Contribution of Intrinsic Lactate to Maintenance of Seizure Activity in Neocortical Slices from Patients with Temporal Lobe Epilepsy and in Rat Entorhinal Cortex


Abstract
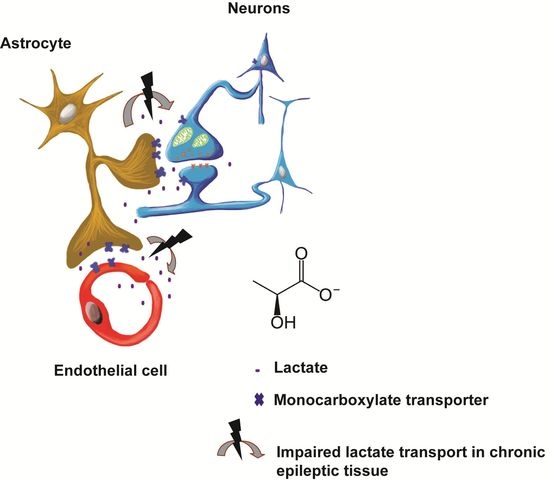
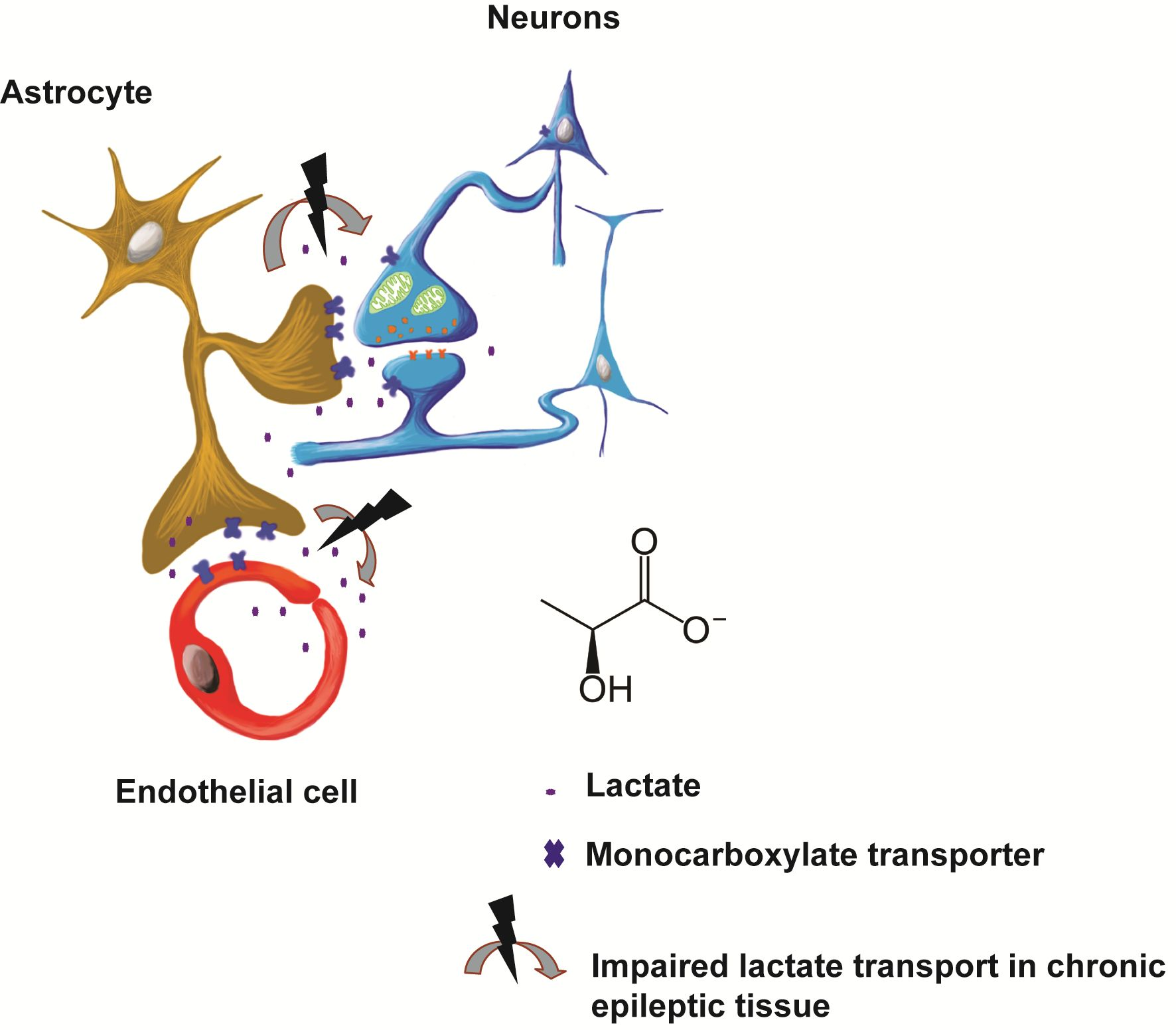
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
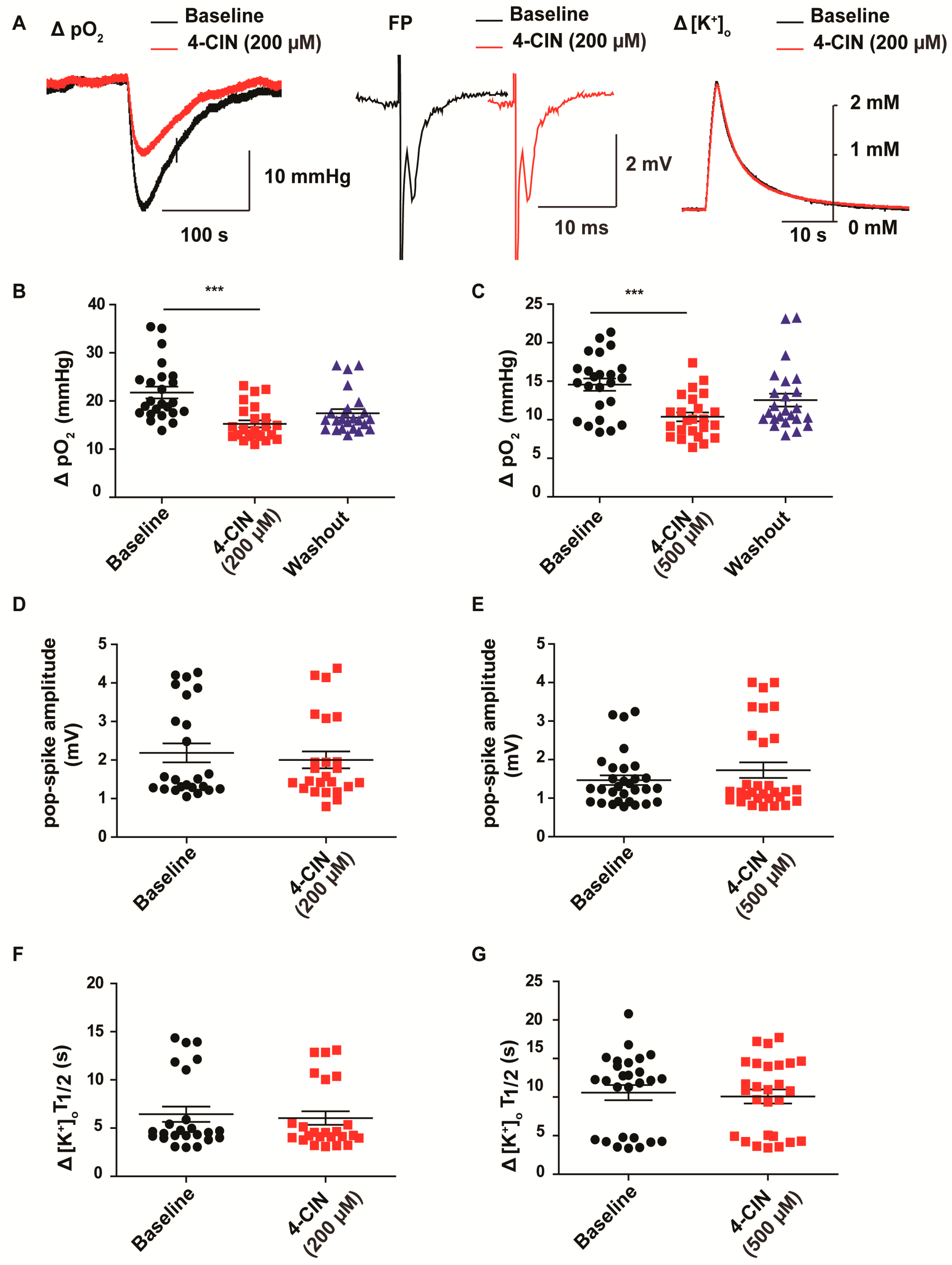
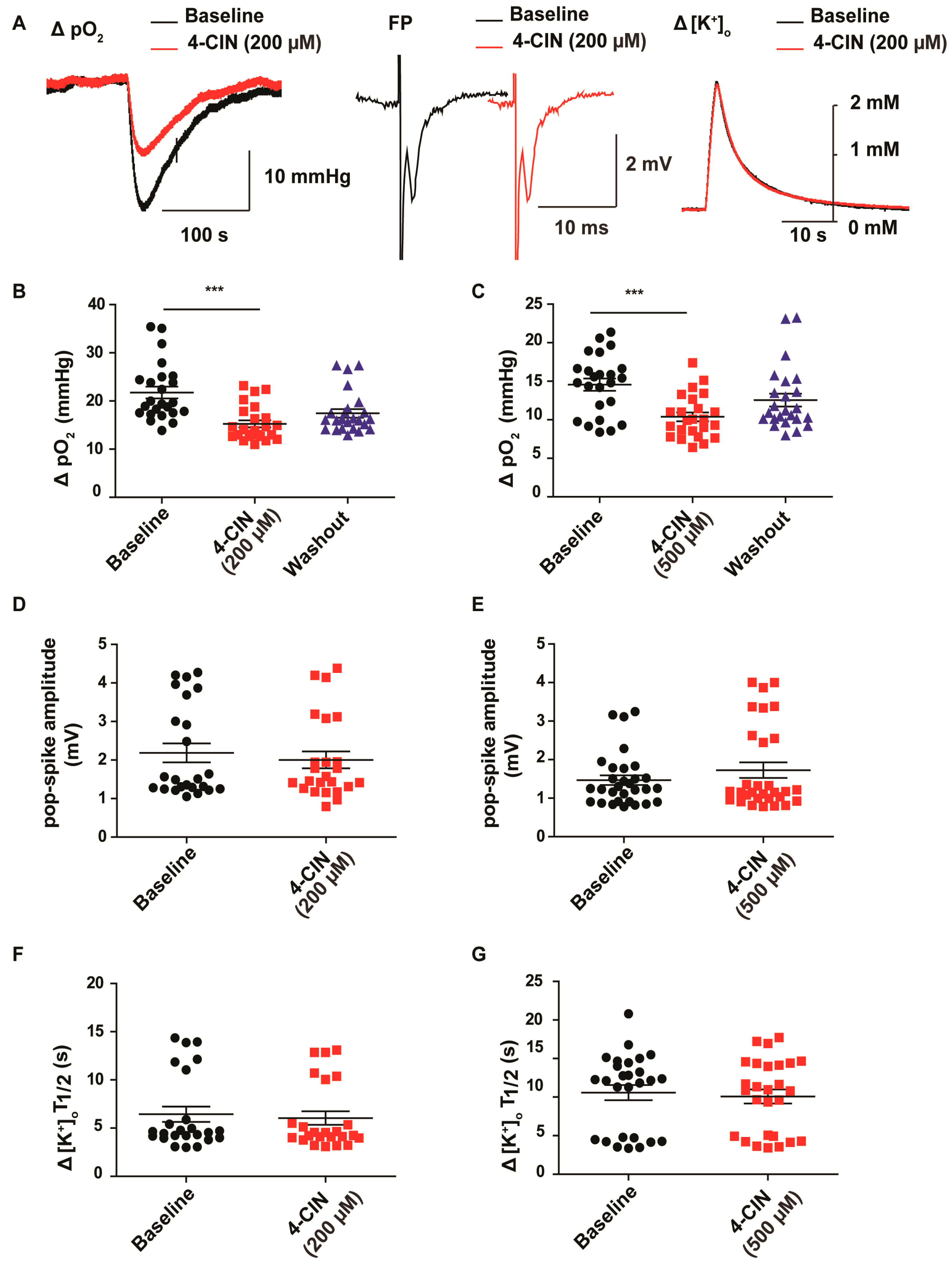
2.1. α-cyano-4-hydroxycinnamic acid (4-CIN) Decreased Stimulus Induced pO2 in Neocortical Slices from Patients with mesial Temporal Lobe Epilepsy (mTLE)
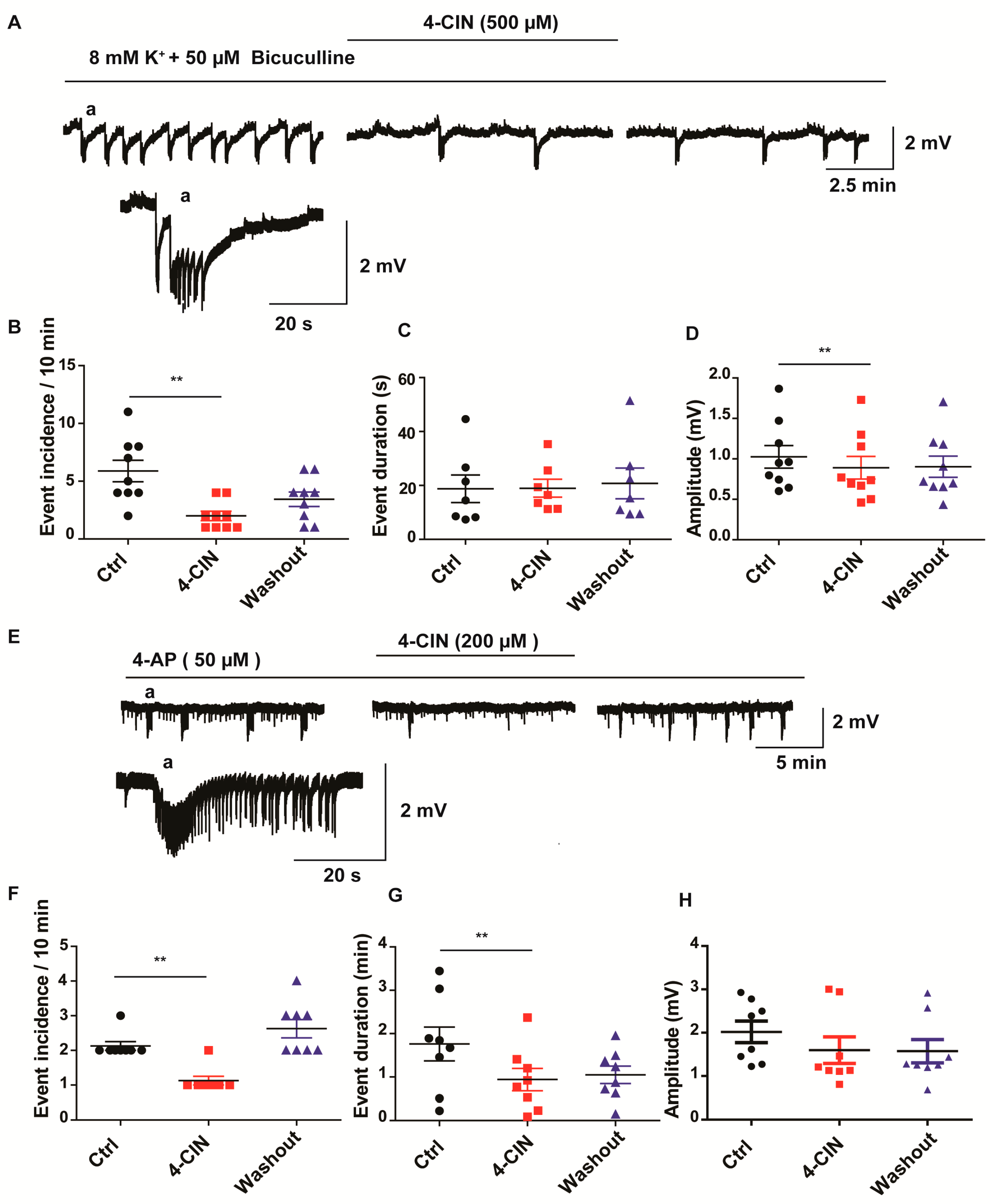
2.2. 4-CIN Reduced Incidence of Seizure-Like Events (SLEs) in Neocortical Slices from Patients with mTLE
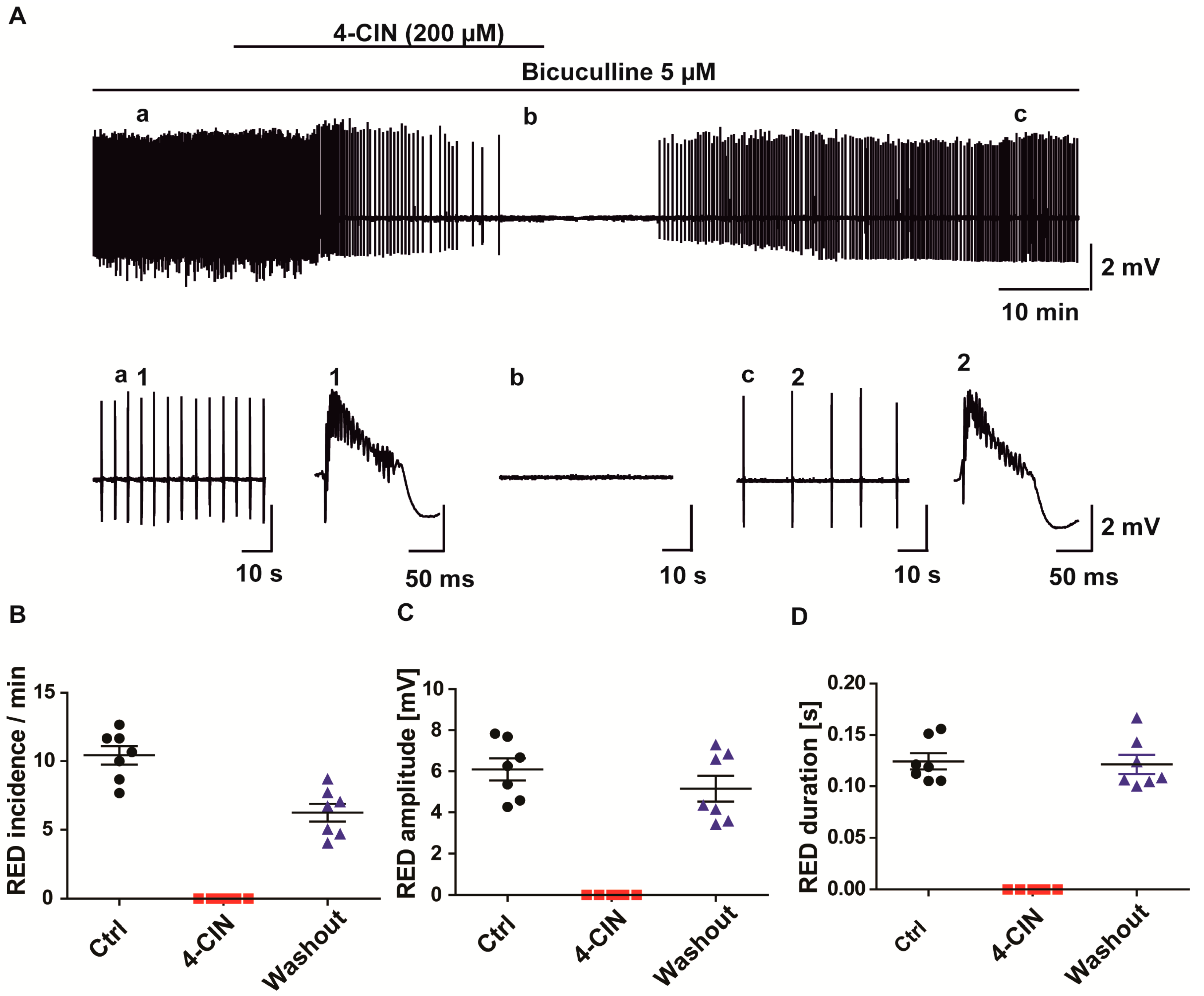
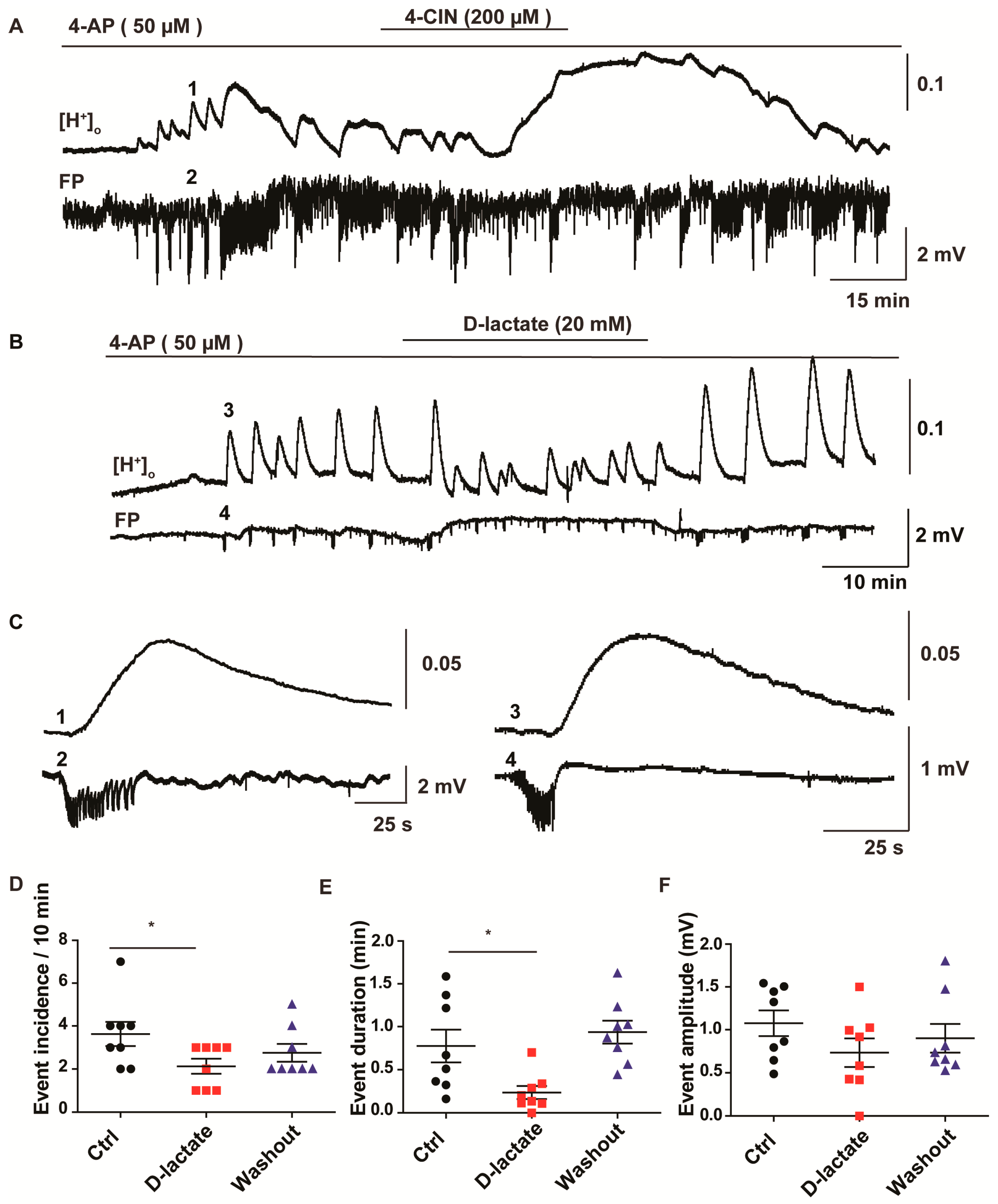
2.3. Lactate Uptake Inhibitors Decreased the Incidence of Pharmacologically Induced Burst Discharges and SLEs in Rat Entorhinal Cortex-Hippocampus Slices
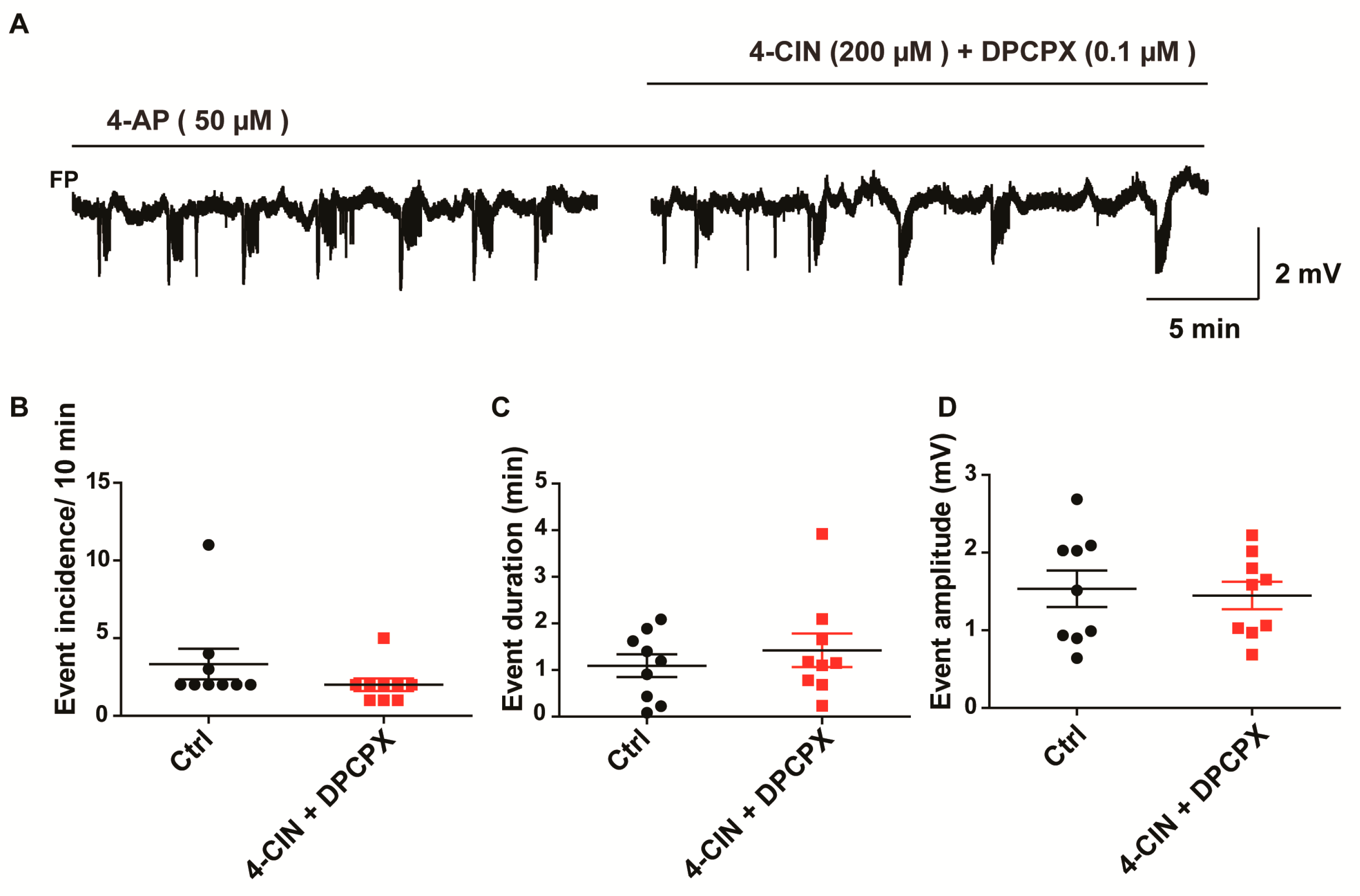
2.4. Anti-Epileptic Effect of 4-CIN Is Mediated by Adenosine through A1 Receptor But Not by Acidosis
3. Discussion
4. Material and Methods
4.1. Slice Preparation
4.2. Electrophysiology and Oxygen Recordings
4.3. Pharmacology
4.4. Data Analysis
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Rahman, B.; Pellegri, G.; Pellerin, L.; Martin, J.L.; Verleysdonk, S.; Hamprecht, B.; Magistretti, P.J. Comparison of lactate transport in astroglial cells and monocarboxylate transporter 1 (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J. Biol. Chem. 1997, 272, 30096–30102. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, A.; Sylantyev, S.; Hadjihambi, A.; Hosford, P.S.; Kasparov, S.; Gourine, A.V. Hemichannel-mediated release of lactate. J. Cereb. Blood Flow Metab. 