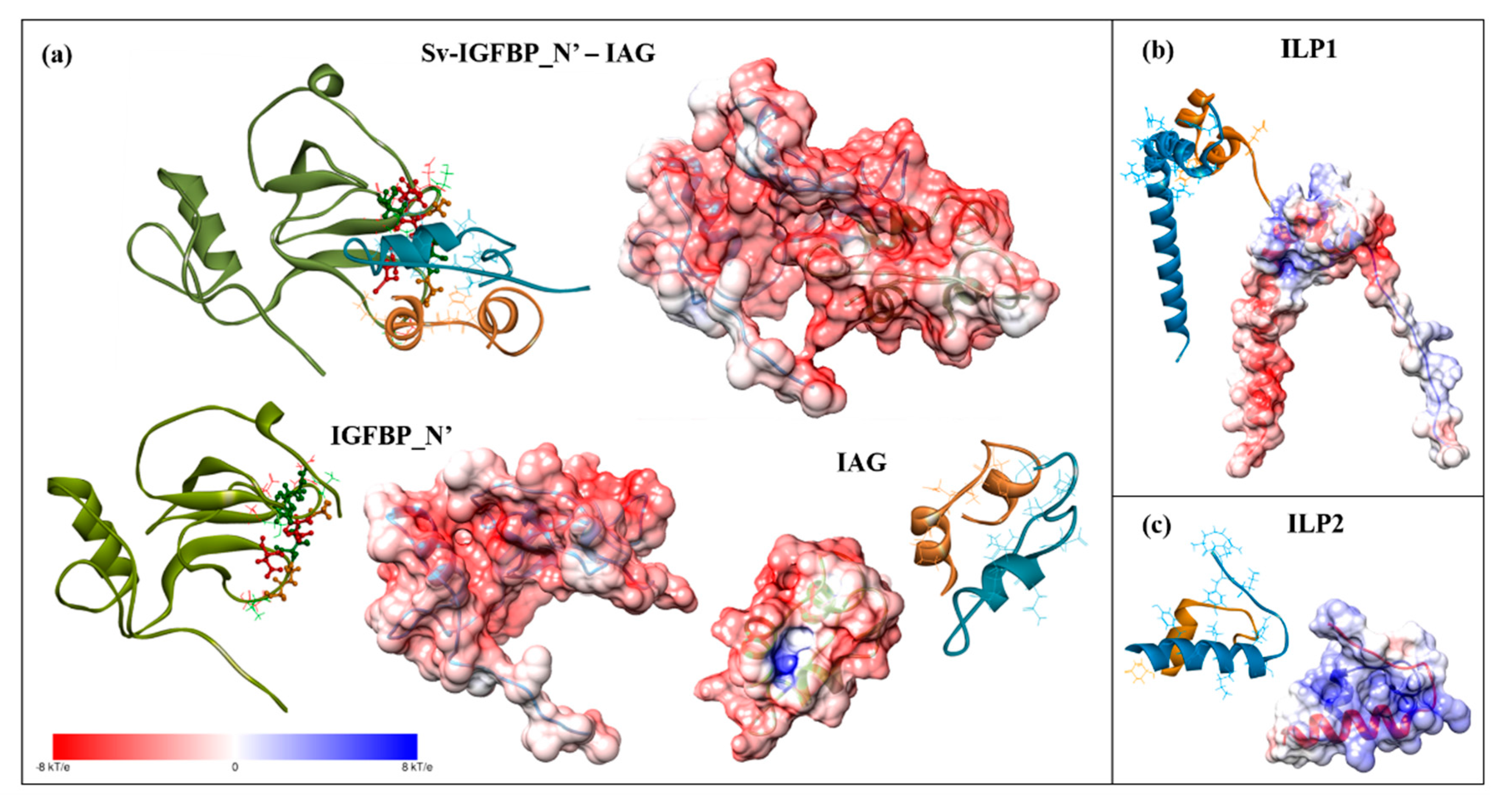
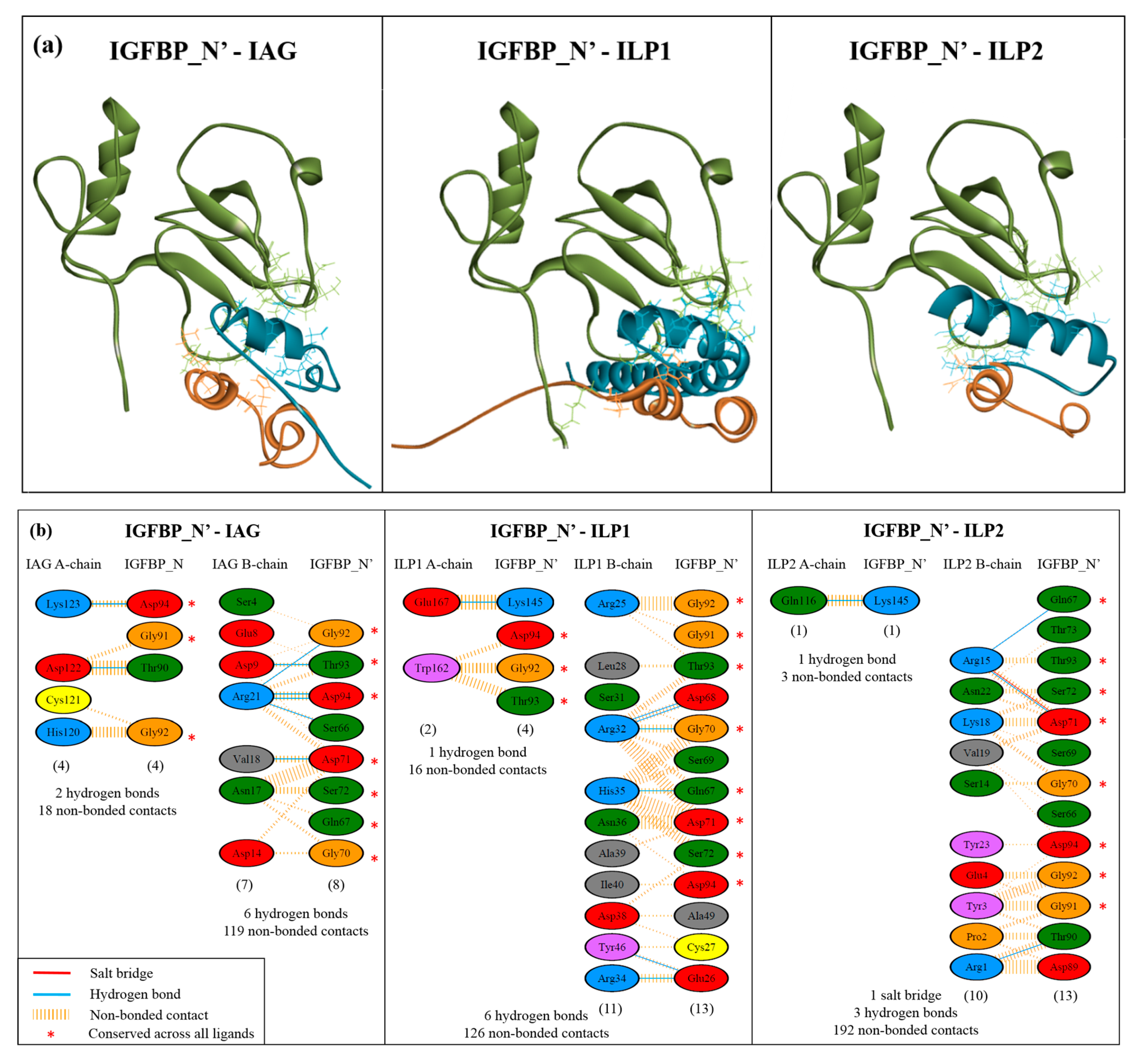
2.4. Complex Formation
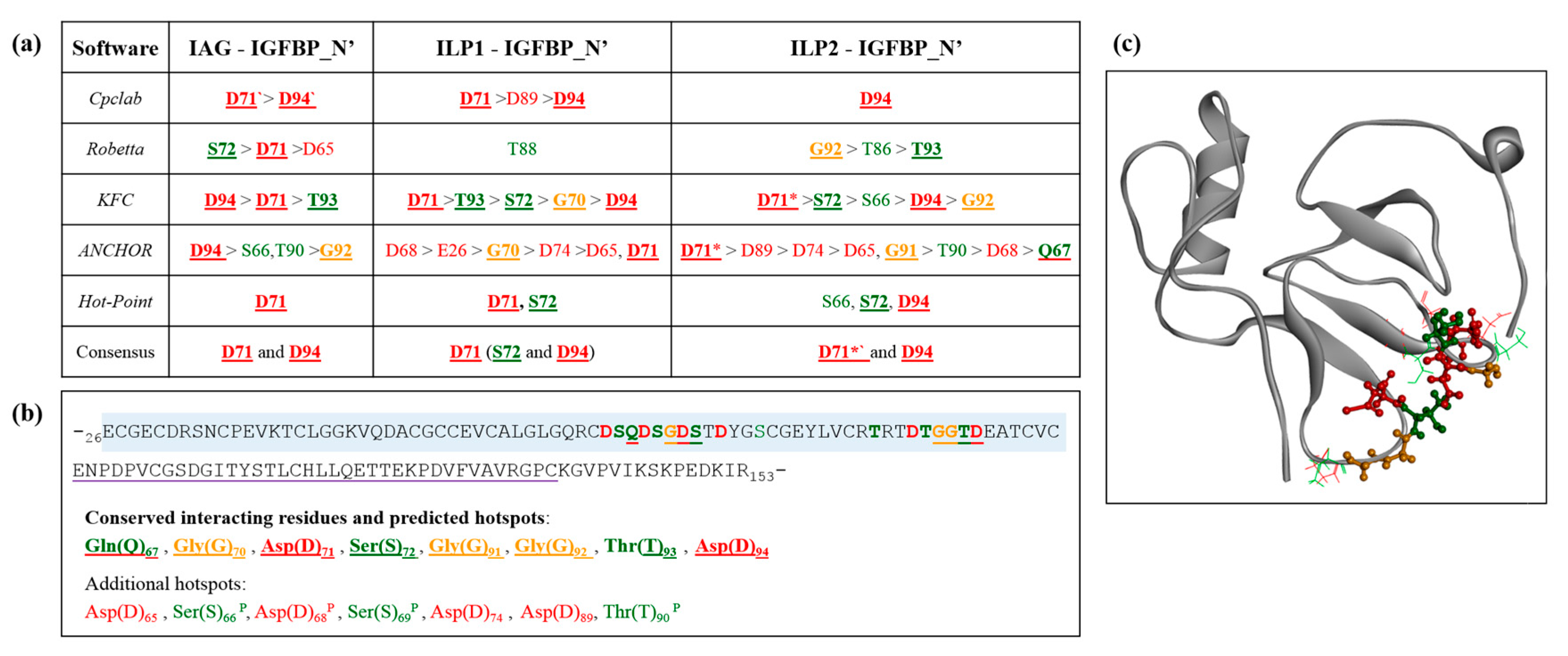
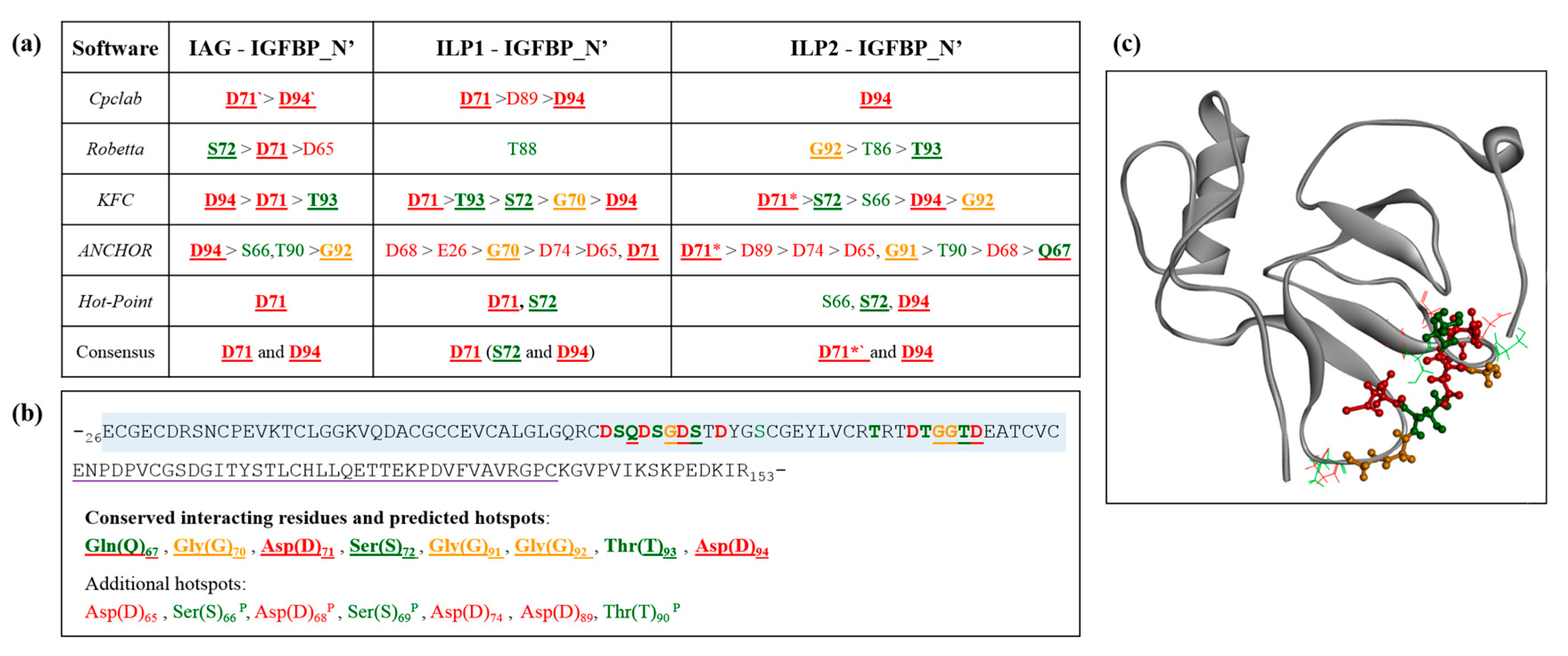
2.5. Interaction Hotspots
2.6. Electrostatic Potential Molecular Surfaces
3. Discussion
4. Materials and Methods
4.1. Sequences
4.2. Protein Structure Modelling
4.3. Molecular Docking and Binding Studies
4.4. Alanine Scanning and Hotspot Residues
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AG | Androgenic gland |
| C’ | C terminal |
| Cq | Cherax quadricarinatus (red-claw crayfish) |
| Dilp7 | Drosophila ILP7 |
| Dilp8 | Drosophila ILP8 |
| IAG | Insulin-like androgenic gland peptide |
| IGF | Insulin-like growth factor |
| IGFBP | Insulin-like growth factor binding protein |
| IGFBP-rP1 | Insulin-like growth factor binding- related protein |
| ILP | Insulin-like peptide |
| N’ | N terminal |
| RPKM | Reads per kilobase per million reads |
| Sv | Sagmariasus verreauxi (Eastern spiny lobster) |
| TKIR | Tyrosine kinase insulin receptor |
References
- Hwa, V.; Oh, Y.; Rosenfeld, R.G. The Insulin-Like Growth Factor-Binding Protein (IGFBP) Superfamily. Endocr. Rev. 1999, 20, 761–787. [Google Scholar] [CrossRef] [PubMed]
- Denley, A.; Cosgrove, L.J.; Booker, G.W.; Wallace, J.C.; Forbes, B.E. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005, 16, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Forbes, B.; McCarthy, P.; Norton, R. Insulin-like growth factor binding proteins: A structural perspective. Front. Endocrinol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Weiss, I.M.; Göhring, W.; Fritz, M.; Mann, K. Perlustrin, a Haliotis laevigata (Abalone) Nacre Protein, Is Homologous to the Insulin-like Growth Factor Binding Protein N-Terminal Module of Vertebrates. Biochem. Biophys. Res. Commun. 2001, 285, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Siwanowicz, I.; Popowicz, G.M.; Wisniewska, M.; Huber, R.; Kuenkele, K.