1. Introduction
Aberrant activation of c-Met, a receptor tyrosine kinase, is frequently observed in various cancer types via a hepatocyte growth factor (HGF)-dependent or -independent manner [
1,
2,
3,
4]. c-Met activation via its ligand, HGF, promotes various tumorigenic potentials including malignant progression, angiogenesis, mesenchymal–epithelial transition (MET), invasion, and metastasis [
5,
6,
7]. In addition to HGF-induced c-Met activation, the c-Met pathway is also stimulated through HGF-independent mechanisms such as c-Met transcriptional upregulation, genomic amplifications, mutations, and structural variations including gene fusions [
1,
2]. c-Met amplification and constitutive kinase activation have been reported in a number of human primary tumors including gastric cancer and non-small cell lung cancer (NSCLC) [
8,
9]. Moreover, elevated HGF expression levels and overexpression of c-Met are often associated with dismal prognosis due to highly aggressive tumor behaviors and increased tumor metastasis [
1,
2,
10,
11]. Based on these characteristics, c-Met and HGF have been considered as ideal therapeutic targets in cancer-specific treatments; thus, numerous pre-clinical studies targeting the HGF/c-Met axis have been reported [
12,
13,
14].
HGF/c-Met axis targeting therapeutic antibodies have been developed to prevent molecular interaction between c-Met and its ligand HGF or to inactivate c-Met via protein degradation or shedding (release from the cell surface) [
13,
14]. Rilotumumab (AMG102) and ficlatuzumab (AV-299) are HGF-neutralizing antibodies that only inhibit HGF-dependent c-Met activation by disrupting the HGF/c-Met interaction [
15,
16,
17]. DN30 is a bivalent c-Met-targeted antibody that has been developed to deplete c-Met by shedding (proteolytic cleavage) the extracellular domain of c-Met, but an interaction between DN30 and c-Met results in the activation of the c-Met signaling pathway, inducing c-Met receptor homodimerization [
18,
19]. Bivalent c-Met-targeted antibodies impairing both HGF-dependent and independent activation rendered disappointing results, as their mechanisms could potentially induce agonistic effects via receptor dimerization [
20]. Through extensive research and development to minimize the agonist activity of c-Met antibodies, onartuzumab (MetMab, formerly huOA5D5.v2), emibetuzumab (LY2875358), and SAIT301 were developed. Onartuzumab is an engineered monovalent c-Met antibody that was developed to prevent agonist activity with knob-into-hole technology, which allows the antibody to directly interact with c-Met in a one-on-one fashion [
21,
22]. Emibetuzumab and SAIT301 are humanized bivalent anti-c-Met monoclonal antibodies that were generated to prevent HGF from binding to c-Met and to induce antibody internalization, subsequently depleting c-Met receptors from the cell surface without inducing functional agonist activity [
23,
24]. However, clinical trials of onartuzumab were terminated after the drug failed to exhibit tumor inhibitory effect in NSCLC [
25]. Other c-Met-targeting antibodies also failed to portray any favorable clinical outcomes.
Heterotopic or orthotopic tumor mouse models have been used extensively for the pre-clinical evaluation of human c-Met-specific therapeutic antibodies [
15,
16,
17,
18,
19,
20,
21,
22,
23,
24]. However, a precise assessment of the therapeutic efficacy of anti-human c-Met antibodies in mouse xenograft models could be hampered, as the reactivity of antibodies is restricted to the engrafted human tumor cells. Consistently, the abovementioned c-Met antibodies bind specifically to human c-Met, but do not engage with the mouse c-Met ortholog of mouse normal tissues [
18,
19,
21,
22,
23,
24], thus resulting in misleading interpretations in pre-clinical mouse models [
26]. Previous reports have shown cross-reactive therapeutic antibodies to both the human protein, and mouse ortholog exhibited more accurate tumor inhibitory effects in mouse models, suggesting the necessity of developing cross-reactive antibodies [
27,
28,
29].
In an effort to overcome this potential problem, we developed IRCR201—a novel human and mouse cross-reactive c-Met-targeting antibody. IRCR201 is a bivalent fully-human antibody that binds to the plexin-semaphorin-integrin (PSI) domain of c-Met and exhibits a tumor inhibitory effect by regulating tumor cellular growth, proving to have ideal effects in cancer treatment. IRCR201 also demonstrates low agonistic activity and disrupts c-Met signaling pathway activation induced by both HGF-dependent and independent mechanisms in various cancer types, eliciting distinct molecular traits compared to other c-Met-targeting antibodies. In addition, IRCR201 provides exceptional anti-tumor activity in both gastric cancer and NSCLC xenograft models. Collectively, our results demonstrate that targeting the PSI domain of c-Met provides a promising therapeutic approach, and its human and mouse cross-reactivity allows successful preclinical studies to assess precise therapeutic efficacy, pharmacokinetics (PK), and toxicity profiles, resulting in the further development of IRCR201 as an ideal cancer-targeted therapeutic agent.
3. Discussion
Aberrant activation of the HGF/c-Met signaling pathway has been reported in numerous human cancers [
1,
2]. In c-Met dysregulated malignancy, tumor progression is facilitated largely by three mechanisms: ligand-dependent c-Met activation; genomic amplification; and oncogenic mutations [
5,
6,
7,
8,
9]. Additionally, c-Met amplification has been associated with acquired resistance to epidermal growth factor receptor (EGFR)- and vascular endothelial growth factor (VEGF)-targeted therapies [
39,
40]. Therefore, targeting the HGF/c-Met signaling axis could offer a promising therapeutic approach for the treatment of c-Met-expressing tumors. In the present study, we demonstrated the development and characterization of IRCR201—novel fully-human anti-c-Met IgG1 which specifically binds to a distinct epitope on c-Met and effectively disrupts the c-Met signaling pathway. In contrast to previously developed c-Met-targeting antibodies with lack of mouse cross-reactivity, IRCR201 binds to both human and mouse c-Met with high affinity. Cross-reactivity with the mouse c-Met ortholog enables the precise evaluation of the tumor inhibitory efficacy of IRCR201 in mouse xenograft models during preclinical studies.
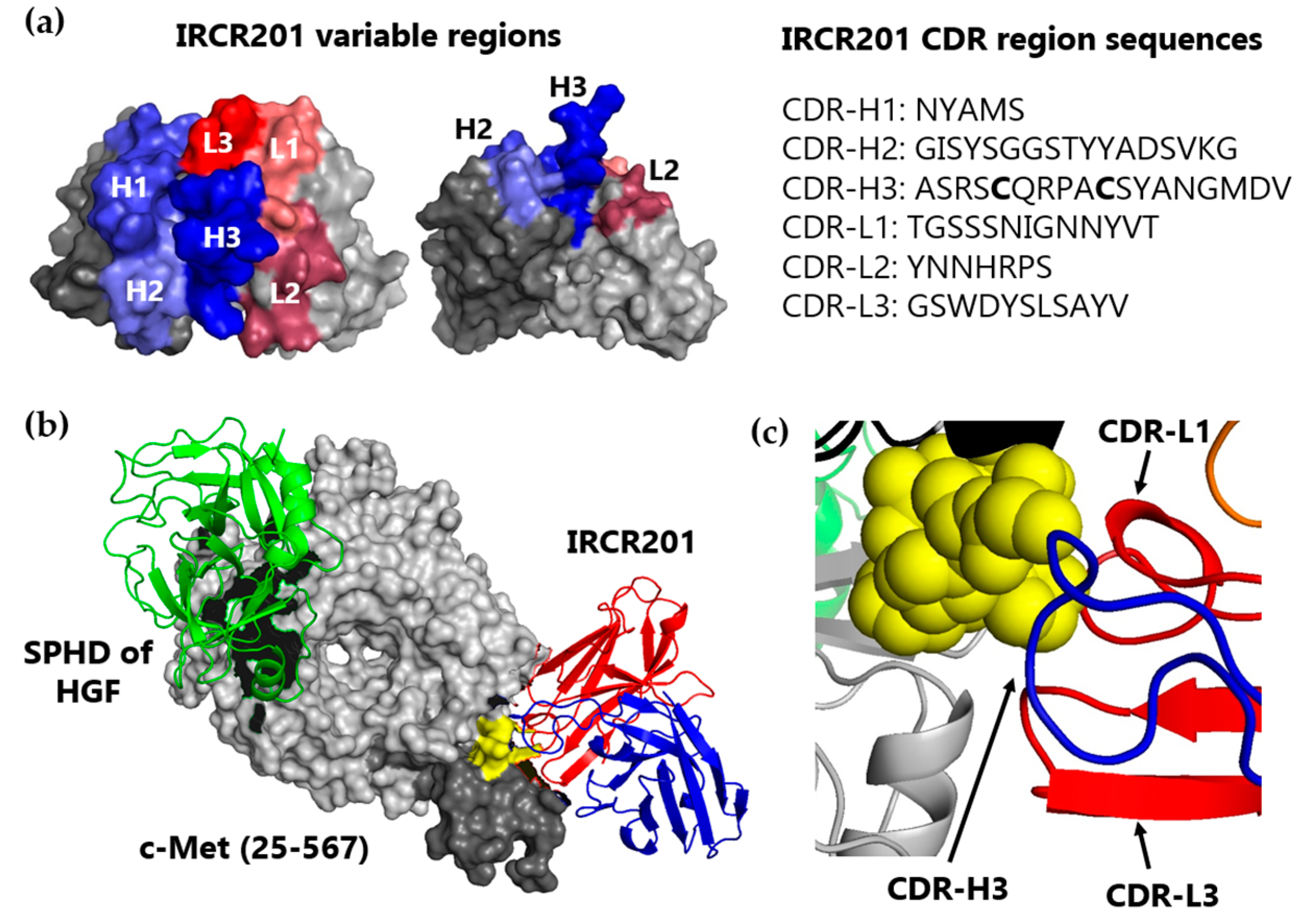
IRCR201 does not mimic the role of HGF and exhibits lower agonist activity compared to huOA5D5.v2 as it binds to the SAPPFVQ amino acid sequence of the PSI domain, as distinct from the Sema domain, which is an HGF binding site of c-Met. The PSI domain containing the SAPPFVQ sequence—the IRCR201 epitope—acts as a domain that gives flexibility to c-Met, promoting molecular interaction between c-Met and HGF [
41,
42]. Although the biological insights of the PSI domain remain relatively unexplored, IRCR201 interacts with the PSI domain of c-Met, thus resulting in an inhibitory effect without agonist activity. Basilico and colleagues also developed specific antibodies that bind to the PSI domain and disrupt the interaction between HGF and c-Met [
43], resulting in the inhibition of HGF-induced c-Met autophosphorylation, distinct from IRCR201, which does not impair the interaction between c-Met and HGF. Previous studies have reported that bivalent c-Met-targeting antibodies with different epitopes could elicit different levels of biochemical functional activity [
18,
43,
44]. The differential epitope of c-Met antibodies may account for the distinct functional activity of IRCR201.