2015, 36, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Sotelo-Hitschfeld, T.; Niemeyer, M.I.; Machler, P.; Ruminot, I.; Lerchundi, R.; Wyss, M.T.; Stobart, J.; Fernández-Moncada, I.; Valdebenito, R.; Garrido-Gerter, P.; et al. Channel-Mediated Lactate Release by K+-Stimulated Astrocytes. J. Neurosci. 2015, 35, 4168–41478. [Google Scholar] [CrossRef] [PubMed]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Bergersen, L.H.; Halestrap, A.P.; Pierre, K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 2005, 79, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Chih, C.P.; He, J.; Sly, T.S.; Roberts, E.L., Jr. Comparison of glucose and lactate as substrates during NMDA-induced activation of hippocampal slices. Brain Res. 2001, 893, 143–154. [Google Scholar] [CrossRef]
- Yamanishi, S.; Katsumura, K.; Kobayashi, T.; Puro, D.G. Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2005, 290, H925–H934. [Google Scholar] [CrossRef] [PubMed]
- Galow, L.V.; Schneider, J.; Lewen, A.; Ta, T.T.; Papageorgiou, I.E.; Kann, O. Energy substrates that fuel fast neuronal network oscillations. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Galeffi, F.; Foster, K.A.; Sadgrove, M.P.; Beaver, C.J.; Turner, D.A. Lactate uptake contributes to the NAD(P)H biphasic response and tissue oxygen response during synaptic stimulation in area CA1 of rat hippocampal slices. J. Neurochem. 2007, 103, 2449–6241. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Mukhtarov, M.; Bregestovski, P.; Mukhtarov, M.; Bregestovski, P.; Zilberter, Y. Lactate Effectively Covers Energy Demands during Neuronal Network Activity in Neonatal Hippocampal Slices. Front. Neuroenerg. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Takahashi, Y.; Watabe, A.M.; Kubo, Y.; Kato, F. On-site energy supply at synapses through monocarboxylate transporters maintains excitatory synaptic transmission. J. Neurosci. 