Detection of Bacterial Pathogens from Broncho-Alveolar Lavage by Next-Generation Sequencing

 and
and
Abstract
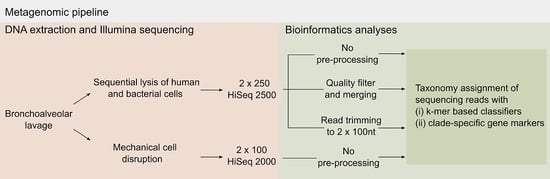
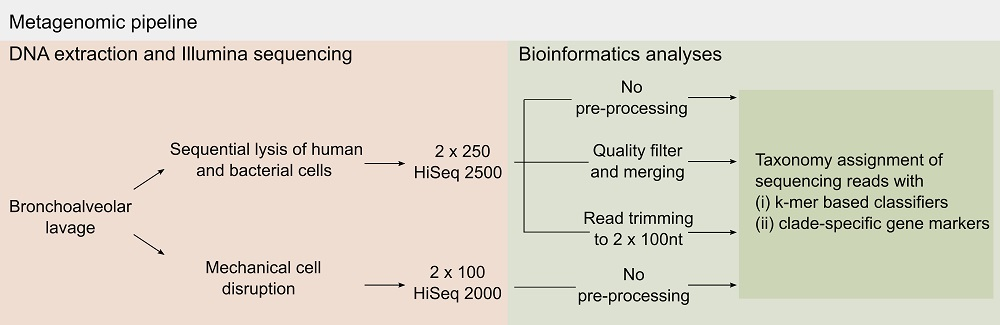
:
1. Introduction
2. Results and Discussion
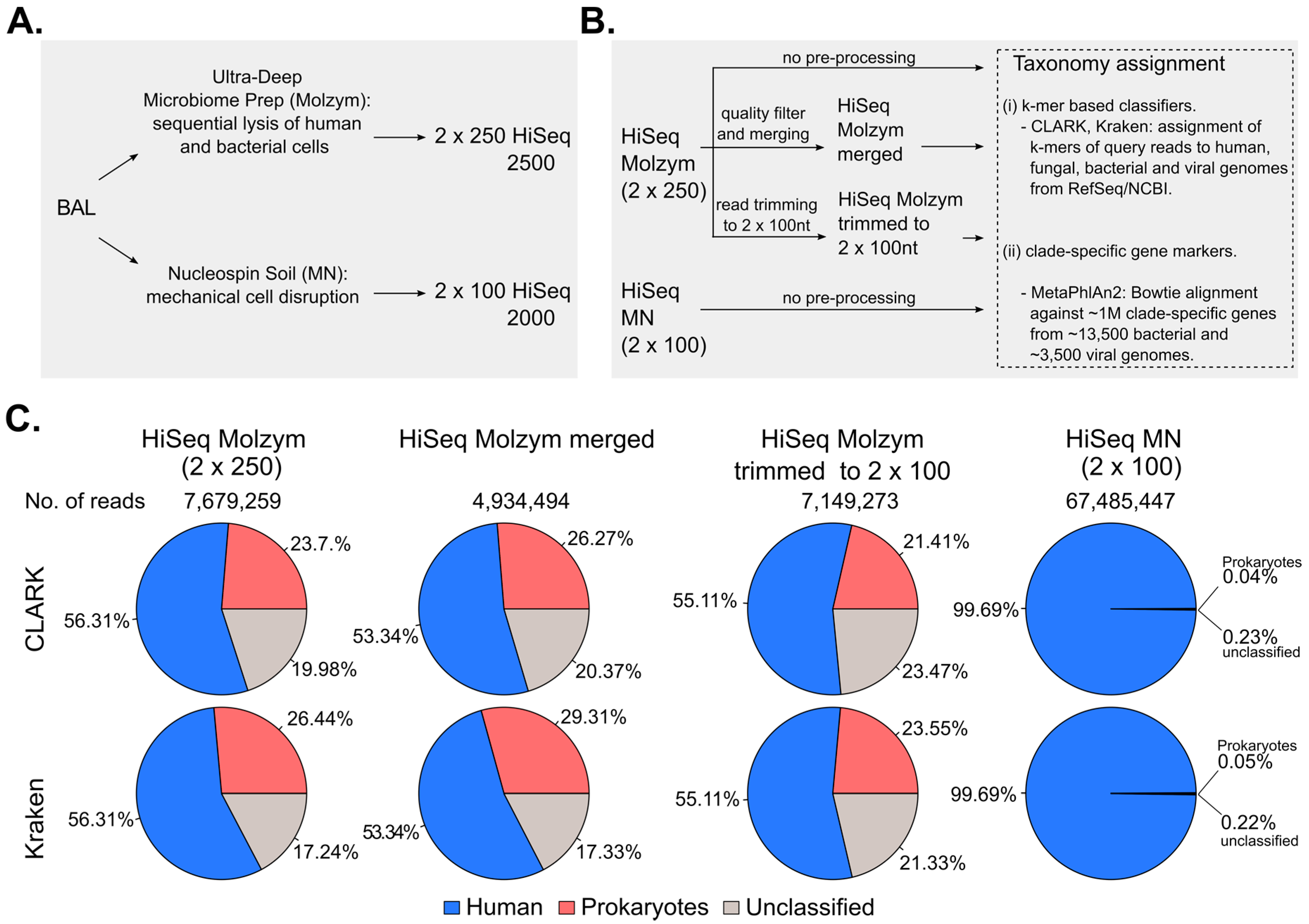
2.1. Pre-Processing of Sequencing Data
2.2. Effect of Host DNA Depletion on the Proportions of Bacterial Reads
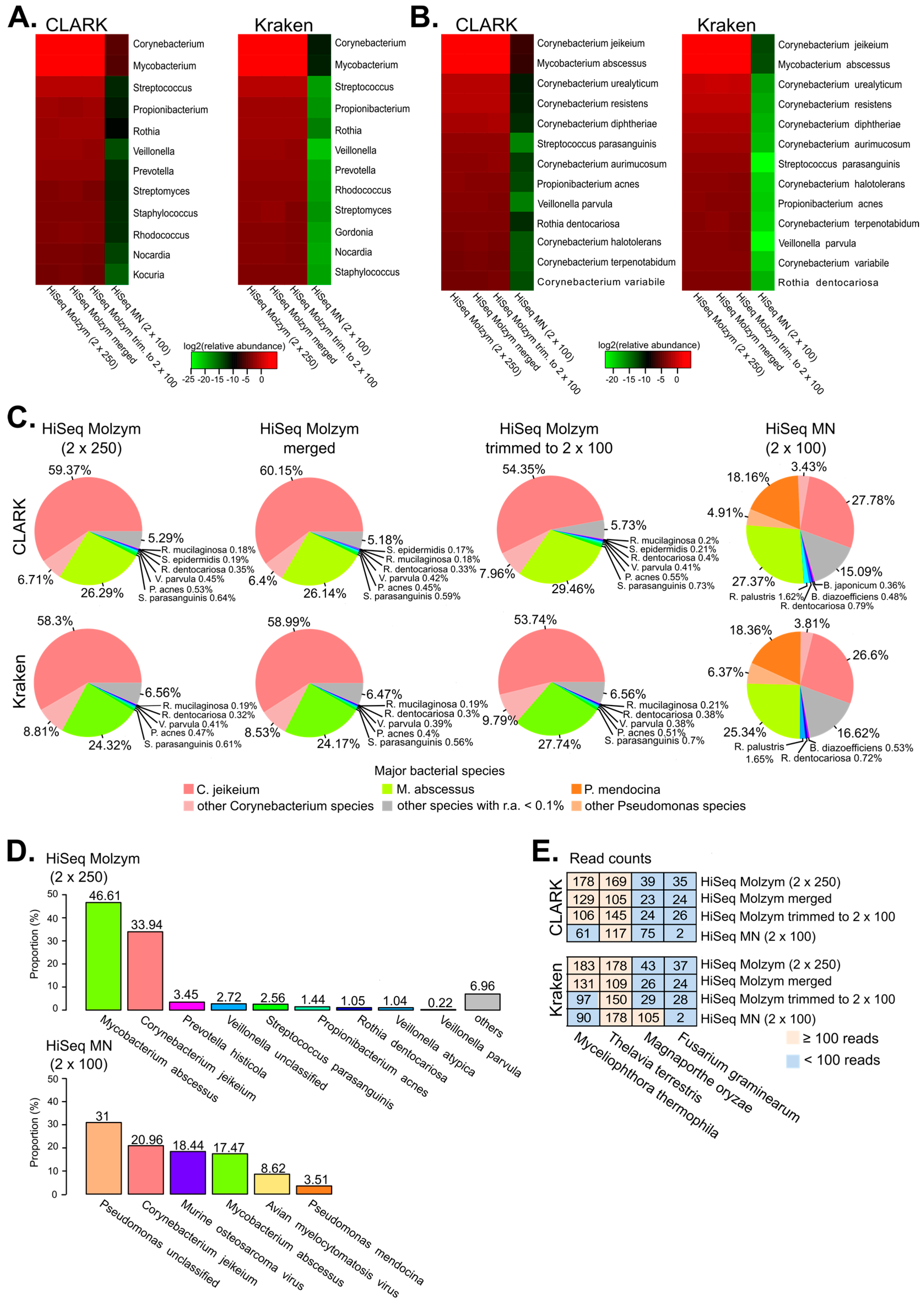
2.3. Identification of Bacterial Species by Metagenomics
2.4. Detection of Fungi and Viruses
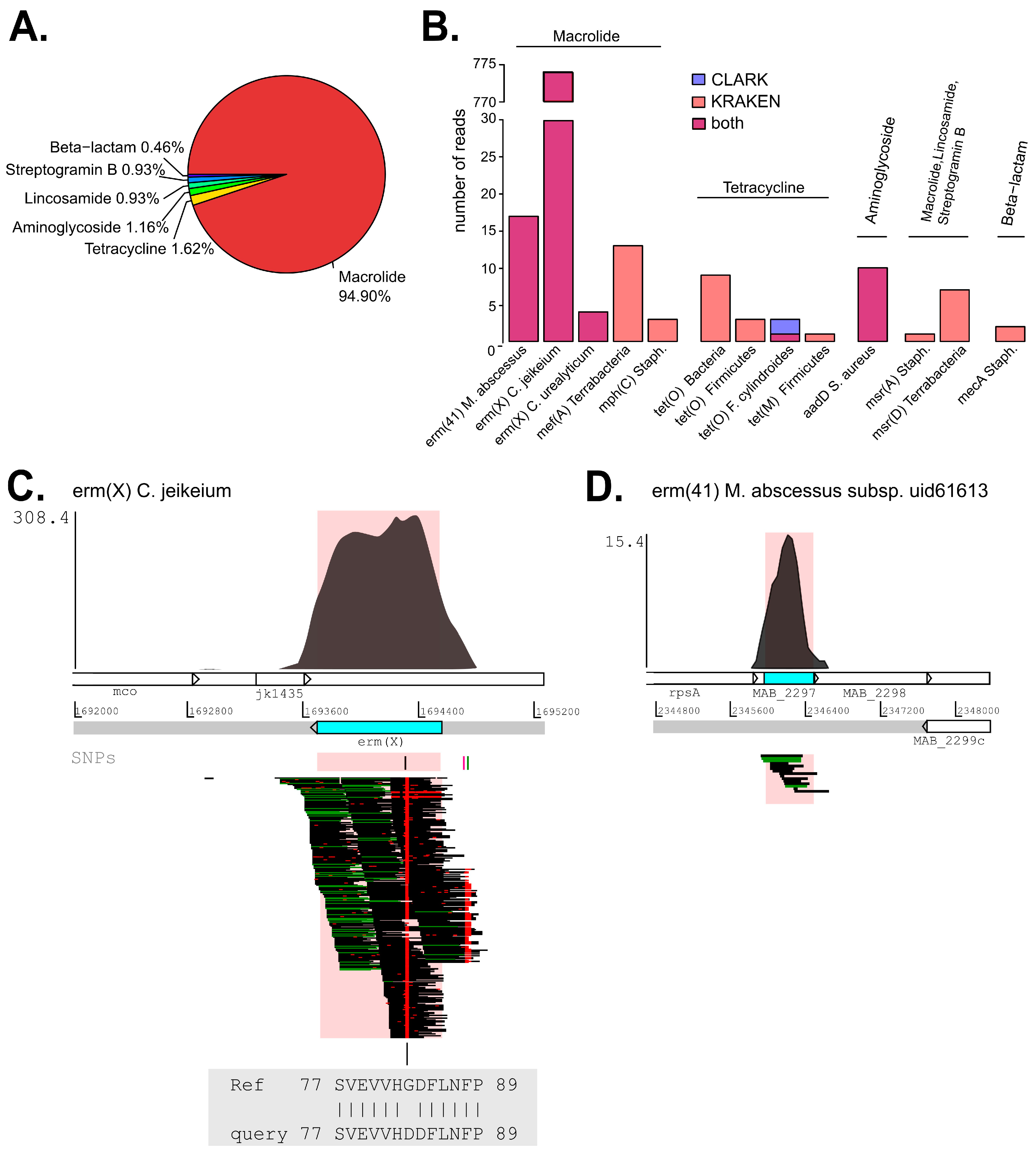
2.5. Analysis of Antibiotic Resistance Determinants (ARDs)
3. Materials and Methods
3.1. Patient Description
3.2. Culture Methods and Antibiograms
3.3. DNA Extraction without Selective Bacterial DNA Enrichment and Sequencing
3.4. Enrichment of Bacterial/Fungal DNA and Sequencing
3.5. qPCR Assays
3.6. Bioinformatics Analyses
3.7. Genome Databases
3.8. Data Availability
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BAL | Broncho-alveolar lavage |
| WMGS | Whole-metagenome shotgun |
| AST | Antibiotic susceptibility test |
| ARD | Antibiotic resistance determinant |
References