-P.; Lang, K.; Engh, R.A.; Holak, T.A. Structural Basis for the Regulation of Insulin-like Growth Factors by IGF Binding Proteins. Structure 2005, 13, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Sitar, T.; Popowicz, G.M.; Siwanowicz, I.; Huber, R.; Holak, T.A. Structural basis for the inhibition of insulin-like growth factors by insulin-like growth factor-binding proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 13028–13033. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.C.; Aizen, J.; Elizur, A.; Hollander-Cohen, L.; Battaglene, S.C.; Ventura, T. Discovery of a novel insulin-like peptide and insulin binding proteins in the Eastern rock lobster Sagmariasus verreauxi. Gen. Comp. Endocrinol. 2015, 215, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.; Weil, S.; Manor, R.; Roth, Z.; Khalaila, I.; Sagi, A. A crayfish insulin-like-binding protein: Another piece in the androgenic gland insulin-like hormone puzzle is revealed. J. Biol. Chem. 2013, 288, 22289–22298. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Bai, H.; Xiong, Y.; Fu, H.; Jiang, S.; Jiang, F.; Jin, S.; Sun, S.; Qiao, H.; Zhang, W. Molecular characterization of insulin-like androgenic gland hormone-binding protein gene from the oriental river prawn Macrobrachium nipponense and investigation of its transcriptional relationship with the insulin-like androgenic gland hormone gene. Gen. Comp. Endocrinol. 2015, 216, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ye, H.; Feng, B.; Huang, H. Insights into insulin-like peptide system in invertebrates from studies on IGF binding domain-containing proteins in the female mud crab, Scylla paramamosain. Mol. Cell. Endocrinol. 2015, 416, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Bae, S.H.; Bachvaroff, T.R.; Schott, E.J.; Ye, H.; Chung, J.S. Does a blue crab putative insulin-like peptide binding protein (ILPBP) play a role in a virus infection? Fish Shellfish Immunol. 2016, 58, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Nagalla, S.R.; Yamanaka, Y.; Kim, H.-S.; Wilson, E.; Rosenfeld, R.G. Synthesis and Characterization of Insulin-like Growth Factor-binding Protein (IGFBP)-7: Recombinant Human MAC25 protein specifically binds IGF-I and -II. J. Biol. Chem. 1996, 271, 30322–30325. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, Y.; Wilson, E.M.; Rosenfeld, R.G.; Oh, Y. Inhibition of Insulin Receptor Activation by Insulin-like Growth Factor Binding Proteins. J. Biol. Chem. 1997, 272, 30729–30734. [Google Scholar] [CrossRef] [PubMed]