2014, 34, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Bergersen, L.H.; Magistretti, P.J.; Pellerin, L. Selective Postsynaptic Co-localization of MCT2 with AMPA Receptor GluR2/3 Subunits at Excitatory Synapses Exhibiting AMPA Receptor Trafficking. Cereb. Cortex 2005, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Hyman, S. Blood-brain barrier permeability to small and large molecules. Adv. Drug Deliv. Rev. 1999, 36, 145–163. [Google Scholar] [CrossRef]
- Gerhart, D.Z.; Enerson, B.E.; Zhdankina, O.Y.; Leino, R.L.; Drewes, L.R. Expression of the monocarboxylate transporter MCT2 by rat brain glia. Glia 1998, 22, 272–281. [Google Scholar] [CrossRef]
- Hanu, R.; McKenna, M.; O’Neill, A.; Resneck, W.G.; Bloch, R.J. Monocarboxylic acid transporters, MCT1 and MCT2, in cortical astrocytes in vitro and in vivo. Am. J. Physiol. Cell Physiol. 2000, 278, C921–C930. [Google Scholar] [PubMed]
- Bergersen, L.; Waerhaug, O.; Helm, J.; Thomas, M.; Laake, P.; Davies, A.J.; Wilson, M.C.; Halestrap, A.P.; Ottersen, O.P. A novel postsynaptic density protein: The monocarboxylate transporter MCT2 is co-localized with delta-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp. Brain Res. 2001, 136, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Angamo, E.A.; Rösner, J.; Liotta, A.; Kovács, R.; Heinemann, U. A neuronal lactate uptake inhibitor slows recovery of extracellular ion concentration changes in the hippocampal CA3 region by affecting energy metabolism. J. Neurophysiol. 2016, 116, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Kuhl, D.E.; Phelps, M.E. Patterns of human local cerebral glucose metabolism during epileptic seizures. Science 1982, 218, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Cavus, I.; Kasoff, W.S.; Cassaday, M.P.; Jacob, R.; Gueorguieva, R.; Sherwin, R.S.; Krystal, J.H.; Spencer, D.D.; Abi-Saab, W.M. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol. 2005, 57, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, F.; de Lanerolle, N.C.; Lee, T.S.; Spencer, D.D.; Kim, J.H.; Bergersen, L.H.; Eid, T. Monocarboxylate transporter 1 is deficient on microvessels in the human epileptogenic hippocampus. Neurobiol. Dis. 2011, 41, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, F.; Perez, E.L.; Melillo, E.R.; Roh, J.M.; Zaveri, H.P.; Lee, T.S.; Wang, Y.; Bergersen, L.H.; Eid, T. Altered expression of brain monocarboxylate transporter 1 in models of temporal lobe epilepsy. Neurobiol. Dis. 2012, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Niu, L.; Shen, M.Z.; Gao, L.; Wang, C.; Li, J.; Song, L.J.; Tao, Y.; Meng, Q.; Yang, Q.L.; Gao, G.D.; Zhang, H. Decreased Astroglial Monocarboxylate Transporter 4 Expression in Temporal Lobe Epilepsy. Mol. Neurobiol. 2014, 50, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, F.; Heuser, K.; de Lanerolle, N.C.; Lee, T.S.; Spencer, D.D.; Kim, J.H.; Gjedde, A.; Eid, T.; Bergersen, L.H. Redistribution of monocarboxylate transporter 2 on the surface of astrocytes in the human epileptogenic hippocampus. Glia 2012, 60, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- During, M.J.; Fried, I.; Leone, P.; Katz, A.; Spencer, D.D. Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J. Neurochem. 1994, 62, 2356–2361. [Google Scholar] [CrossRef] [PubMed]
- Antonio, L.L.; Anderson, M.L.; Angamo, E.A.; Gabriel, S.; Klaft, Z.J.; Liotta, A.; Salar, S.; Sandow, N.; Heinemann, U. In vitro seizure like events and changes in ionic concentration. J. Neurosci. Methods 2016, 260, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Liotta, A.; Caliskan, G.; ul Haq, R.; Hollnagel, J.O.; Rösler, A.; Heinemann, U.; Behrens, C.J. Partial Disinhibition Is Required for Transition of Stimulus-Induced Sharp Wave-Ripple Complexes Into Recurrent Epileptiform Discharges in Rat Hippocampal Slices. J. Neurophysiol. 