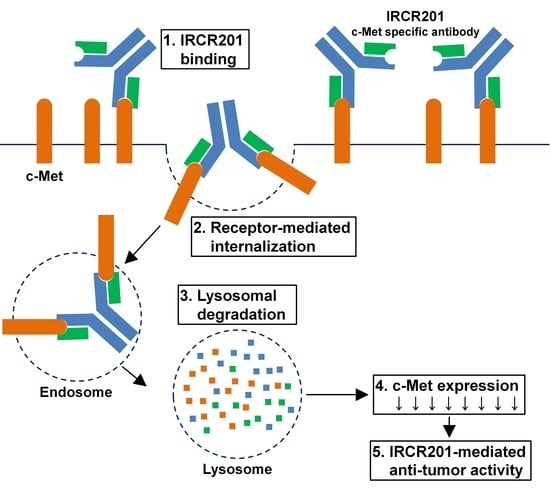
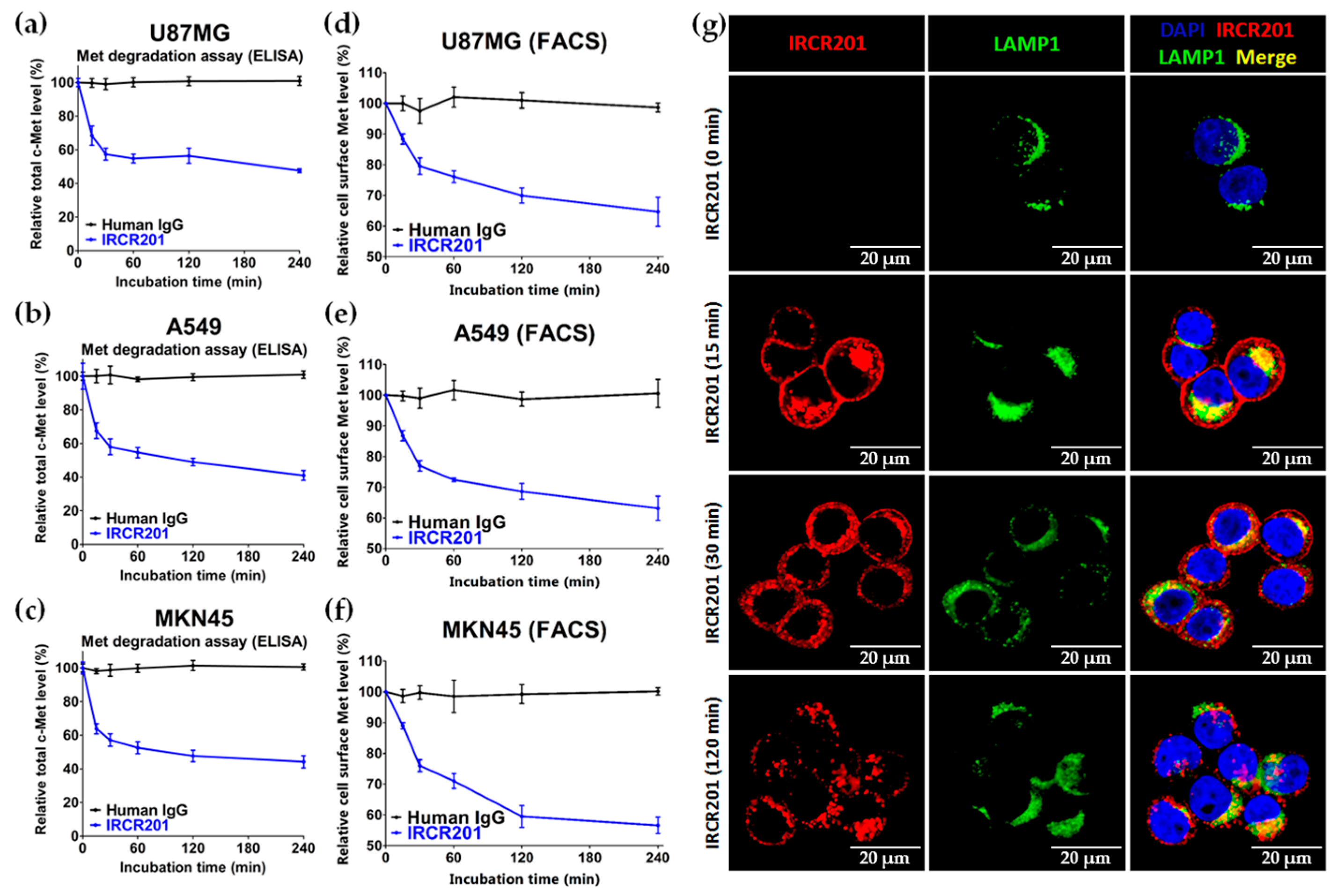
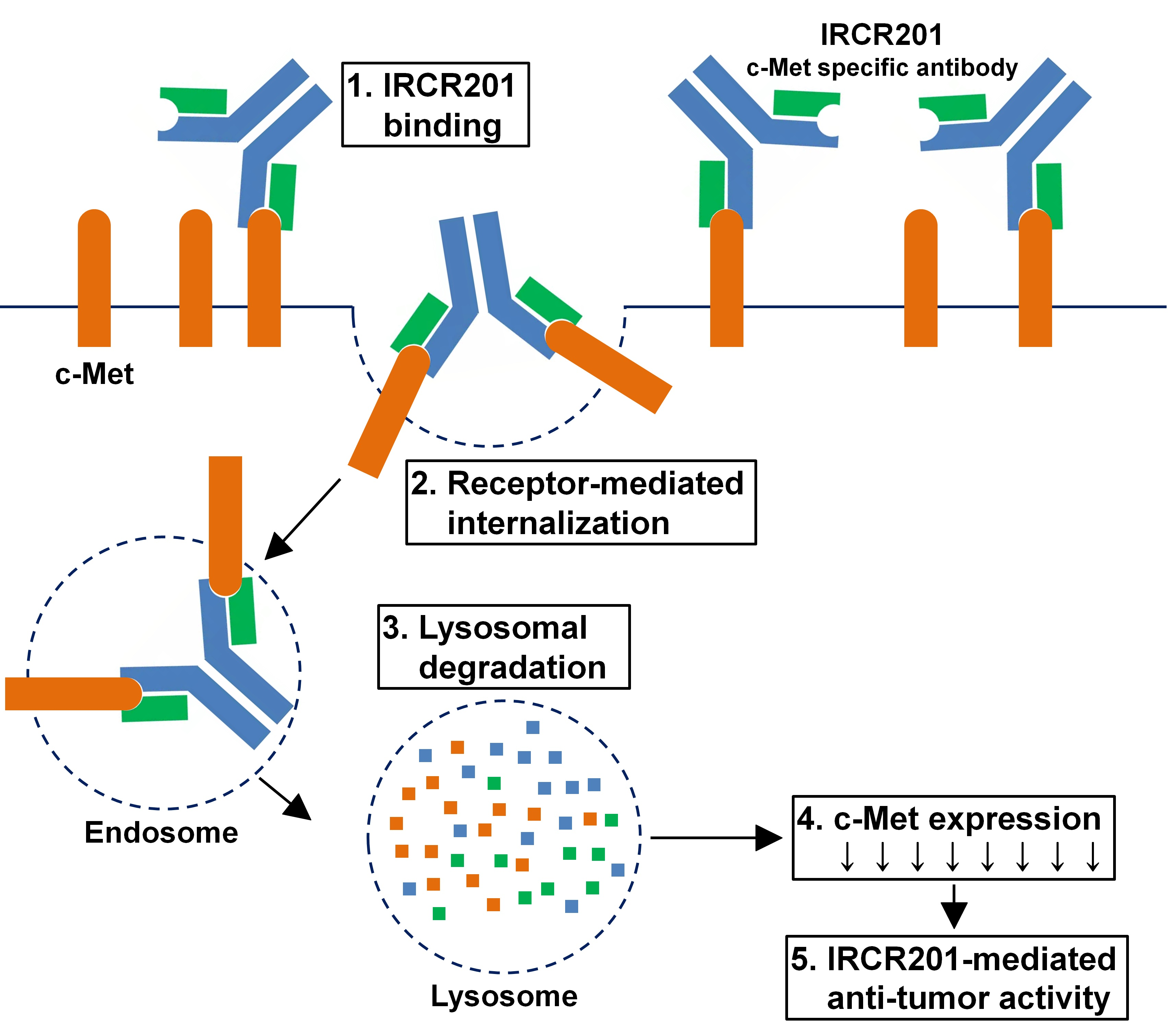
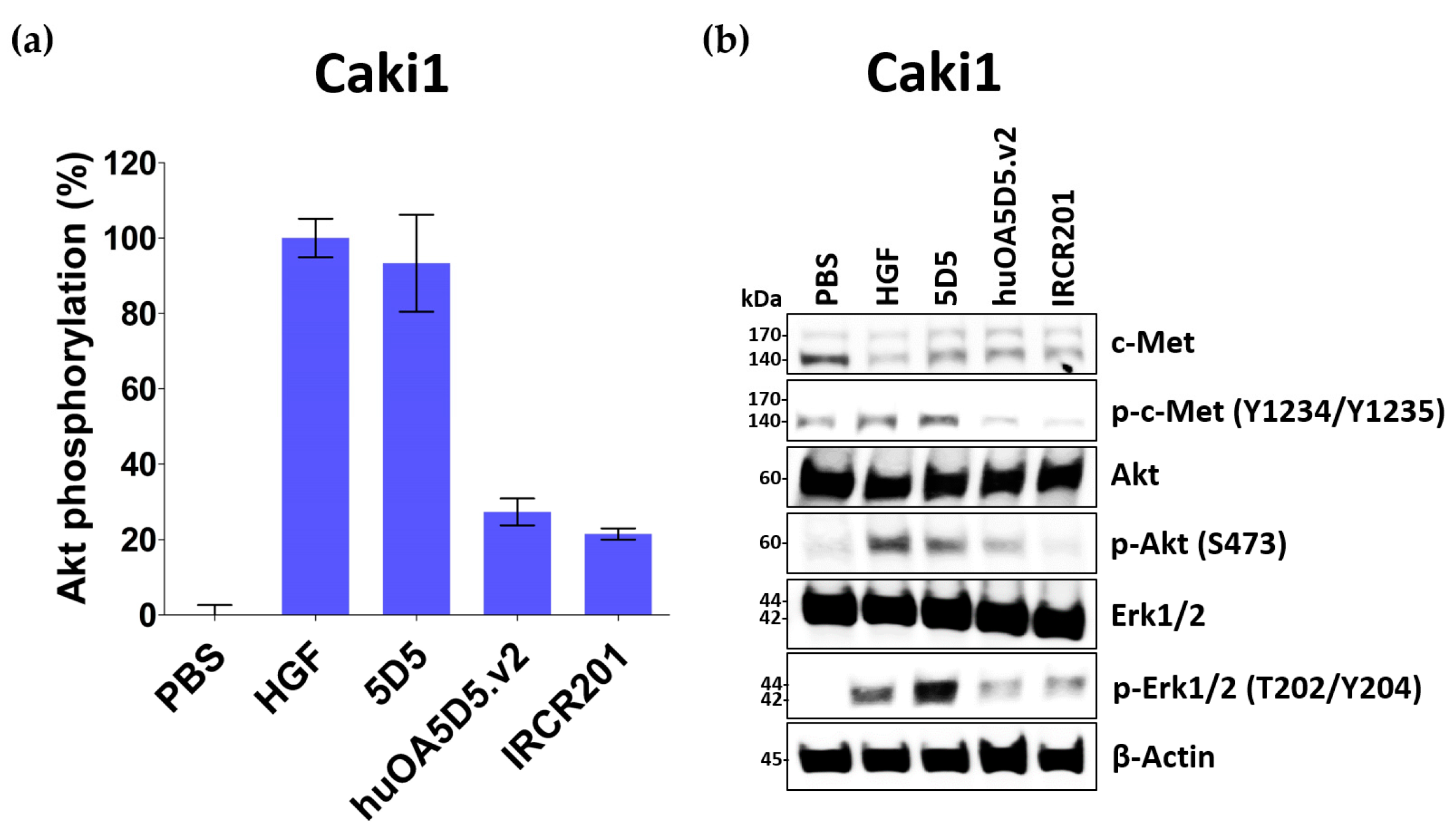
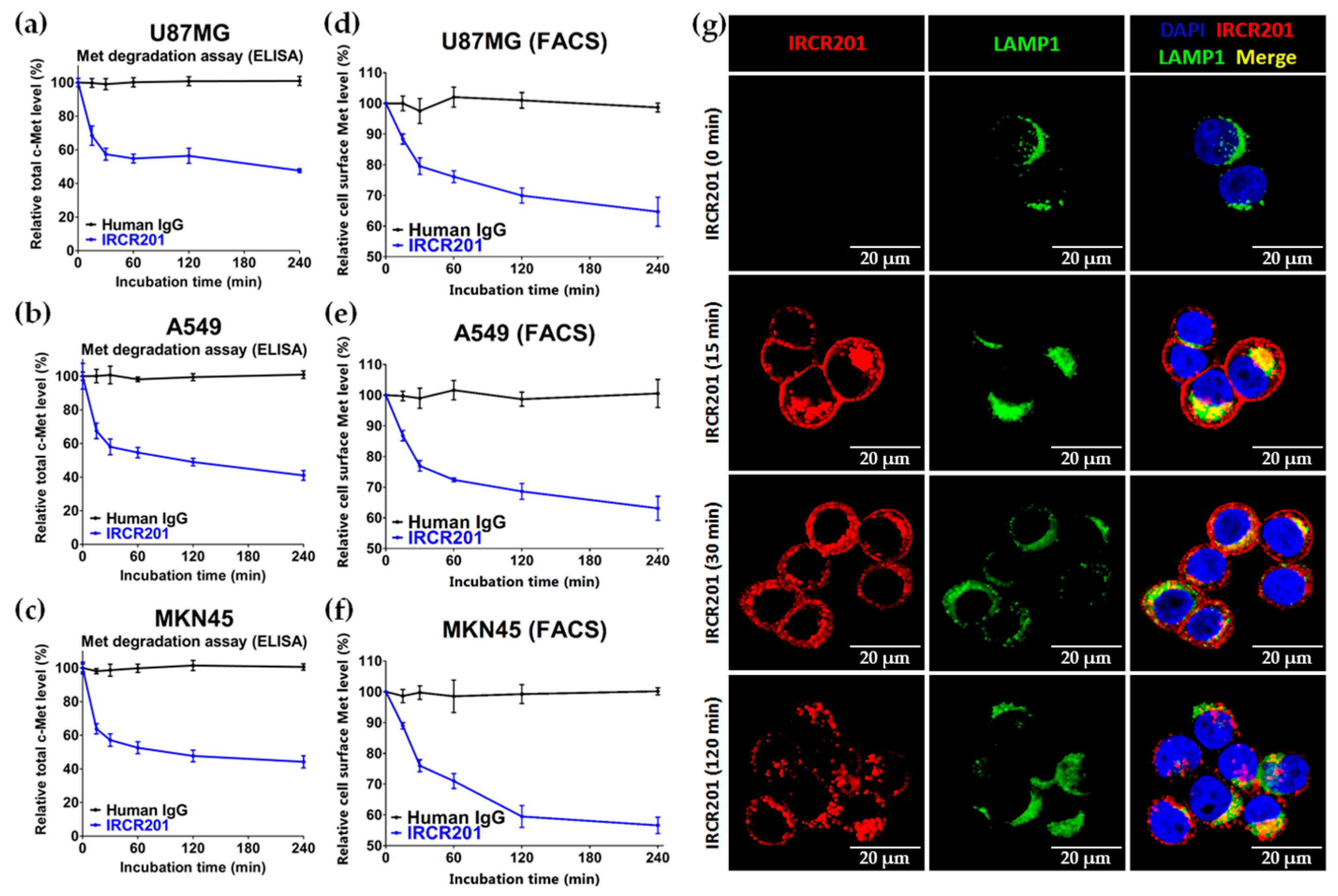
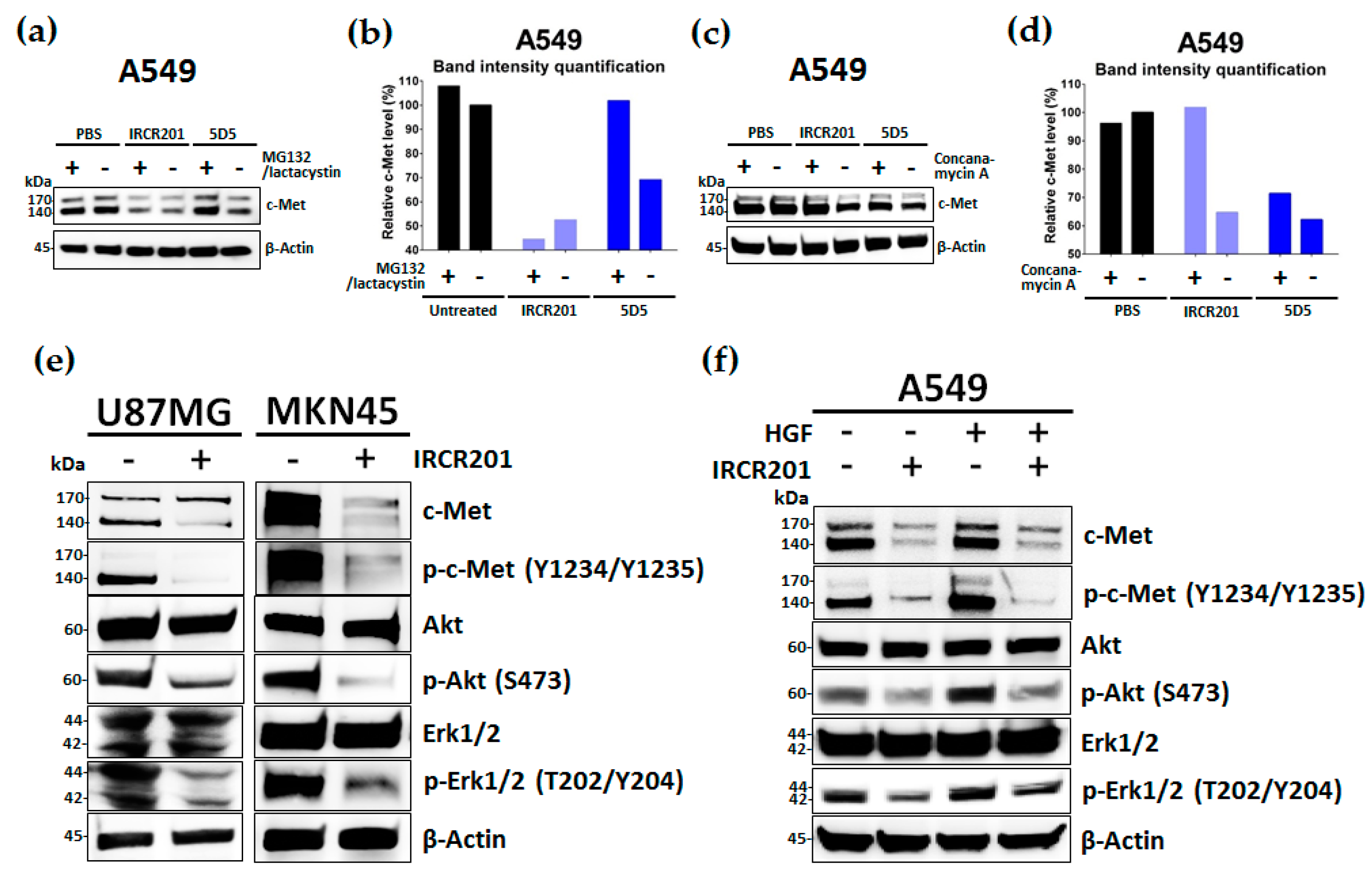
IRCR201 possesses a different binding epitope from previously developed c-Met inhibitory antibodies, and rapidly promoted c-Met depletion primarily through the lysosomal degradation pathway, thereby abrogating downstream signaling cascades in U87MG, A549, and MKN45. To further ascertain the effect of IRCR201 at a cell surface receptor level, a flow cytometry assay was configured to detect cell surface c-Met of U87MG, A549, and MKN45 following the antibody treatment. Similar results were observed throughout various cell lines, as the rapid degradation of cell surface c-Met was observed at the 15-min mark. Total receptor expression levels in the membrane were decreased by