- Akaogi, K.; Sato, J.; Okabe, Y.; Sakamoto, Y.; Yasumitsu, H.; Miyazaki, K. Synergistic growth stimulation of mouse fibroblasts by tumor-derived adhesion factor with insulin-like growth factors and insulin. Cell Growth Differ. 1996, 7, 1671–1677. [Google Scholar] [PubMed]
- Eigenbrot, C.; Ultsch, M.; Lipari, M.T.; Moran, P.; Lin, S.J.; Ganesan, R.; Quan, C.; Tom, J.; Sandoval, W.; van Lookeren Campagne, M.; et al. Structural and Functional Analysis of HtrA1 and its Subdomains. Structure 2012, 20, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, P.; Hohmann, B.; Oh, Y.; Rosenfeld, R.G.; Shymko, R.M. Binding properties of insulin-like growth factor binding protein-3 (IGFBP-3), IGFBP-3 N- and C-terminal fragments, and structurally related proteins MAC25 and connective tissue growth factor measured using a biosensor. Endocrinology 2002, 143, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.E.; Kim, H.-S.; Yang, Y.-F.; Oh, Y. IGF-independent effects of the IGFBP superfamily. In Insulin-Like Growth Factor Receptor Signalling; Leroith, D., Zumkeller, W., Baxter, R.C., Eds.; Plenum Publishers: New York, NY, USA, 2003; pp. 262–274. [Google Scholar]
- Wu, Q.; Brown, M.R. Signaling and function of insulin-like peptides in insects. Annu. Rev. Entomol. 2006, 51, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Brogiolo, W.; Stocker, H.; Ikeya, T.; Rintelen, F.; Fernandez, R.; Hafen, E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 2001, 11, 213–221. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Okamoto, N. Insulin-like and IGF-like peptides in the silkmoth Bombyx mori: Discovery, structure, secretion and function. Front. Physiol. 2013, 4, 217. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.S. An insulin-like growth factor found in hepatopancreas implicates carbohydrate metabolism of the blue crab Callinectes sapidus. Gen. Comp. Endocrinol. 