IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types


1
Key Laboratory for Neuro-Information of Ministry of Education, School of Life Science and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China
2
Development and Planning Department, Inner Mongolia University, Hohhot 010021, China
3
Department of Physics, School of Sciences, and Center for Genomics and Computational Biology, North China University of Science and Technology, Tangshan 063000, China
4
Department of Pathophysiology, Southwest Medical University, Luzhou 646000, China
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(9), 1838; https://doi.org/10.3390/ijms18091838
Submission received: 7 August 2017
/
Revised: 21 August 2017
/
Accepted: 21 August 2017
/
Published: 24 August 2017
(This article belongs to the Special Issue Special Protein Molecules Computational Identification)
Abstract
:Ion channels (IC) are ion-permeable protein pores located in the lipid membranes of all cells. Different ion channels have unique functions in different biological processes. Due to the rapid development of high-throughput mass spectrometry, proteomic data are rapidly accumulating and provide us an opportunity to systematically investigate and predict ion channels and their types. In this paper, we constructed a support vector machine (SVM)-based model to quickly predict ion channels and their types. By considering the residue sequence information and their physicochemical properties, a novel feature-extracted method which combined dipeptide composition with the physicochemical correlation between two residues was employed. A feature selection strategy was used to improve the performance of the model. Comparison results of in jackknife cross-validation demonstrated that our method was superior to other methods for predicting ion channels and their types. Based on the model, we built a web server called IonchanPred which can be freely accessed from http://lin.uestc.edu.cn/server/IonchanPredv2.0.
1. Introduction
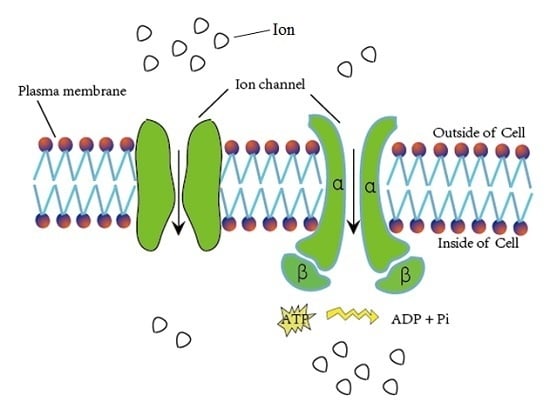
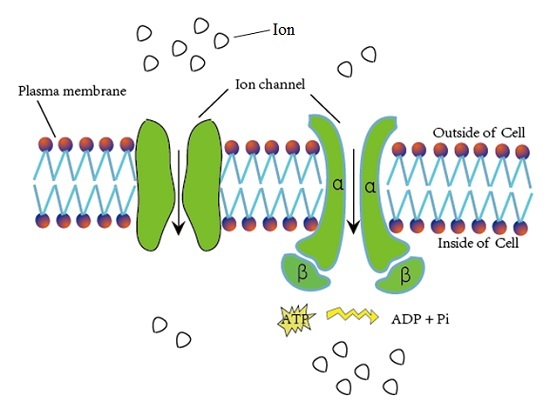
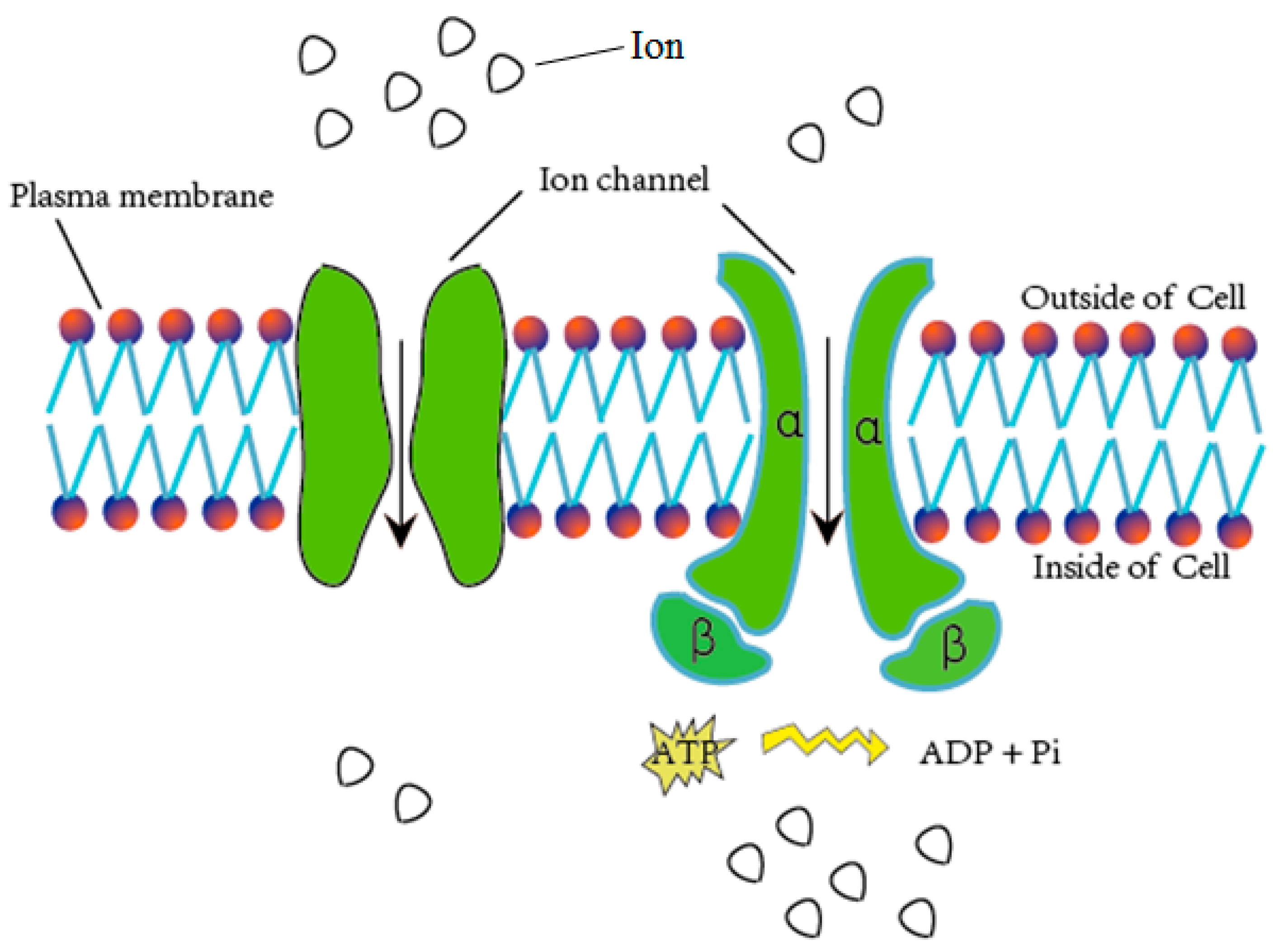