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid Whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Singh, H.; Li, W.; Moncera, K.; Torralba, M.; Yu, Y.; Manuel, O.; Biggs, W.; Venter, J.C.; Nelson, K.; et al. Microbial metagenome of urinary tract infection. bioRxiv 2017. [Google Scholar] [CrossRef]
- Schmidt, K.; Mwaigwisya, S.; Crossman, L.C.; Doumith, M.; Munroe, D.; Pires, C.; Khan, A.M.; Woodford, N.; Saunders, N.J.; Wain, J.; et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J. Antimicrob. Chemother. 2017, 72, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Ruppé, E.; Lazarevic, V.; Girard, M.; Mouton, W.; Ferry, T.; Laurent, F.; Schrenzel, J. Clinical metagenomics of bone and joint infections: A proof of concept study. Sci. Rep. 2017, 7, 7718. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Peggs, K.S.; Mattes, F.M.; Phadke, R.; Garson, J.A.; Grant, P.; Samayoa, E.; Federman, S.; Miller, S.; Lunn, M.P.; et al. Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin. Infect. Dis. 2015, 60, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Aoki, K.; Okuma, S.; Sato, T.; Ishii, Y.; Tateda, K. Metagenomic analysis for detecting pathogens in culture-negative infective endocarditis. J. Infect. Chemother. 2015, 21, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Joanna María, O.-A.; José Miguel, S.-C.; Fabiola, G.-A.; Elizabeth, G.-D.; Araceli, R.-C.; Patricia, A.-P.; Claudia, W.-A.; Maribel, G.