2014, 199, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ye, H.; Huang, H.; Yang, Y.; Gong, J. An insulin-like androgenic gland hormone gene in the mud crab, Scylla paramamosain, extensively expressed and involved in the processes of growth and female reproduction. Gen. Comp. Endocrinol. 2014, 204, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Charniaux-Cotton, H. Discovery in, an amphipod crustacean (Orchestia gammarella) of an endocrine gland responsible for the differentiation of primary and secondary male sex characteristics. C. R. Hebd. Seances Acad. Sci. 1954, 239, 780–782. [Google Scholar] [PubMed]
- Charniaux-Cotton, H. The androgenic gland of some decapodal crustaceans and particularly of Lysmata seticaudata, a species with functional protandrous hermaphroditism. C. R. Hebd. Seances Acad. Sci. 1958, 246, 2814–2817. [Google Scholar] [PubMed]
- Sagi, A.; Snir, E.; Khalaila, I. Sexual differentiation in decapod crustaceans: Role of the androgenic gland. Invertebr. Reprod. Dev. 1997, 31, 55–61. [Google Scholar] [CrossRef]
- Martin, G.; Sorokine, O.; Moniatte, M.; Bulet, P.; Hetru, C.; Van Dorsselaer, A. The structure of a glycosylated protein hormone responsible for sex determination in the isopod, Armadillidium vulgare. Eur. J. Biochem. 1999, 262, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Okuno, A.; Hasegawa, Y.; Ohira, T.; Katakura, Y.; Nagasawa, H. Characterization and cDNA cloning of androgenic gland hormone of the terrestrial isopod Armadillidium vulgare. Biochem. Biophys. Res. Commun. 1999, 264, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Manor, R.; Weil, S.; Oren, S.; Glazer, L.; Aflalo, E.D.; Ventura, T.; Chalifa-Caspi, V.; Lapidot, M.; Sagi, A. Insulin and gender: An insulin-like gene expressed exclusively in the androgenic gland of the male crayfish. Gen. Comp. Endocrinol. 