Ion channels are pore-forming membrane proteins for the transmembrane exchange of inorganic ions (as shown in Figure 1). Ion channels exist in the membranes of all cells and are required in numerous physiological and pathological processes, such as regulating neuronal and cardiac excitability, muscle contraction, hormone secretion, fluid movement, and immune cell activation [1]. Due to their important role in biological processes, ion channels are often used as targets for disease diagnosis and drug development. There are over 300 types of ion channels in living cells [2], and they differ in their structure and function. According to the different gating mechanisms, the ion channels can be mainly divided into two categories, namely voltage-gated ion channels (VGIC) and ligand-gated ion channels (LGIC) [3]. The opening and closing of the voltage-gated ion channels depends on the change of the membrane potential, whereas the state of the ligand channels is closely related to the binding of the ligand. The voltage-gated ion channels can be further classified into the following four subclasses: potassium (K+), sodium (Na+), calcium (Ca2+), and anion channels.
In view of the important role and multiple types of ion channels, the structures and functions of ion channels have continued to attract the attention of numerous researchers in recent years [4,5,6,7,8,9,10]. Due to the rapid growth of proteomic data, it is particularly important to develop bioinformatics tools to quickly predict and identify ion channels and their types. Consequently, many computational methods based on machine learning algorithm have been developed in the last 10 years [11,12,13,14,15,16,17]. Liu et al. [11] proposed a method to identify voltage-gated potassium channels, and indicated that the local sequence information-based method was better than the global sequence information-based method. Saha et al. [12] developed a support vector machine (SVM)-based method by using amino acid composition and dipeptide composition to predict voltage-gated ion channels and their subtypes. In 2011, our group [13] developed a more generalized predictive tool, called IonchanPred, and identified ion channels and their types accurately. Recently, Tiwari et al. [16] proposed a random forest based methods and Gao et al. [17] proposed a model to predict ion channels and their subfamilies by combining a SVM-based model with BLAST sequence similarity search. Although many predictors for identifying ion channels are available, three essential issues remain elusive. Firstly, the use of high similarity sequences may overestimate the performance of a model. Secondly, the long-range effect is lost in most published models. Thirdly, web servers should be improved.