-V.; Gloria, L.-Á.; Adda Jeanette, G.-C.; et al. Fatal Psychrobacter sp. infection in a pediatric patient with meningitis identified by metagenomic next-generation sequencing in cerebrospinal fluid. Arch. Microbiol. 2016, 198, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Blanco-Guzman, M.; Wylie, T.N.; Lawrence, S.J.; Ghobadi, A.; DiPersio, J.F.; Storch, G.A. High-throughput sequencing of cerebrospinal fluid for diagnosis of chronic Propionibacterium acnes meningitis in an allogeneic stem cell transplant recipient. Transpl. Infect. Dis. 2016, 18, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Gaïa, N.; Emonet, S.; Girard, M.; Renzi, G.; Despres, L.; Wozniak, H.; Yugueros Marcos, J.; Veyrieras, J.-B.; Chatellier, S.; et al. Challenges in the culture-independent analysis of oral and respiratory samples from intubated patients. Front. Cell. Infect. Microbiol. 2014, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Aas, J.A.; Paster, B.J.; Stokes, L.N.; Olsen, I.; Dewhirst, F.E. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 2005, 43, 5721–5732. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripoll, F.; Pasek, S.; Schenowitz, C.; Dossat, C.; Barbe, V.; Rottman, M.; Macheras, E.; Heym, B.; Herrmann, J.-L.; Daffé, M.; et al. Non Mycobacterial Virulence Genes in the Genome of the Emerging Pathogen Mycobacterium abscessus. PLoS ONE 2009, 4, e5660. [Google Scholar] [CrossRef] [PubMed]