2011, 105, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Sebastião, A.M.; Ribeiro, J.A. Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J. Neurosci. 1998, 18, 1987–1995. [Google Scholar] [PubMed]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, R.; Schuchmann, S.; Gabriel, S.; Kardos, J.; Heinemann, U. Ca2+ signalling and changes of mitochondrial function during low-Mg2+-induced epileptiform activity in organotypic hippocampal slice cultures. Eur. J. Neurosci. 2001, 13, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, R.; Schuchmann, S.; Gabriel, S.; Kann, O.; Kardos, J.; Heinemann, U. Free Radical-Mediated Cell Damage after Experimental Status Epilepticus in Hippocampal Slice Cultures. J. Neurophysiol. 2002, 88, 2909–2918. [Google Scholar] [CrossRef] [PubMed]
- Van Gompel, J.J.; Bower, M.R.; Worrell, G.A.; Stead, M.; Chang, S.Y.; Goerss, S.J.; Kim, I.; Bennet, K.E.; Meyer, F.B.; Marsh, W.R.; et al. Increased cortical extracellular adenosine correlates with seizure termination. Epilepsia 2014, 55, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, G.K.; Cruz, N.F.; Ball, K.K.; Dienel, G.A. Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J. Neurochem. 2009, 111, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R.; Njunting, M.; Behrens, C.J.; Otáhal, J.; Lehmann, T.N.; Gabriel, S.; Heinemann, U. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain 2005, 128, 2396–2407. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blümcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Ziemann, A.E.; Schnizler, M.K.; Albert, G.W.; Severson, M.A.; Howard, M.A.; Welsh, M.J.; Wemmie, J.A. Seizure termination by acidosis depends on ASIC1a. Nat. Neurosci. 