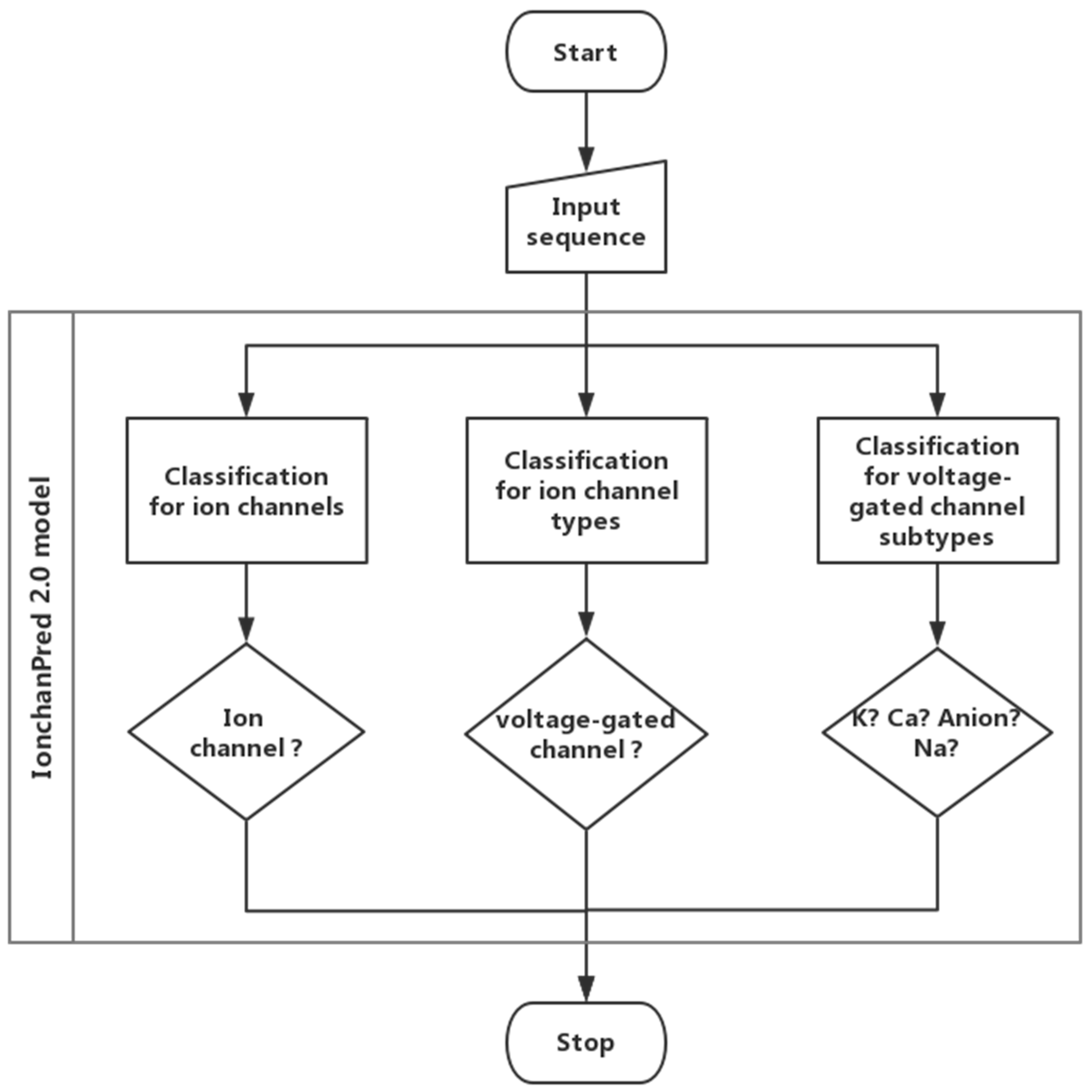
In this paper, a support vector machine-based model was constructed to quickly identify ion channels and their types. In this model, a novel feature extraction method called pseudo-dipeptide composition was employed. The analysis of variance (ANOVA) [18] was introduced to rank features. The incremental feature selection (IFS) was employed to find an optimized feature set which can produce the maximum accuracy. Finally, a web server called IonchanPred 2.0 was established. The flow chart is shown in Figure 2.
2. Results and Discussion
2.1. Parameter Optimization
The establishment of our proposed model depends on two important parameters: and . factor denotes the rank of correlation and the larger may contain more global sequence-order information. represents the weight of the correlation of residues’ physiochemical properties compared to the traditional dipeptide component. To obtain the optimal value for the two parameters, a serial of experiments was performed according to the following standard:
In view of this, a total of individual combinations were obtained. Then, we can investigate the accuracy of SVM with the jackknife test. The optimal parameter combinations corresponding to the three individual datasets are shown in Table 1. It shows that the highest overall accuracy can be up to 87.5% when and for the dataset including ion channels and non-ion channels (NIC). For the benchmark dataset VGIC vs. LGIC, the maximum accuracy is 93.9% when and . The best model for four types of VGIC prediction can produce overall accuracy of 89.1%. After the parameters are optimized, the samples for the three individual datasets can be respectively formulated as follows: a 589-dimensional vector involving 400 dimensions for traditional dipeptide composition and dimensions for correlation information for IC vs. NIC prediction, a vector involving dimensions for VGIC vs. LGIC, and a vector involving dimensions for four types of voltage-gated ion channels datasets.