approximately 40% post IRCR201 treatment. Furthermore, we found that IRCR201 resulted in c-Met co-localization within the lysosome, suggesting an underlying mechanism behind IRCR201-induced c-Met degradation. The c-Met depletion activity of IRCR201 was inhibited by concanamycin A, supporting the notion that IRCR201 deteriorates c-Met through the lysosomal degradation pathway. These findings demonstrate that IRCR201 induces receptor-mediated internalization and lysosomal degradation, providing a potential underlying mechanism behind the loss of total c-Met protein.
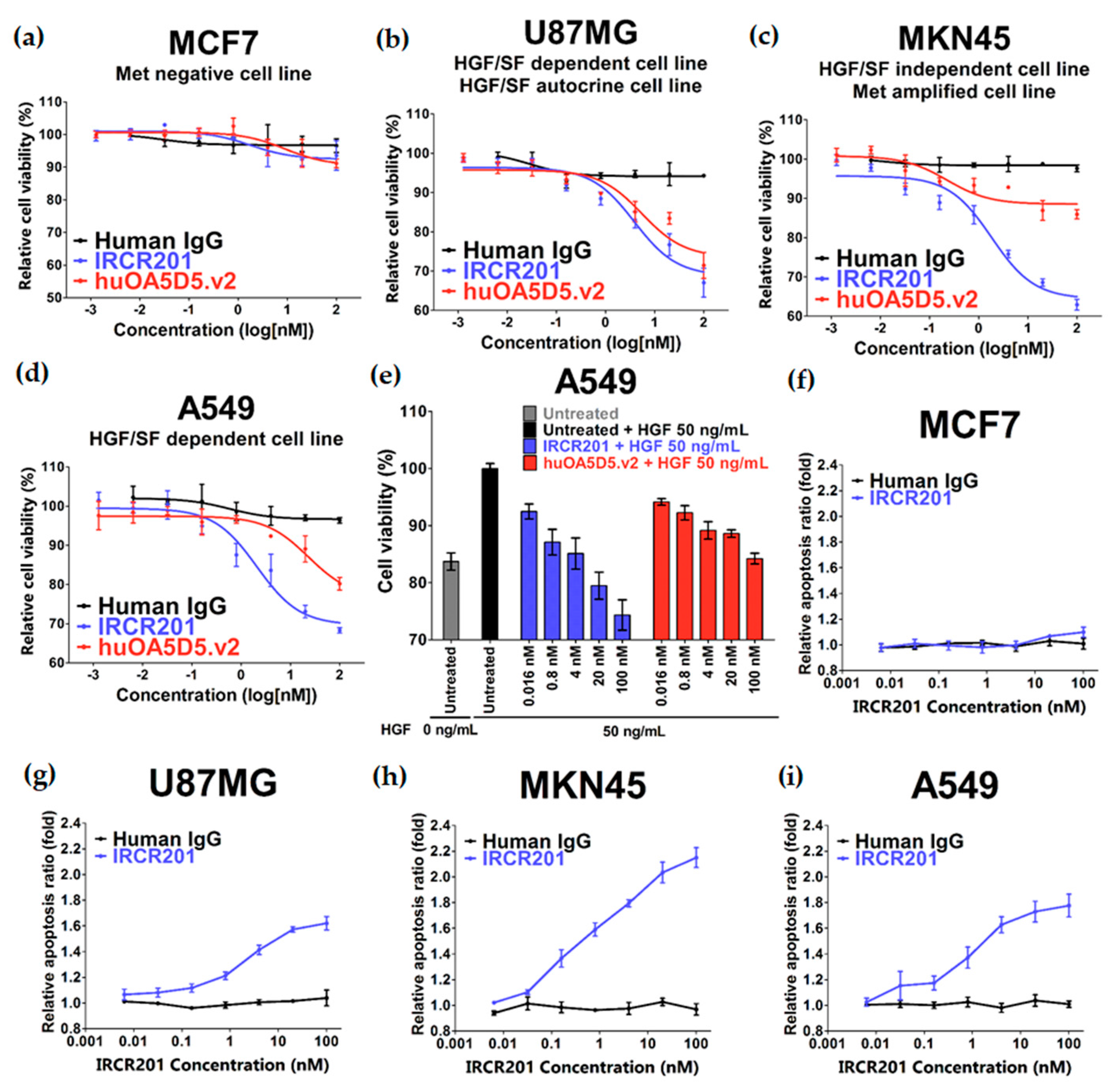
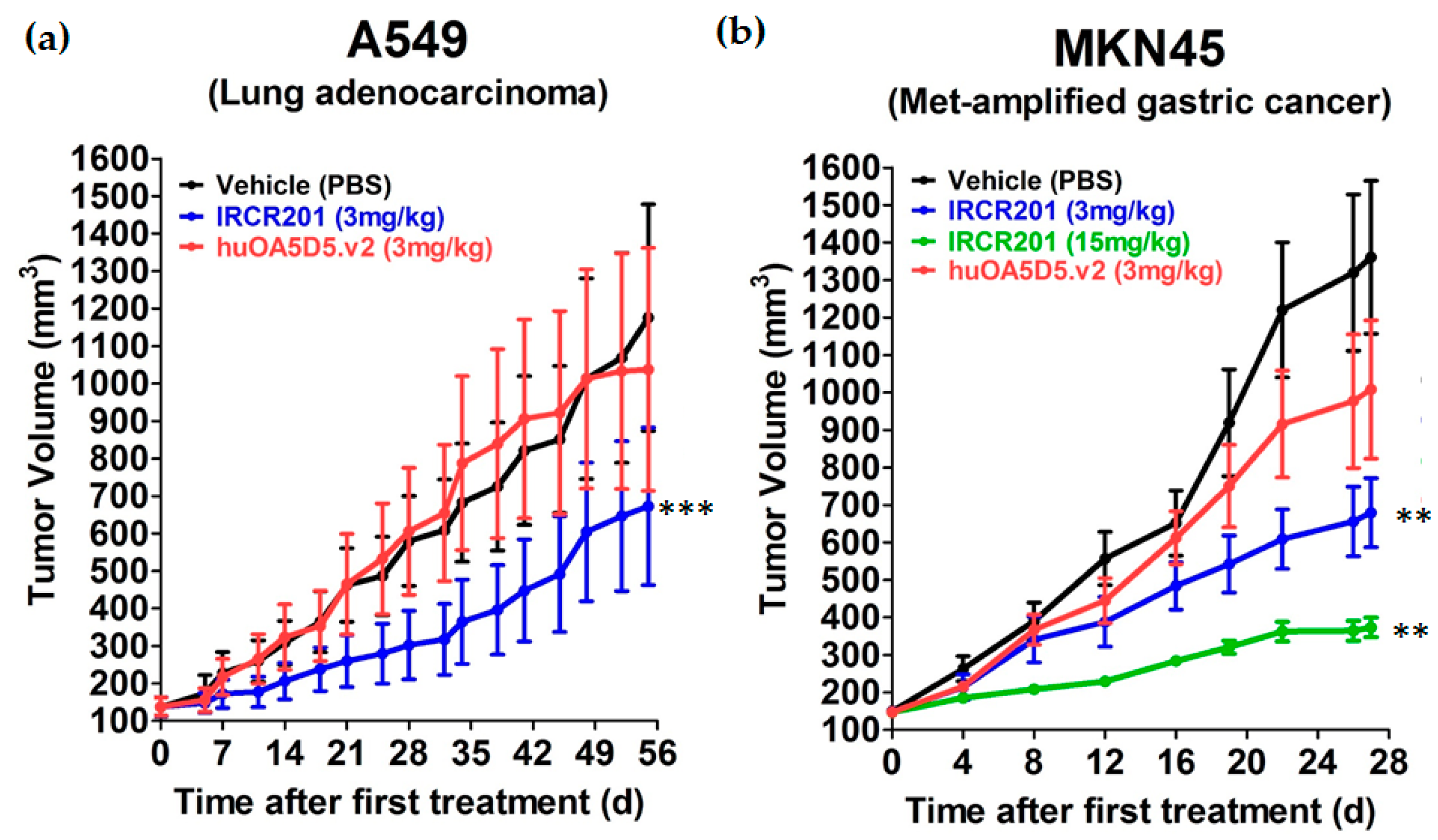
IRCR201 does not inhibit the interaction between HGF and c-Met, but it exhibited excellent growth inhibition capacity in various types of c-Met-expressing cancer cell lines compared to huOA5D5.v2 in vitro. IRCR201-induced c-Met degradation demonstrated a therapeutic effect mediated by the downregulation of tyrosine kinase activity and the subsequent inhibition of cellular proliferation in U87MG, A549, and MKN45. IRCR201 also showed significant antitumor effect in an A549 NSCLC xenograft tumor model. Since c-Met amplification leads to shorter survival in patients with gastric cancer and NSCLC [
45,
46], we assessed the tumor inhibitory capability of IRCR201 in a c-Met-amplified tumor model to provide clinical benefit in patients. Treatment with IRCR201 dramatically decreased tumor growth in the c-Met-amplified MKN45 gastric xenograft model, suggesting that it may portray antitumor activity in a broader range of cancer classes with constitutive c-Met activation via genomic amplification or mutations. These results suggest that IRCR201 could be a promising therapeutic agent to inhibit tumor growth driven by constitutively active c-Met through overexpression, gene amplification, or genomic mutations.