2007, 150, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Manor, R.; Aflalo, E.D.; Weil, S.; Raviv, S.; Glazer, L.; Sagi, A. Temporal silencing of an androgenic gland-specific insulin-like gene affecting phenotypical gender differences and spermatogenesis. Endocrinology 2009, 150, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.; Manor, R.; Weil, S.; Gafni, O.; Linial, A.; Aflalo, E.D.; Ventura, T.; Sagi, A. A sexual shift induced by silencing of a single insulin-like gene in crayfish: Ovarian upregulation and testicular degeneration. PLoS ONE 2010, 5, e15281. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Rosen, O.; Sagi, A. From the discovery of the crustacean androgenic gland to the insulin-like hormone in six decades. Gen. Comp. Endocrinol. 2011, 173, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, J.A. Similarities between decapod and insect neuropeptidomes. Peer J. 2016, 4, e2043. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Nakamori, R.; Murai, T.; Yamauchi, Y.; Masuda, A.; Nishimura, T. A secreted decoy of InR antagonizes insulin/IGF signaling to restrict body growth in Drosophila. Genes Dev. 2013, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Aizen, J.; Chandler, J.C.; Fitzgibbon, Q.P.; Sagi, A.; Battaglene, S.C.; Elizur, A.; Ventura, T. Production of recombinant insulin-like androgenic gland hormones from three decapod species: In vitro testicular phosphorylation and activation of a newly identified tyrosine kinase receptor from the Eastern spiny lobster, Sagmariasus verreauxi. Gen. Comp. Endocrinol. 2016, 229, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, O.; Manor, R.; Weil, S.; Aflalo, E.D.; Lezer, Y.; Levy, T.; Aizen, J.; Ventura, T.; Mather, P.B.; Khalaila, I.; et al. Identification and characterization of an insulin-like receptor involved in crustacean reproduction. Endocrinology 2016, 157, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Redfern, O.; Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 2007, 8, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.L.; Akhtar, S.; Bajpai, P. In silico protein modeling: Possibilities and limitations. EXCLI J. 2014, 13, 513–515. [Google Scholar] [PubMed]