2008, 11, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Velísek, L.; Dreier, J.P.; Stanton, P.K.; Heinemann, U.; Moshé, S.L. Lowering of extracellular pH suppresses low-Mg(2+)-induces seizures in combined entorhinal cortex-hippocampal slices. Exp. Brain Res. 1994, 101, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.M.; Dichter, M.; Morad, M. Modulation of the N-methyl-d-aspartate channel by extracellular H+. Proc. Natl. Acad. Sci. USA 1990, 87, 6445–6449. [Google Scholar] [CrossRef] [PubMed]
- Dulla, C.G.; Dobelis, P.; Pearson, T.; Frenguelli, B.G.; Staley, K.J.; Masino, S.A. Adenosine and ATP Link PCO2 to Cortical Excitability via pH. Neuron 2005, 48, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate Receptor Sites Link Neurotransmission, Neurovascular Coupling, and Brain Energy Metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, L.; Puyal, J.; Chatton, J.Y. Lactate Modulates the Activity of Primary Cortical Neurons through a Receptor-Mediated Pathway. PLoS ONE 2013, 8, e71721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- During, M.J.; Spencer, D.D. Adenosine: A potential mediator of seizure arrest and postictal refractoriness. Ann. Neurol. 1992, 32, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Schubert, P.; Heinemann, U. The anticonvulsive action of adenosine: A postsynaptic, dendritic action by a possible endogenous anticonvulsant. Brain Res. 1984, 321, 160–164. [Google Scholar] [CrossRef]
- Dulla, C.G.; Frenguelli, B.G.; Staley, K.J.; Masino, S.A. Intracellular Acidification Causes Adenosine Release During States of Hyperexcitability in the Hippocampus. J. Neurophysiol. 2009, 102, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Frenguelli, B.G.; Wall, M.J. Combined electrophysiological and biosensor approaches to study purinergic regulation of epileptiform activity in cortical tissue. J. Neurosci. Methods 2016, 260, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, B.E.; Shuttleworth, C.W. Adenosine receptor activation is responsible for prolonged depression of synaptic transmission after spreading depolarization in