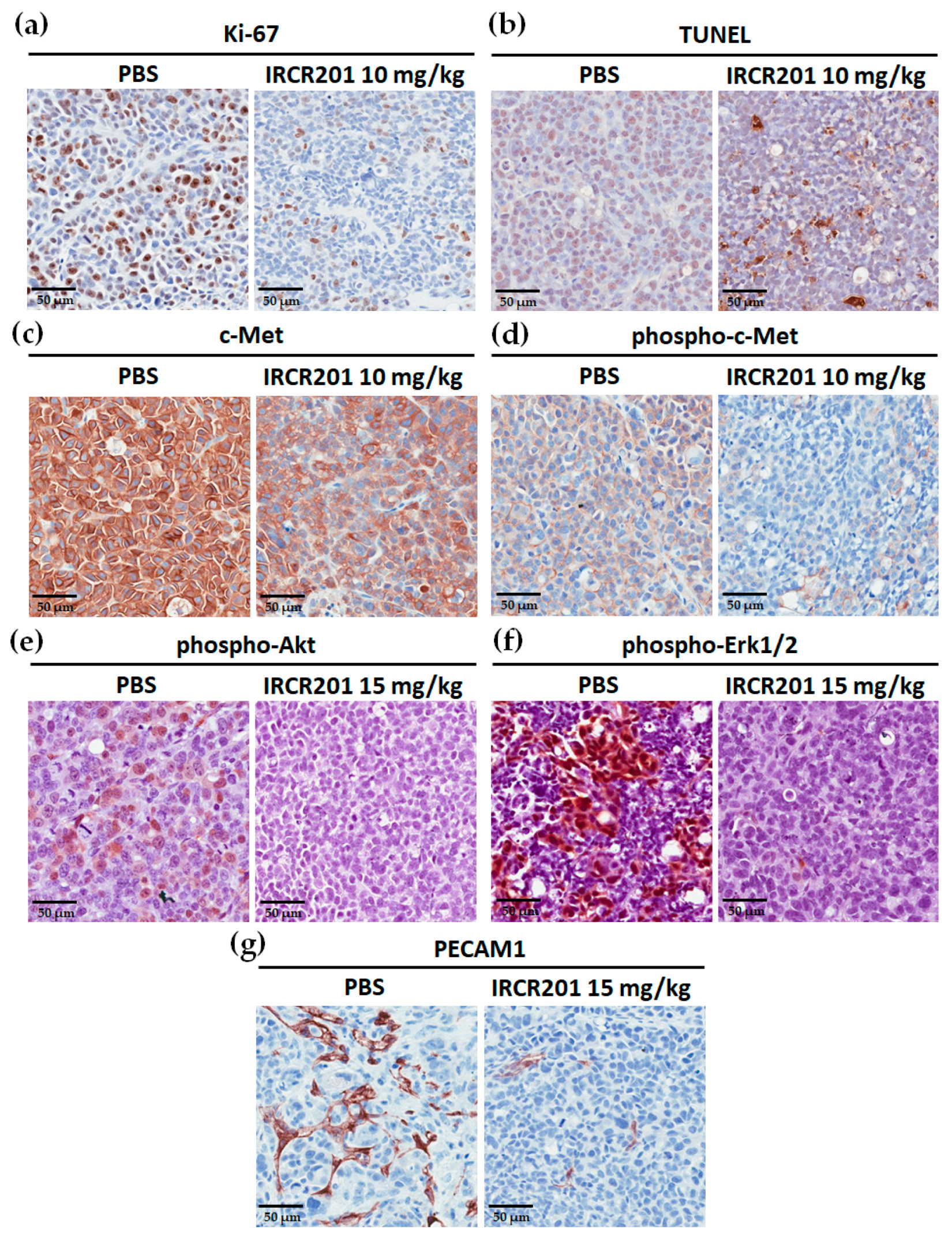
The HGF/c-Met signaling pathway is a pivotal component of tumor-associated angiogenesis that leads to tumor progression and metastasis via a sufficient supply of oxygen and nutrients through blood vessels [
1,
2]. Immunohistochemistry analysis also revealed that IRCR201 significantly inhibited the proliferation of tumor cells and angiogenesis of tumor-associated blood vessels, suggesting that IRCR201 suppresses the HGF/c-Met pathway in the tumor microenvironment.
Previous efforts to develop bivalent c-Met antagonistic antibodies were unsuccessful, as the antibodies incited agonistic effects [
20]. Through the comprehensive research of c-Met biology, numerous research groups have developed various c-Met antibodies such as onartuzumab (huOA5D5.v2), emibetuzumab (LY2875358), telisotuzumab (ABT-700), SAIT301, ARGX-111, and DN30 [
21,
22,
23,
24,
43,
44,
47,
48], which have diverse molecular mechanisms of action in downregulating the functional activity of c-Met. Among these, emibetuzumab and SAIT301—the c-Met inhibitory antibodies—have similar c-Met degradation activity to that of IRCR201; however, they have completely different binding epitopes from IRCR201.
By impairing both HGF-dependent and c-Met-amplified downstream activation in various cancer types, IRCR201 is differentiated from other therapeutic antibodies by securing a wide range of drug response groups. The anti-cancer activity of IRCR201 may be further enhanced by antibody-mediated cell cytotoxicity (ADCC), as the antibody’s isotype is human IgG1, which binds to the Fc receptors on immune cells. IRCR201 is also applicable to antibody–drug conjugate (ADC) platforms for toxin delivery, as it demonstrated the lysosomal degrading traits of c-Met.
This study currently has an accompanying research program to investigate the specific degradation pathway responsible for IRCR201-induced c-Met depletion. IRCR201 is also being used in toxicological studies using mouse models to confirm that its mouse cross-reactivity has an adverse effect on mouse health.
In summary, the newly identified traits of IRCR201 allow the evaluation of precise therapeutic efficacy in mouse models. Our comprehensive results show that IRCR201 is capable of inhibiting a variety of cancer types with HGF-dependent c-Met activation or gene amplification-driven constitutive c-Met activation. Taken together, IRCR201 represents a promising therapeutic antibody to treat cancer patients suffering from dysregulation of the HGF/c-Met signaling pathway.
4. Materials and Methods
4.1. Generation of Antibodies
Biopanning was performed using phage displayed synthetic human scFv libraries according to basic panning protocol [
49]. Briefly, scFv displaying phage libraries were enriched on 1 µg immobilized human c-Met ECD-Fc (Sino Biological, 10692-H03H, Beijing, China) and mouse c-Met ECD-Fc (Sino Biological, 50622-M02H, Beijing, China) in MaxiSorp™ immune-tubes (Nunc, 444202, Roskilde, Denmark). c-Met specific binders were screened and selected by ELISA using produced scFvs in human and mouse c-Met-coated 96-well enzyme immunoassay/radioimmunoassay (EIA/RIA) plates (Costar, #3590, Corning, NY, USA). Finally, the selected scFv was reformatted to human IgG1, and named IRCR201. Antibody expression vectors of onartuzumab (huOA5D5.v2) were made using the amino acid sequences shown in patent US7892550 and in the published literature [
22]. IRCR201 and huOA5D5.v2 were produced in an Expi293™ transient mammalian expression system (Gibco, A14635, Carlsbad, CA, USA) and purified by HiTrap™ Mabselect SuRe (GE Healthcare Life Sciences, 11-0034-93, Uppsala, Sweden). The anti-human c-Met agonistic antibody 5D5 was purchased in the form of hybridoma (American Type Culture Collection (ATCC), HB-11895, Manassas, VA, USA) and purified from hybridoma supernatant by HiTrap™ Protein G HP (GE Healthcare Life Sciences, 17-0405-01, Uppsala, Sweden). Control human IgG was purchased from Sigma (Sigma, I2511, St. Louis, MO, USA).