2.2. Model Establishment
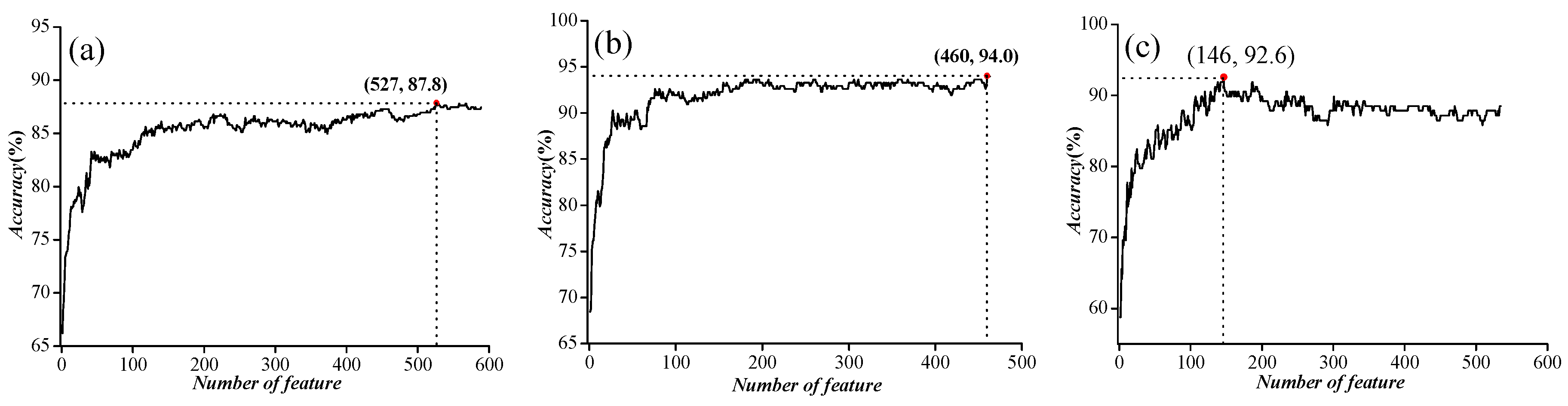
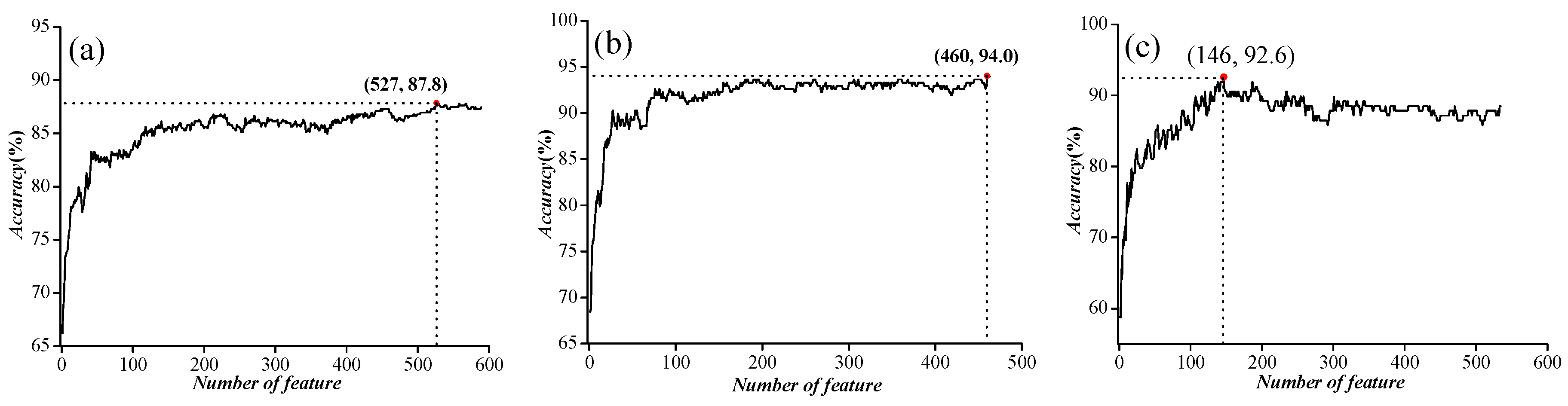
In order to further improve the accuracy, we used ANOVA to exclude noise or redundant information. After the feature selection, the features were sorted according to the decreasing order of the F values described in Section 3.3 Feature Selection to obtain the feature list. Then, we used the IFS to determine the optimal number of features, as described below. The feature subset starts from a feature ranking first in the feature list. A new feature subset was composed when the second feature of this list was added. We repeated this process until all candidate features were added. In this case, we obtained 589, 463, and 535 feature subsets, respectively, for the three benchmark datasets mentioned above. The performance of each feature subset was examined by using SVM with the jackknife test. We plotted the relationship between the overall accuracy and the numbers of features in Figure 3. We noticed that the prediction performances were the best when the top ranked 527, 460, and 147 features were used for the three datasets, respectively.
In order to further evaluate the predictive performance of our model, we also calculated the average accuracies for the three datasets. A comparison of the results with the previous model [13] are shown in Table 2. It is clear that the predictive performance of our proposed model is better than the previous model.