- Ventura, T.; Fitzgibbon, Q.P.; Battaglene, S.C.; Sagi, A.; Elizur, A. Identification and characterization of androgenic gland specific insulin-like peptide-encoding transcripts in two spiny lobster species: Sagmariasus verreauxi and Jasus edwardsii. Gen. Comp. Endocrinol. 2014, 214, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.C.; Aizen, J.; Fitzgibbon, Q.P.; Elizur, A.; Ventura, T. Applying the Power of Transcriptomics: Understanding Male Sexual Development in Decapod Crustacea. Integr. Comp. Biol. 2016, 56, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Moralez, A.; Andag, U.; Clarke, J.B.; Busby, W.H.; Clemmons, D.R. Substitutions for Hydrophobic Amino Acids in the N-terminal Domains of IGFBP-3 and -5 Markedly Reduce IGF-I Binding and Alter Their Biologic Actions. J. Biol. Chem. 2000, 275, 18188–18194. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhang, G.; Dong, F.; Rechler, M.M. Insulin-like Growth Factor (IGF)-binding Protein-3 Mutants That Do Not Bind IGF-I or IGF-II Stimulate Apoptosis in Human Prostate Cancer Cells. J. Biol. Chem. 2002, 277, 10489–10497. [Google Scholar] [CrossRef] [PubMed]
- Żesławski, W.; Beisel, H.-G.; Kamionka, M.; Kalus, W.; Engh, R.A.; Huber, R.; Lang, K.; Holak, T.A. The interaction of insulin-like growth factor-I with the N-terminal domain of IGFBP-5. EMBO J. 2001, 20, 3638–3644. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M.J.J. Contacts-based prediction of binding affinity in protein-protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein—Protein interface determinant amino-acid residues. Proteins Struct. Funct. Bioinf. 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Computational alanine scanning mutagenesis—An improved methodological approach. J. Comput. Chem. 2006, 28, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Kalus, W.; Zweckstetter, M.; Renner, C.; Sanchez, Y.; Georgescu, J.; Grol, M.; Demuth, D.; Schumacher, R.; Dony, C.; Lang, K. Structure of the IGF-binding domain of the insulin-like growth factor-binding protein-5 (IGFBP-5): Implications for IGF and IGF-I receptor interactions. EMBO J. 1998, 17, 6558–6572. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Strong, D.D.; Baylink, D.J.; Mohan, S. Structure-Function Analysis of the Human Insulin-like Growth Factor Binding Protein-4. J. Biol. Chem. 1998, 273, 23509–23516. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Forbes, B.E.; McNeil, K.A.; Baxter, R.C.; Firth, S.M. Role of N- and C-terminal Residues of Insulin-like Growth Factor (IGF)-binding Protein-3 in Regulating IGF Complex Formation and Receptor Activation. J. Biol. Chem. 2004, 279, 53232–53240. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Thompson, J.A.; Bach, L.A. Promotion of cancer cell migration: An insulin-like growth factor (IGF)-independent action of IGF-binding protein-6. J. Biol. Chem. 2007, 282, 22298–22306. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Duan, D.; Zhu, S.; Zhang, J. Investigation of alanine mutations affecting insulin-like growth factor (IGF) I binding to IGF binding proteins. Growth Factors 2015, 33, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Renteria, M.E.; Gandhi, N.S.; Vinuesa, P.; Helmerhorst, E.; Mancera, R.L. A comparative structural bioinformatics analysis of the insulin receptor family ectodomain based on phylogenetic information. PLoS ONE 2008, 3, e3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.M.; Blundell, T.L.; Fernandez-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Manor, R.; Aflalo, E.D.; Weil, S.; Rosen, O.; Sagi, A. Timing sexual differentiation: Full functional sex reversal achieved through silencing of a single insulin-like gene in the prawn, Macrobrachium rosenbergii. Biol. Reprod. 2012, 86, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lezer, Y.; Aflalo, E.D.; Manor, R.; Sharabi, O.; Abilevich, L.K.; Sagi, A. On the safety of RNAi usage in aquaculture: The case of all-male prawn stocks generated through manipulation of the insulin-like androgenic gland hormone. Aquaculture 2015, 435, 157–166. [Google Scholar] [CrossRef]
- Borth, W. Alpha 2-macroglobulin, a multifunctional binding protein with targeting characteristics. FASEB J. 1992, 6, 3345–3353. [Google Scholar] [PubMed]