4.2. Cells and Cell Cultures
The MCF7 (ATCC, HTB-22), U87MG (ATCC, HTB-14), A549 (ATCC, CCL-185), and Caki-1 (ATCC, HTB-46) cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), and the MKN45 (JCRB Cell Bank, JCRB0254) cell line was purchased from the Japanese Collection of Research Bioresources Cell Bank (JCRB Cell Bank, Osaka, Japan). All cell lines were tested for mycoplasma, cross-contamination, and genetic fingerprints. U87MG were cultured in Eagle’s Minimum Essential Medium (EMEM; ATCC, 30-2003, Manassas, VA, USA) containing 10% (v/v) fetal bovine serum (FBS; Gibco, 26140079, Carlsbad, CA, USA) and 1% (v/v) penicillin/streptomycin (Gibco, 15140122, Carlsbad, CA, USA). MCF7, MKN45, A549, and Caki1 were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, 11875093, Carlsbad, CA, USA) containing 10% (v/v) FBS (Gibco, 26140079, Carlsbad, CA, USA) and 1% (v/v) penicillin/streptomycin (Gibco, 15140122, Carlsbad, CA, USA). All cells were cultured according to the manufacturer's manual and had been passaged for fewer than three months after thawing. Cell counting was performed with the Scepter™ 2.0 automated cell counter (Millipore, Billerica, MA, USA) and c-Met expression was confirmed by Western blot using cell lysates.
4.3. Animals
BALB/c-nude mice (female) were used for the in vivo studies. All animals were obtained from Orient Bio Inc. (Seongnam, Korea), and were maintained under specific pathogen-free conditions in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal (AAALAC) International in accordance with the current regulations and guidelines of the Laboratory Animal Research Center (LARC) at the Samsung Medical Center (Project identification code: NTX1161851-20160324002). Mice were acclimated for at least a week before they entered the studies.
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
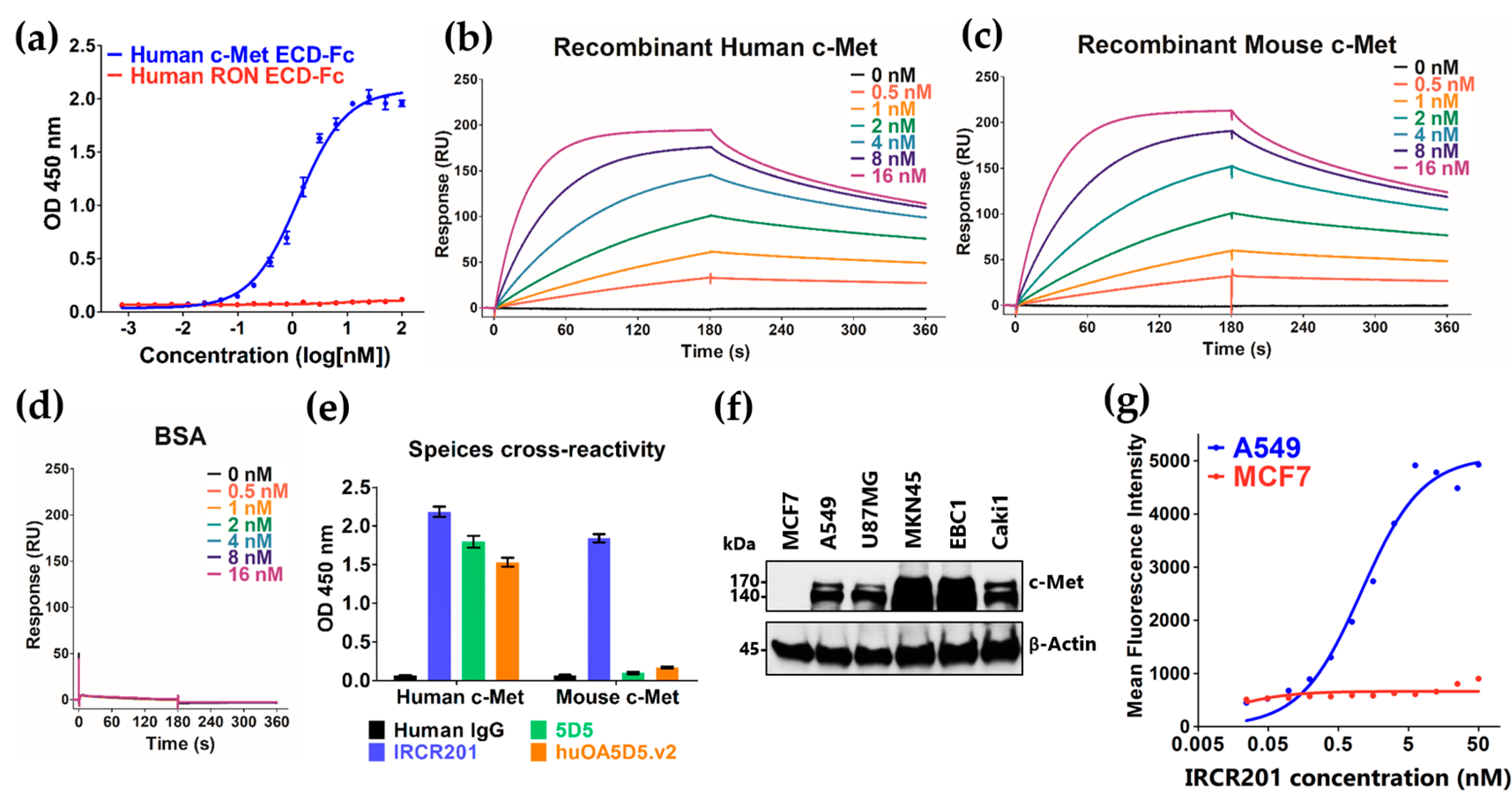
To analyze the specificity and selectivity of IRCR201, 0.1 µg human c-Met protein (Sino Biological, 10692-H03H, Beijing, China) or 0.1 µg human RON protein (Sino Biological, 11608-H08H, Beijing, China) was coated in 96-well EIA/RIA plates (Costar, #3590, Corning, NY, USA) at 4 °C overnight. Following three washes with 1× phosphate-buffered saline with Tween 20 (PBST; Cell Signaling Technology, #9809, Danvers, MA, USA), they were blocked with 3% (w/v) skim milk (BD Difco, 232100, Sparks, MD, USA) for 1 h at room temperature. The wells of the plates were washed three times with 1× PBST and then incubated with 100 µL of IRCR201 diluted to the appropriate concentration for 1 h at room temperature. After three washes with 1× PBST, the wells of the plates were incubated with 100 µL of 1:10,000 goat anti-human IgG F(ab’)2 conjugated horseradish peroxidase (HRP) (Thermo Scientific, 31482, Waltham, MA, USA) for 1 h at room temperature, and then again washed three times with 1× PBST. 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate solution (Thermo Scientific, N301, Waltham, MA, USA) was added to each well, and then reactions were quenched by STOP solution (Cell Signaling Technology, #7002, Danvers, MA, USA).
To investigate the species cross-reactivity of c-Met antibodies to human and mouse c-Met, 0.1 µg/well human c-Met ECD-Fc (Sino Biological, 10692-H03H, Beijing, China) or 0.1 µg/well mouse c-Met ECD-Fc (Sino Biological, 50622-M02H, Beijing, China) was coated in 96-well EIA/RIA plates (Costar, #3590, Corning, NY, USA). The subsequent steps were performed as described above. Since 5D5 was a mouse antibody, goat anti-mouse Fab conjugated HRP (Abcam, ab6823, Cambridge, UK) was used as a secondary antibody.