3. Materials and Methods
3.1. Benchmark Databases
The data used to establish the prediction model in this paper were collected from Lin et al. [13]. The sequences of ion channels were collected from the Universal Protein Resource (UniProt) [19] and the Ligand-Gated Ion channel database [20]. To construct a high-quality benchmark dataset, some sequences were removed according to three characteristics. Firstly, a sequence that contained some ambiguous residues (such as “X”, “B”, “Z”). Secondly, a sequence that was the fragment of other proteins. Thirdly, a sequence that was annotated based on homology or prediction. Then, redundant sequences were removed by using the CD-HIT [21] program with a sequence identity threshold of 40%, which has been widely used to filter out redundant samples in genomics and proteomics [22,23,24,25,26].
After the raw data were preprocessed, we finally obtained 298 ion channels including 148 voltage-gated ion channels and 150 ligand-gated ion channels. These voltage-gated ion channels can be classified into four subtypes as follows: 81 potassium (K+), 29 calcium (Ca2+), 12 sodium (Na+), and 26 voltage-gated anion channels. Here, all the 300 non-ion channel proteins were randomly selected from the membrane proteins which were not marked as ion channels in the UniProt database. Moreover, any two sequences in these non-ion channels should guarantee that the identity between them is less than 40%.
3.2. Feature Extraction of Samples
In order to characterize each protein sequence as accurately as possible, the order effect of sequence was usually selected as a method for generating effective feature vectors. Therefore, PseAAC [27,28] incorporating dipeptide composition was selected as the method for feature extraction of protein samples in this paper.
Assuming that there is a protein sequence of L amino acid residues:
where represents the amino acid residue at -th sequence position. Therefore, we can get a set of feature vectors with the dimension of from any sequence like Equation (1)
where the first 400 features represent the effect of the classical dipeptide composition; the elements in addition to the 400 components represent the sequence order effect of protein samples, namely the first tier to -th tier correlation factors of protein sequence. These features can be calculated by:
where is the normalized occurrence frequencies of the 400 dipeptides in protein P; is the weight factor; is the j-tier sequence-correlation factor computed by:
where is the correlation function of physicochemical properties and can be calculated as:
where denotes the value of -th kind physicochemical property of ; is similar. To obtain the high-quality feature set, all the data of physicochemical properties must be subjected to a standard conversion as below:
where represents the 20-native amino acid according to the alphabetical order of their single-letter codes: A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, and Y. denotes the original value of the -th physicochemical property for residue . The values of each physicochemical property obtained after the standard conversion have two advantages. These values will have a zero-mean over the 20 native amino acids and remain unchanged if they are subjected to the same conversion procedure again. The values of the nine kinds of physicochemical properties used in this paper are from previous results [29].
3.3. Feature Selection
Generally, all features do not equally contribute to an ion channel prediction system. Some features make key contributions, whereas some others make minor contributions [30,31]. Therefore, the selection of features is an important step for establishing an effective prediction model. To analyze these feature vectors, ANOVA was used to choose the optimal feature sets in this paper.
In order to assess the contribution of each feature to the predictive system, the F value was defined as follows:
where and respectively denote the sample variance between groups (also called means square between, MSB) and the sample variable within groups (also called means square within, MSW), and are expressed as:
where K and N respectively denote the number of groups and the total number of samples. represents the frequency of the -th feature of the -th sample in the -th group. denotes the total number of samples in the -th group. Thus, each feature corresponds to an F score.
Obviously, the larger F value means the greater contribution of the corresponding feature to the classification. Thus, according to their F values, we may rank all features. Subsequently, we used the incremental feature selection (IFS) to determine the optimal number of features [32]. Firstly, we examined the accuracy of the first feature subset including a feature with the highest F value in the ranked feature set. Secondly, we investigated the accuracy of the second feature subset which was produced by adding the feature with the second highest F value. This process was repeated from the higher F to the lower F value until all candidate features were added. The performances of all feature subsets were evaluated. Then, we were able to obtain the best feature subset which was capable of producing the maximum accuracy.
3.4. Support Vector Machine
SVM is a kind of classification algorithm that can improve the generalization ability of machine learning and achieve the minimization of experience risk and confidence scope by minimizing the structural risk. Therefore, a good statistical result can be usually achieved even using a small sample. SVM, as a powerful supervised learning method, has been widely used in various fields including bioinformatics [33,34,35,36,37,38]. In this paper, we used LIBSVM 3.21 [39] which could be freely downloaded from http://www.csie.ntu.edu.tw/~cjlin/libsvm/. The radial basis function (RBF) kernel was selected as kernel function and one vs. one (OVO) strategy was used for multiclass classification. For achieving the optimal model, the penalty constant C and the kernel width parameter were tuned by an optimization procedure with a grid search method [39]. The search spaces for C and were [] and [] with steps being 2 and, respectively.
3.5. Performance Evaluation
A cross-validation technique is generally employed to estimate the accuracy of a predictive model. Three cross-validation methods including the independent dataset test, subsampling test, and jackknife test can be used [40,41,42,43]. Among them, the jackknife test is considered to be the most objective and rigorous one. Therefore, the jackknife test was employed to assess the performance of our methods.