brain slices. Neuroscience 2012, 223, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Jarosch, M.S.; Gebhardt, C.; Fano, S.; Huchzermeyer, C.; Ul Haq, R.; Behrens, C.J. Heinemann U Early adenosine release contributes to hypoxia-induced disruption of stimulus-induced sharp wave-ripple complexes in rat hippocampal area CA3. Eur. J. Neurosci. 2015, 42, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Connor, J.A.; Shuttleworth, C.W. Modulation of the amplitude of NAD(P)H fluorescence transients after synaptic stimulation. J. Neurosci. Res. 2007, 85, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Zurolo, E.; Iyer, A.; de Groot, M.; Anink, J.; Carbonell, C.; van Vliet, E.A.; Baayen, J.C.; Boison, D.; Gorter, J.A. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 2011, 52, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Adenosine and Epilepsy: From Therapeutic Rationale to New Therapeutic Strategies. Neuroscientist 2005, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Klaft, Z.J.; Hollnagel, J.O.; Salar, S.; Calişkan, G.; Schulz, S.B.; Schneider, U.C.; Horn, P.; Koch, A.; Holtkamp, M.; Gabriel, S.; et al. Adenosine A1 receptor-mediated suppression of carbamazepine-resistant seizure-like events in human neocortical slices. Epilepsia 2016, 57, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Roopra, A.; Sutula, T.P. Seizure suppression via glycolysis inhibition with 2-deoxy-d-glucose (2DG). Epilepsia 2008, 49, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.R.; Stafstrom, C.E. Glycolytic inhibition by 2-deoxy-d-glucose abolishes both neuronal and network bursts in an in vitro seizure model. J. Neurophysiol. 2017, 118, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Samokhina, E.; Popova, I.; Malkov, A.; Ivanov, A.I.; Papadia, D.; Osypov, A.; Molchanov, M.; Paskevich, S.; Fisahn, A.; Zilberter, M.; et al. Chronic inhibition of brain glycolysis initiates epileptogenesis. J. Neurosci. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sada, N.; Lee, S.; Katsu, T.; Otsuki, T.; Inoue, T. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015, 347, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Rho, J.M. Anticonvulsant Mechanisms of the Ketogenic Diet. Epilepsia 2007, 48, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Löscher, W.; Rho, J.M. Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb. Perspect. Med. 2016, 6, a022780. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.M.; Love, E.R.; Moorhouse, S.R.; Pratt, O.E. The influence of age on the influx of ketone bodies into the brain of the rat. J. Physiol. 1977, 268, 15P–16P. [Google Scholar] [PubMed]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angamo, E.A.; Ul Haq, R.; Rösner, J.; Gabriel, S.; Gerevich, Z.; Heinemann, U.; Kovács, R. Contribution of Intrinsic Lactate to Maintenance of Seizure Activity in Neocortical Slices from Patients with Temporal Lobe Epilepsy and in Rat Entorhinal Cortex. Int. J. Mol. Sci. 2017, 18, 1835. https://doi.org/10.3390/ijms18091835
Angamo EA, Ul Haq R, Rösner J, Gabriel S, Gerevich Z, Heinemann U, Kovács R. Contribution of Intrinsic Lactate to Maintenance of Seizure Activity in Neocortical Slices from Patients with Temporal Lobe Epilepsy and in Rat Entorhinal Cortex. International Journal of Molecular Sciences. 2017; 18(9):1835. https://doi.org/10.3390/ijms18091835
Chicago/Turabian StyleAngamo, Eskedar Ayele, Rizwan Ul Haq, Jörg Rösner, Siegrun Gabriel, Zoltán Gerevich, Uwe Heinemann, and Richard Kovács. 2017. "Contribution of Intrinsic Lactate to Maintenance of Seizure Activity in Neocortical Slices from Patients with Temporal Lobe Epilepsy and in Rat Entorhinal Cortex" International Journal of Molecular Sciences 18, no. 9: 1835. https://doi.org/10.3390/ijms18091835