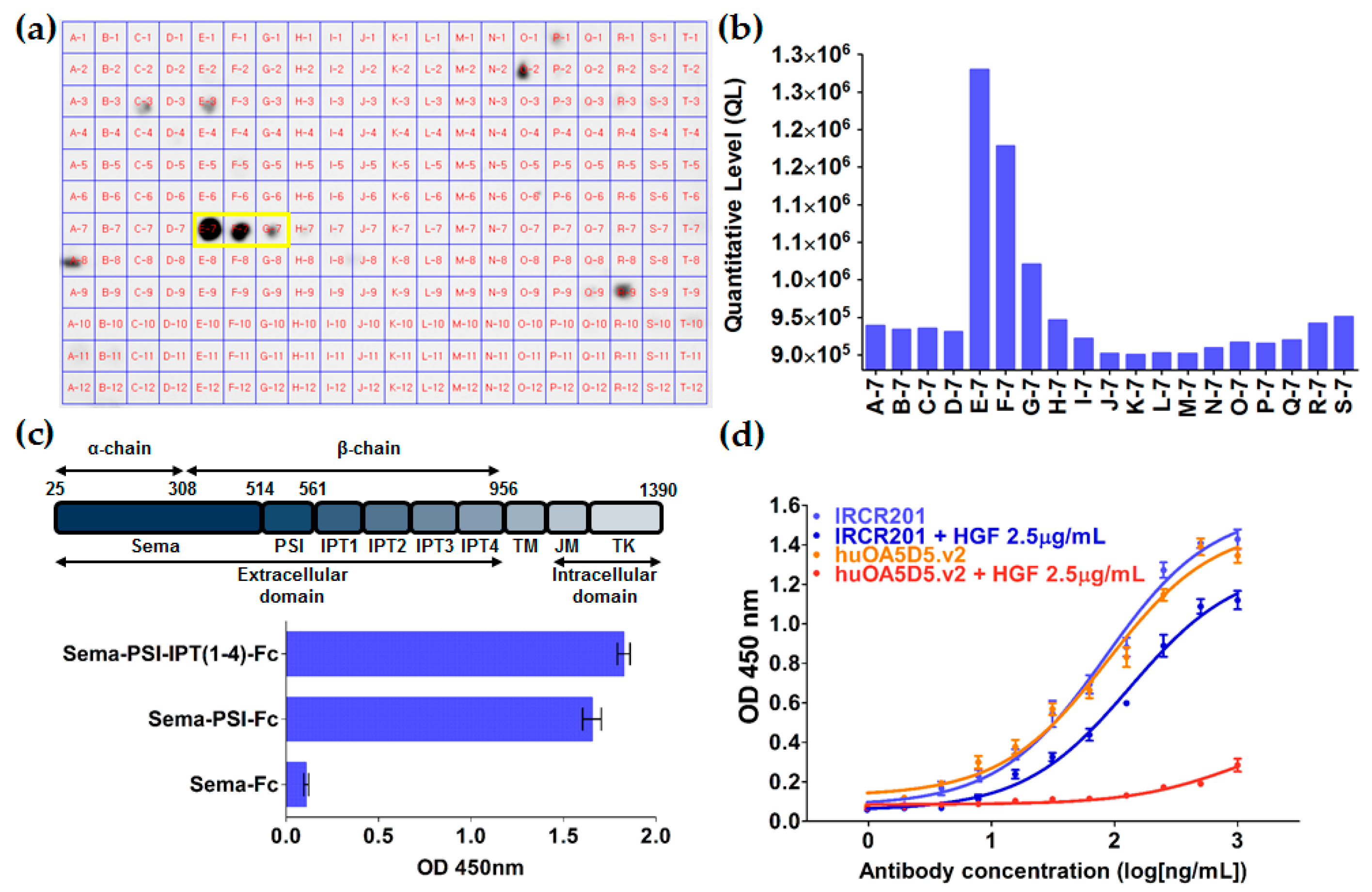
To confirm the binding property of IRCR201 to human c-Met domain proteins, 96-well EIA/RIA plates (Costar, #3590, Corning, NY, USA) were immobilized with 100 µL/well of 0.1 µg recombinant human Sema-Fc (protein consists of amino acids 1 to 515 of human c-Met), 0.1 µg recombinant human Sema-PSI-Fc (protein consists of amino acids 1 to 562 of human c-Met), and 0.1 µg recombinant human whole c-Met ECD-Fc (Sino Biological, 10692-H03H, Beijing, China). Recombinant human c-Met domain proteins with an Fc tag (rhSema-Fc and rhSema-PSI-Fc) were expressed in an Expi293™ system (Gibco, Carlsbad, CA, USA) and purified from HiTrap™ Mabselect SuRe (GE Healthcare Life Sciences, 11-0034-93, Uppsala, Sweden). Then, 100 µL of IRCR201 (1 µg/mL) was used as the primary antibody, and 100 µL of 1:10,000 goat anti-human IgG F(ab’)2 conjugated HRP (Thermo scientific, 31482, Waltham, MA, USA) was used as the secondary antibody. The color of the plates was developed using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution (Thermo scientific, N301, Waltham, MA, USA) and the reaction was terminated with STOP solution (Cell Signaling Technology, #7002, Danvers, MA, USA).
4.5. HGF Ligand-Blocking ELISA
For the HGF/c-Met blocking assay (competitive ELISA), 96-well EIA/RIA plates (Costar, #3590, Corning, NY, USA) were coated with recombinant human c-Met (1 μg/μL, 100 μL) at 4 °C overnight. After washing the wells with 1× PBST, they were blocked with 3% skim milk for 1 h at room temperature. After pre-binding with/without 2.5 μg/mL HGF (R&D Systems, 294-HG, Minneapolis, MN, USA) at room temperature for 1 h, IRCR201 and huOA5D5.v2 were incubated at room temperature for 1 h. After three washes with 1× PBST, the wells were incubated with 100 μL of 1:10,000 goat anti-human IgG F(ab’)2 conjugated HRP (Thermo scientific, 31482, Waltham, MA, USA) for 1 h and then washed three times with 1× PBST. TMB substrate solution (Thermo Scientific, N301, Waltham, MA, USA) was added to each well and incubated at room temperature until color was developed. Afterwards, reactions were quenched by STOP solution (Cell Signaling Technology, #7002, Danvers, MA, USA). Optical density (OD) of the wells was measured at 450 nm with an Infinite® M200 pro (Tecan, Männedorf, Switzerland).
4.6. Surface Plasmon Resonance Assay
The binding affinities and kinetics of IRCR201 for human c-Met and mouse c-Met were measured using a Biacore™ T100 (GE Healthcare Life Sciences, Uppsala, Sweden). Human c-Met, mouse c-Met, or BSA were immobilized on a CM5 sensor chip (GE Healthcare Life Sciences, BR100530, Uppsala, Sweden) using an Amine coupling kit (GE Healthcare Life Sciences, BR100050, Uppsala, Sweden). Different concentrations of purified IRCR201 were injected into immobilized human and mouse c-Met to determine KD values. To obtain the kinetic and affinity constants of IRCR201 to human and mouse c-Met, Biacore™ T100 evaluation software (GE Healthcare Life Sciences, Uppsala, Sweden) was used.
4.7. Epitope Mapping
The epitope mapping of IRCR201 to c-Met was performed using the scFv format of IRCR201. The scFv of IRCR201 was produced using TOP10F' Escherichia coli (E. coli) (Invitrogen, C303003, Carlsbad, CA, USA) and purified from the bacterial lysates by immobilized metal affinity chromatography (IMAC) using nickel–nitrilotriacetic acid (Ni-NTA) agarose (QIAGEN, 30210, Hilden, Germany) according to the instructions of the manufacturer. The c-Met 15-mer peptides corresponding to 1-932 of the c-Met extracellular domain were synthesized at 5 nmol/spot and covalently bound to a Whatman® 50 cellulose support by the C-terminus (JPT Peptide Technologies GmbH, Berlin, Germany). The membrane of c-Met 15-mer peptides was blocked with 5% skim milk for 3 h at room temperature with shaking. The membrane was incubated with the scFv of IRCR201 in 5% skim milk at 4 °C overnight. The scFv of IRCR201 bound to the c-Met 15-mer peptides was transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, IPVH00010, Billerica, MA, USA) using the electroblotting method. The transferred PVDF membranes were washed in 1× TBST (tris-buffered saline with Tween 20) twice for 10 min. The PVDF membrane was blocked with 5% skim milk for 3 h and incubated with anti-HA-peroxidase (Roche Diagnostics, 12013819001, Mannheim, Germany) for 2 h with agitation. The membrane was washed three times with 1× TBST for 5 min and incubated with enhanced chemiluminescence (ECL) detection reagents (GE Healthcare Life Sciences, RPN2106, Little Chalfont, UK) for about 1 min. A sheet of film on the membrane was placed into a film cassette in a darkroom, according to the manufacturer’s instructions. The intensity of the bands was quantified by Multi Gauge software V3.0 (Fuji photo film Co., Ltd., Tokyo, Japan).
4.8. Protein Modeling and Docking
The sequences of the V
H and V
L segment of IRCR201 were assembled in the scFv format by homology modeling using the Rosetta-based computational homology modeling technique [
31]. The IRCR201/c-Met docking model was made using the ZDOCK server [
32]. Structure models were analyzed using PyMOL (DeLano Scientific LLC, Palo Alto, CA, USA).
4.9. Agonism Analysis
Wells of 96-well tissue culture plates (Costar #3595, Corning, NY, USA) were seeded with 5000 Caki1 cells in RPMI1640 medium supplemented with 10% (v/v) FBS. After culture for 24 h, cells were starved in serum-free RPMI1640 medium for another 24 h. The cells were then cultured in the presence of anti-c-Met antibodies and HGF in serum-free medium for 30 min at 37 °C. Next, the medium was removed and the cells were washed once with 1× PBS (pH 7.4). Cells were lysed and p-Akt levels were quantified by PathScan® phospho-Akt1 (Ser473) chemiluminescent sandwich ELISA kit (Cell Signaling Technology, #7134, Danvers, MA, USA) according to the manufacturer’s protocol.
To clarify agonistic activity of IRCR201, Caki1 cells were starved in serum-free RPMI 1640 medium for 24 h and then treated with HGF or c-Met antibodies for 30 min. Cells were lysed and then the Western blotting experiments were proceeded.
4.10. c-Met Degradation Assay
U87MG, A549, and MKN45 cells were treated with 100 nM of IRCR201 for 0 min, 15 min, 30 min, 60 min, 120 min, and 240 min, and then detached by mild trypsinization, fixed using 4% paraformaldehyde solution (Biosesang, P2031, Seongnam, Korea) at 4 °C, and resuspended at a concentration of 5 × 105 cells/mL in flow cytometry buffer (1% (w/v) FBS, 0.03% (w/v) NaN3, and 25 mM HEPES in 1× PBS, pH 7.4). To confirm the expression of c-Met on the cell surface depending on IRCR201 treatment times, fixed cells were incubated with 1:50 Met (D1C2) XP® Rabbit mAb conjugated Alexa Fluor® 488 (Cell Signaling Technology, #8494, Danvers, MA, USA). Cells were run on a FACSAriaTM III (BD Biosciences, Mountainview, CA, USA) and analyzed using the CellQuest™ program (BD Biosciences, Mountainview, CA, USA).