In addition, we also used other assessment criteria to evaluate the effectiveness of our predictive model in this paper. These assessment criteria, including sensitivity (Sn), overall accuracy (OA), and average accuracy (AA), are defined as follows:
where and respectively denote true positives and false negatives of the -th class. N and n represent the total number of samples and number of classes, respectively.
4. Conclusions
We constructed an SVM-based model for the accurate prediction of ion channel proteins and their types. In this model, a pseudo-dipeptide composition was adopted to extract features. The ANOVA was used to exclude noise or redundant information of feature vectors and then IFS was employed to determine the optimal number of features. High accuracies indicated that the proposed method was an effective tool for predicting ion channels and their types. A free web server based on the proposed method presented in this paper has been constructed and is accessible at the website (http://lin.uestc.edu.cn/server/IonchanPredv2.0).
Acknowledgments
This work was supported by the Applied Basic Research Program of Sichuan Province (No. 2015JY0100 and 14JC0121), the Fundamental Research Funds for the Central Universities of China (Nos. ZYGX2015J144; ZYGX2015Z006; ZYGX2016J118; ZYGX2016J125; ZYGX2016J126), Program for the Top Young Innovative Talents of Higher Learning Institutions of Hebei Province (No. BJ2014028), the Outstanding Youth Foundation of North China University of Science and Technology (No. JP201502), China Postdoctoral Science Foundation (No.2015M582533), and the Scientific Research Foundation of the Education Department of Sichuan Province (11ZB122).
Author Contributions
Hao Lin, Wei Chen, and Hua Tang conceived and designed the experiments; Ya-Wei Zhao performed the experiments; Ya-Wei Zhao analyzed the data; Ya-Wei Zhao and Zhen-Dong Su contributed reagents/materials/analysis tools; Ya-Wei Zhao, Wuritu Yang, and Hao Lin wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wulff, H.; Christophersen, P. Recent developments in ion channel pharmacology. Channels 2015, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Gabashvili, I.S.; Sokolowski, B.H.; Morton, C.C.; Giersch, A.B. Ion channel gene expression in the inner ear. J. Assoc. Res. Otolaryngol. 2007, 8, 305–328. [Google Scholar] [CrossRef] [PubMed]
- Ger, M.F.; Rendon, G.; Tilson, J.L.; Jakobsson, E. Domain-based identification and analysis of glutamate receptor ion channels and their relatives in prokaryotes. PLoS ONE 2010, 5, e12827. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, L.M.; Su, T.; He, N.; Lin, Z.J.; Wang, J.; Shi, Y.W.; Yi, Y.H.; Liao, W.P. Ion Channel Genes and Epilepsy: Functional Alteration, Pathogenic Potential, and Mechanism of Epilepsy. Neurosci. Bull. 2017, 33, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Knutson, K.; Alcaino, C.; Linden, D.R.; Gibbons, S.J.; Kashyap, P.; Grover, M.; Oeckler, R.; Gottlieb, P.A.; Li, H.J.; et al. Mechanosensitive ion channel Piezo2 is important for enterochromaffin cell response to mechanical forces. J. Phys. 2017, 595, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Huang, S.; Meynard, D.; Chaine, C.; Michel, R.; Roelfsema, M.R.G.; Guiderdoni, E.; Sentenac, H.; Very, A.A. A Dual Role for the OsK5.2 Ion Channel in Stomatal Movements and K+ Loading into Xylem Sap. Plant Phys. 2017, 174, 2409–2418. [Google Scholar] [CrossRef] [PubMed]