- Chandler, J.C.; Aizen, J.; Elizur, A.; Battaglene, S.C.; Ventura, T. Male Sexual Development and the Androgenic Gland: Novel Insights through the de novo Assembled Transcriptome of the Eastern Spiny Lobster, Sagmariasus verreauxi. Sex. Dev. 2016, 9, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [PubMed]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Jimenez-Garcia, B.; Fernandez-Recio, J. CCharPPI web server: Computational characterization of protein-protein interactions from structure. Bioinformatics 2015, 31, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.M.; Gohlke, H. DrugScorePPI webserver: Fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 2010, 38, W480–W486. [Google Scholar] [CrossRef] [PubMed]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, 219, pl2. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mitchell, J.C. KFC2: A knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins 2011, 79, 2671–2683. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Gursoy, A.; Keskin, O. HotRegion: A database of predicted hot spot clusters. Nucleic Acids Res. 2012, 40, D829–D833. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandler, J.C.; Gandhi, N.S.; Mancera, R.L.; Smith, G.; Elizur, A.; Ventura, T. Understanding Insulin Endocrinology in Decapod Crustacea: Molecular Modelling Characterization of an Insulin-Binding Protein and Insulin-Like Peptides in the Eastern Spiny Lobster, Sagmariasus verreauxi. Int. J. Mol. Sci. 2017, 18, 1832. https://doi.org/10.3390/ijms18091832
Chandler JC, Gandhi NS, Mancera RL, Smith G, Elizur A, Ventura T. Understanding Insulin Endocrinology in Decapod Crustacea: Molecular Modelling Characterization of an Insulin-Binding Protein and Insulin-Like Peptides in the Eastern Spiny Lobster, Sagmariasus verreauxi. International Journal of Molecular Sciences. 2017; 18(9):1832. https://doi.org/10.3390/ijms18091832
Chicago/Turabian StyleChandler, Jennifer C., Neha S. Gandhi, Ricardo L. Mancera, Greg Smith, Abigail Elizur, and Tomer Ventura. 2017. "Understanding Insulin Endocrinology in Decapod Crustacea: Molecular Modelling Characterization of an Insulin-Binding Protein and Insulin-Like Peptides in the Eastern Spiny Lobster, Sagmariasus verreauxi" International Journal of Molecular Sciences 18, no. 9: 1832. https://doi.org/10.3390/ijms18091832