To analyze the total c-Met degradation of c-Met-expressing cells by IRCR201, an ELISA-based quantification method was used. When U87MG, A549, and MKN45 cells in 96-well cell culture plates (Costar, #3595, Corning, NY, USA) reached approximately 70% confluency, cells were treated with 100 nM of IRCR201 for 0 min, 15 min, 30 min, 60 min, 90 min, and 120 min. After washing with 1× PBS (pH 7.4), cells were resuspended with lysis buffer (Roche Diagnostics, 04719956001, Mannheim, Germany) supplemented with protease inhibitor cocktail tablets (Roche Diagnostics, 04719956001, Mannheim, Germany) and phosphatase inhibitor (Roche Diagnostics, 4906845001, Mannheim, Germany). The changes in c-Met protein were analyzed in a timely manner by a human HGFR/c-MET DuoSet® ELISA kit (R&D systems, DY358, Minneapolis, MN, USA) according to the manufacturer’s protocol.
4.11. Proliferation Assay
MCF7, U87MG, A549, and MKN45 cells were seeded into 96-well white plates (Costar, #3610, Corning, NY, USA) at a density of 3,000 cells/well and cultured for 72 h with 0–100 nM of IRCR201, huOA5D5.v2, or human IgG control at 37 °C in a humidified 5% CO2 atmosphere. The number of viable cells was estimated by a CellTiter-Glo® luminescent cell viability assay kit (Promega, G7573, Madison, WI, USA). The luminescent signal intensity of the plates was read by an Infinite® m200 pro (Tecan, Männedorf, Switzerland) and normalized to the untreated group.
4.12. Caspase 3/7 Activity Assay
The caspase 3/7 activity was detected using the Caspase-Glo® 3/7 Luciferase assay kit (Promega, G8091, Madison, WI, USA) in accordance with the manufacturer's recommendations. Cells in appropriate complete medium were incubated for 24 h with the untreated group, Human IgG, or IRCR201. After the addition of Caspase-Glo® 3/7 reagent, the luminescent signal intensity was measured by an Infinite® m200 pro (Tecan, Männedorf, Switzerland) and normalized to the untreated group.
4.13. Western Blot Analysis
When U87MG, A549, and MKN45 cells reached 70% confluency in six-well tissue culture plates (Nunc, 140675, Roskilde, Denmark), cells were incubated with IRCR201 (100 nM) or PBS in full-serum media for 24 h at 37 °C in a humidified 5% CO2 atmosphere. In the case of A549, conditions for treatment with HGF (50 ng/mL) were added. Cells were then lysed with Lysis-M reagent (Roche Diagnostics, 04719956001, Mannheim, Germany) supplemented with protease inhibitor cocktail tablets (Roche Diagnostics, 04719956001, Mannheim, Germany) and phosphatase inhibitor (Roche Diagnostics, 4906845001, Mannheim, Germany). Samples were denatured with 4× lithium dodecyl sulfate (LDS) sample buffer (Invitrogen, NP0007, Carlsbad, CA, USA) and 10× reducing reagent (Invitrogen, NP0009, Carlsbad, CA, USA) at 70 °C for 10 min. For Western blotting, cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 4–20% Mini-PROTEAN® TGX™ gels (Bio-Rad, #4561094 or #4561096, Hercules, CA, USA) and transferred to nitrocellulose membranes (Bio-Rad, #1704158, Hercules, CA, USA) using a Trans-Blot® Turbo™ transfer system (Bio-Rad, Hercules, CA, USA). Blots were blocked with 5% BSA solution for 1 h at room temperature and then incubated overnight with appropriate primary antibodies at 4 °C with agitation. Primary antibodies used include the Met (D1C2) XP® Rabbit mAb (Cell Signaling Technology, #8198, Danvers, MA, USA), Phospho-Met (Tyr1234/1235) (D26) XP® Rabbit mAb (Cell Signaling Technology, #3077, Danvers, MA, USA), Akt Rabbit mAb (Cell Signaling Technology, #9272, Danvers, MA, USA), Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb (Cell Signaling Technology, #4060, Danvers, MA, USA), p44/42 MAPK (Erk1/2) Rabbit mAb (Cell Signaling Technology, #9102, Danvers, MA, USA), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Rabbit mAb (Cell Signaling Technology, #4370, Danvers, MA, USA), and Actin (13E5) Rabbit mAb (Cell Signaling Technology, #4970, Danvers, MA, USA), all used according to the manufacturer’s recommendations. Following overnight incubation with primary antibodies, blots were washed three times with 1× PBST for 20 min, and then incubated with 1:2000 goat anti-rabbit IgG-HRP (Abcam, ab6721) as a secondary antibody for 1 h at room temperature. Blots were then washed three times with 1× PBST for 20 min, and immune-reactive bands were visualized using ECL Western blotting detection reagents (GE Healthcare Life Sciences, RPN2106 or RPN2232, Little Chalfont, UK) and scanned by ImageQuant™ LAS 4000 (GE Healthcare Life Sciences, Uppsala, Sweden).
To confirm the degradation mechanism of c-Met in A549, 100 nM concanamycin A (Abcam, ab144227, Cambridge, UK) or 5 μM MG132 (Selleckchem, S2619, Houston, TX, USA)/5 μM lactacystin (Abcam, ab141411, Cambridge, UK) mixture was pre-treated for 2 h, and IRCR201 or 5D5 was treated for 4 h at 37 °C in a humidified 5% CO2 atmosphere. Cells were lysed, and the Western blotting steps were conducted as described above.
4.14. Laser Scanning Microscopy
MKN45 cells were cultured on eight-well chamber 15 µ-slide (ibidi GmbH, #80826, Planegg, Germany). MKN45 cells were incubated with 10 µg/mL IRCR201 at 37 °C in a humidified 5% CO2 atmosphere. After being washed with 1× PBS (pH 7.4), the cells were fixed by 4% paraformaldehyde (Biosesang, P2031, Seongnam, Korea), permeabilized with 0.1% Triton™ X-100 (Sigma-Aldrich, X100, St. Louis, MO, USA), and blocked by adding 1% BSA. IRCR201 binding to c-Met were stained by anti-Human IgG secondary antibody conjugated Alexa Fluor® 647 (Invitrogen, A-21445, Carlsbad, CA, USA), and LAMP1 was detected by anti-LAMP1 antibody conjugated Alexa Fluor® 488 (Abcam, ab187591, Cambridge, UK). MKN45 cells stained fluorescence dyes were mounted with VECTASHIELD® Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, H-1200, Burlingame, CA, USA). All fluorescence images were obtained by LSM780 (Zeiss, Jena, Germany) and analyzed by ZEN2012 (Zeiss, Jena, Germany).
4.15. In Vivo Therapeutic Efficacy
The efficacy of IRCR201 was evaluated in various tumor xenograft models. A549 and MKN45 single cells suspended in Hank’s Balanced Salt Solution (HBSS; Gibco, 24020117, Carlsbad, CA, USA) with Matrigel® (Corning, 354234, Lowell, MA, USA) were subcutaneously injected into the right flank region of female BALB/c-nu mice (1 × 106 cells per mouse in 100 μL). When the mean tumor volume reached approximately 150 mm3 (day 0), animals were randomized according to tumor volume to minimize intragroup and intergroup variation. After regrouping, IRCR201, huOA5D5.v2, or vehicle was intravenously administered twice per week. Each treatment group consisted of six mice. Tumor volumes were measured using three-dimensional calipers. Body weights were measured as general confirmation of toxicity.
4.16. Immunohistochemistry
For the immunostaining of Ki-67, c-Met, and phospho-c-Met, phospho-Akt, phospho-Erk1/2, and PECAM1, antigen retrieval was processed in formalin-fixed and subsequently paraffin-embedded MKN45 tumor tissues. After blocking with 5% bovine serum albumin in 1× PBS (pH 7.4) for 1 h at room temperature, the sections were incubated overnight with primary antibodies at 4 °C. Primary antibodies used include Ki-67 (D2H10) Rabbit mAb (Cell Signaling Technology, #9027, Danvers, MA, USA), Met (D1C2) XP® Rabbit mAb (Cell Signaling Technology, #8198, Danvers, MA, USA), Phospho-Met (Tyr1234/1235) (D26) XP® Rabbit mAb (Cell Signaling Technology, #3077, Danvers, MA, USA), Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb (Cell Signaling Technology, #4060, Danvers, MA, USA), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Rabbit mAb (Cell Signaling Technology, #4370, Danvers, MA, USA), and PECAM1 (D8V9E) XP® Rabbit mAb (Cell Signaling Technology, #77699, Danvers, MA, USA); all were used according to the manufacturer’s recommendations. After slides were washed twice in 1× PBST, tumor sections were incubated with 1:200 biotinylated anti-rabbit IgG (Vector Laboratories, BA-1000, Burlingame, CA, USA). Detection and visualization of the tumor sections were performed using the avidin/biotin/peroxidase complex (Vector Laboratories, PK-4000, Burlingame, CA, USA) and 3,3′-diaminobenzidine (Invitrogen, 750118, Carlsbad, CA, USA). For TUNEL assay, the ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Millipore, S7100, Billerica, MA, USA) was used according to the manufacturer’s protocol. The IHC images of the tumor sections were obtained using the Aperio imaging system (Leica Biosystems, Wetzlar, Germany).
4.17. Statistical Analysis
All data were analyzed with GraphPad Prism® V5.01 (GraphPad Software, Inc., La Jolla, CA, USA) and expressed as the mean ± SEM unless otherwise noted. The statistical significance in tumor growth between different groups was analyzed by one-tailed two sample t-test.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}