- Zubcevic, L.; Herzik, M.A., Jr.; Chung, B.C.; Liu, Z.; Lander, G.C.; Lee, S.Y. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 2016, 23, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Linsdell, P. Metal bridges to probe membrane ion channel structure and function. Biomol. Concepts 2015, 6, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Prindle, A.; Liu, J.; Asally, M.; Ly, S.; Garcia-Ojalvo, J.; Suel, G.M. Ion channels enable electrical communication in bacterial communities. Nature 2015, 527, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Li, M.L.; Tan, F.Y.; Lu, M.C.; Wang, K.L.; Guo, Y.Z.; Wen, Z.N.; Jiang, L. Local sequence information-based support vector machine to classify voltage-gated potassium channels. Acta Biochim. Biophys. Sin. 2006, 38, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Zack, J.; Singh, B.; Raghava, G.P. VGIchan: Prediction and classification of voltage-gated ion channels. Genom. Proteom. Bioinform. 2006, 4, 253–258. [Google Scholar] [CrossRef]
- Lin, H.; Ding, H. Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J. Theor. Biol. 2011, 269, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, H. Identification of voltage-gated potassium channel subfamilies from sequence information using support vector machine. Comput. Biol. Med. 2012, 42, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Deng, E.Z.; Chen, W.; Lin, H. Identifying the subfamilies of voltage-gated potassium channels using feature selection technique. Int. J. Mol. Sci. 2014, 15, 12940–12951. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Srivastava, R. An efficient approach for the prediction of ion channels and their subfamilies. Comput. Biol. Chem. 2015, 58, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cui, W.; Sheng, Y.; Ruan, J.; Kurgan, L. PSIONplus: Accurate Sequence-Based Predictor of Ion Channels and Their Types. PLoS ONE 2016, 11, e0152964. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, W.X.; He, J.; Liu, X.H.; Ding, H.; Chen, W. Predicting cancerlectins by the optimal g-gap dipeptides. Sci. Rep. 2015, 5, 16964. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, C. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Donizelli, M.; Djite, M.A.; le Novere, N. LGICdb: A manually curated sequence database after the genomes. Nucleic Acids Res. 2006, 34, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics 2016, 107, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-AI: Identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 2017, 8, 4208–4217. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Mao, Y.; Hu, L.; Wu, Y.; Ji, Z. miRClassify: An advanced web server for miRNA family classification and annotation. Comput. Biol. Med. 2014, 45, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lin, H. Prediction of midbody, centrosome and kinetochore proteins based on gene ontology information. Biochem. Biophys. Res. Commun. 2010, 401, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Lin, H. Prediction of ketoacyl synthase family using reduced amino acid alphabets. J. Ind. Microbiol. Biotechnol. 2012, 39, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Chou, K.C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using Chou′s pseudo amino acid composition with feature selection technique. Mol. BioSyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.W.; Lai, H.Y.; Tang, H.; Chen, W.; Lin, H. Prediction of phosphothreonine sites in human proteins by fusing different features. Sci. Rep. 2016, 6, 34817. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, J.; Wang, X. Protein remote homology detection by combining Chou′s distance-pair pseudo amino acid composition and principal component analysis. Mol. Genet. Genom. 2015, 290, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Feng, P.M.; Chen, W.; Lin, H. Identification of bacteriophage virion proteins by the ANOVA feature selection and analysis. Mol. BioSyst. 2014, 10, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Ju, Y.; Zou, Q. Prediction of G-protein-coupled receptors with SVM-Prot features and random forest. Scientifica 2016, 2016, 8309253. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ju, Y.; Zou, Q. Protein Folds Prediction with Hierarchical Structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.-C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Chen, X.X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169–28175. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines. Acm Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Chou, K.C.; Zhang, C.T. Prediction Of Protein Structural Classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, H.; Wang, X.; Chou, K.-C. Pse-Analysis a python package for DNA, RNA and protein peptide sequence analysis based on pseudo components and kernel methods. Oncotarget 2017, 8, 13338–13343. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liang, Z.Y.; Tang, H.; Chen, W. Identifying σ70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Tang, H.; Li, W.C.; Lin, H.; Chen, W.; Chou, K.C. iOri-Human: Identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 2016, 7, 69783–69793. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of material exchange through ion channels.

Figure 2.
Workflow of the IonchanPred 2.0 model.
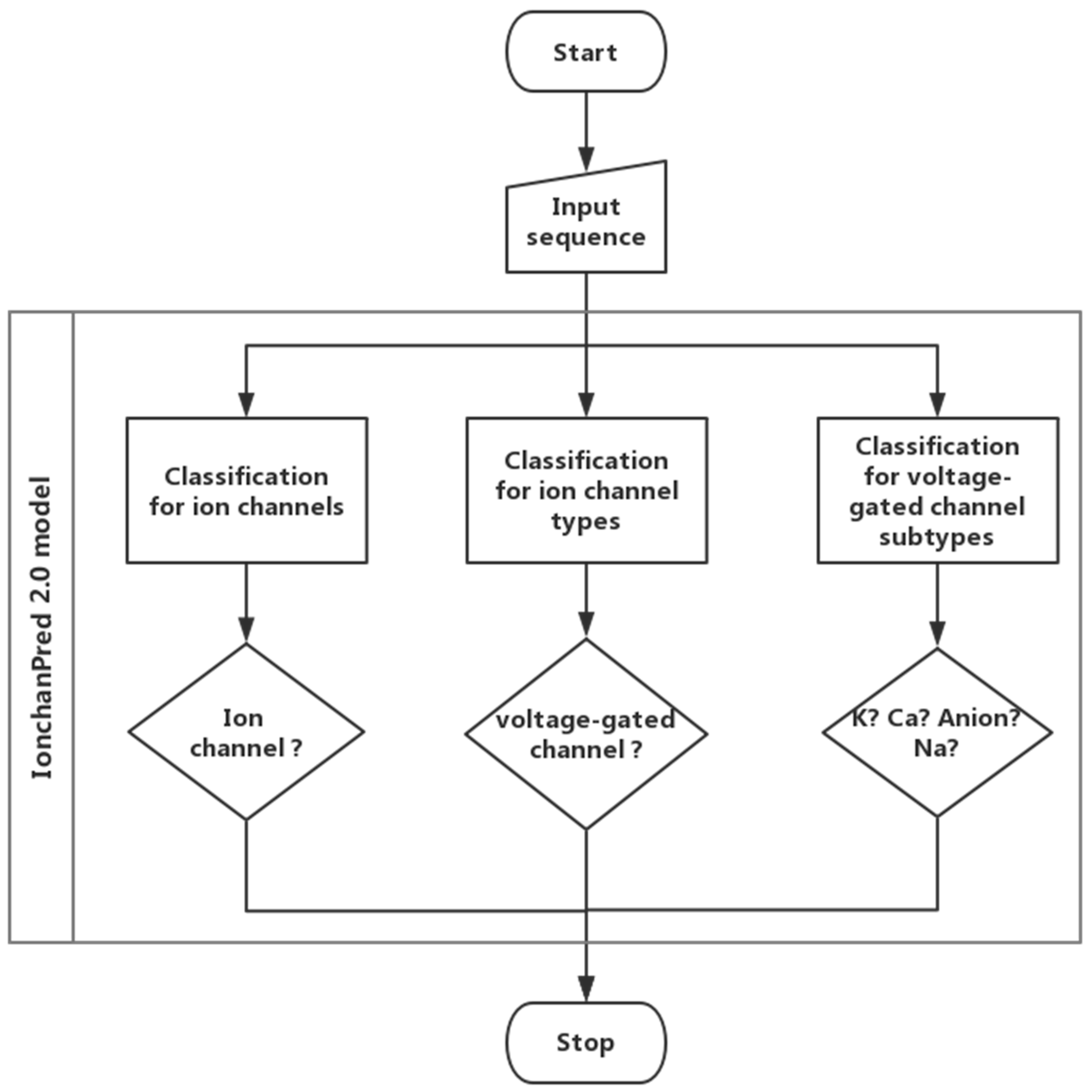
Figure 3.
The feature selection results for three independent datasets. (a) Incremental feature selection (IFS) curve for ion channel (IC) vs. non-ion channel (NIC) prediction; (b) IFS curve for voltage-gated ion channels (VGIC) vs. ligand-gated ion channels (LGIC) prediction; (c) IFS curve for four types of VGIC prediction.
Figure 3.
The feature selection results for three independent datasets. (a) Incremental feature selection (IFS) curve for ion channel (IC) vs. non-ion channel (NIC) prediction; (b) IFS curve for voltage-gated ion channels (VGIC) vs. ligand-gated ion channels (LGIC) prediction; (c) IFS curve for four types of VGIC prediction.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimal parameters for the three datasets.
| Database | λ | ω | OA (%) |
|---|---|---|---|
| IC vs. NIC | 21 | 0.20 | 87.5 |
| VGIC vs. LGIC | 7 | 0.30 | 93.9 |
| four types of VGIC | 9 | 0.15 | 89.1 |
IC: ion channels; NIC: non-ion channels; VGIC: voltage-gated ion channels; LGIC: ligand-gated ion channels; OA: overall accuracy.
Table 2.
Performance evaluation parameters of our proposed model and a previous model.
| Datasets | Our Model | Previous Model [13] | |||||
|---|---|---|---|---|---|---|---|
| Sn | OA | AA | Sn | OA | AA | ||
| IC vs. NIC | IC | 80.2 | 87.8 | 87.8 | 85.9 | 86.6 | 86.6 |
| NIC | 95.3 | 87.3 | |||||
| VGIC vs. LGIC | VGIC | 94.7 | 94.0 | 94.0 | 94.6 | 92.6 | 92.7 |
| LGIC | 93.2 | 90.7 | |||||
| Types of VGIC | K+ | 97.5 | 92.6 | 87.7 | 92.6 | 87.8 | 83.7 |
| Ca2+ | 89.7 | 82.8 | |||||
| Na+ | 75.0 | 75.0 | |||||
| An− | 88.5 | 84.6 | |||||
Sn: sensitivity; AA: average accuracy; OA: overall accuracy; IC: ion channels; NIC: non-ion channels; VGIC: voltage-gated ion channels; LGIC: ligand-gated ion channels.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhao, Y.-W.; Su, Z.-D.; Yang, W.; Lin, H.; Chen, W.; Tang, H. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. Int. J. Mol. Sci. 2017, 18, 1838. https://doi.org/10.3390/ijms18091838
AMA Style
Zhao Y-W, Su Z-D, Yang W, Lin H, Chen W, Tang H. IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types. International Journal of Molecular Sciences. 2017; 18(9):1838. https://doi.org/10.3390/ijms18091838
Chicago/Turabian StyleZhao, Ya-Wei, Zhen-Dong Su, Wuritu Yang, Hao Lin, Wei Chen, and Hua Tang. 2017. "IonchanPred 2.0: A Tool to Predict Ion Channels and Their Types" International Journal of Molecular Sciences 18, no. 9: 1838. https://doi.org/10.3390/ijms18091838
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.