- Tauch, A.; Kaiser, O.; Hain, T.; Goesmann, A.; Weisshaar, B.; Albersmeier, A.; Bekel, T.; Bischoff, N.; Brune, I.; Chakraborty, T.; et al. Complete Genome sequence and analysis of the multiresistant nosocomial pathogen Corynebacterium jeikeium K411, a lipid-requiring bacterium of the human skin flora. J. Bacteriol. 2005, 187, 4671–4682. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Gaïa, N.; Girard, M.; Schrenzel, J. Decontamination of 16S rRNA gene amplicon sequence datasets based on bacterial load assessment by qPCR. BMC Microbiol. 2016, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Brinkmann, A.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Kurth, A. Protocol for metagenomic virus detection in clinical specimens. Emerg. Infect. Dis. 2015, 21, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:1303.3997. [Google Scholar]
- Hays, C.; Lienhard, R.; Auzou, M.; Barraud, O.; Guérin, F.; Ploy, M.-C.; Cattoir, V. Erm(X)-mediated resistance to macrolides, lincosamides and streptogramins in Actinobaculum schaalii. J. Antimicrob. Chemother. 2014, 69, 2056–2060. [Google Scholar] [CrossRef] [PubMed]
- Torelli, R.; Sanguinetti, M.; Moody, A.; Pagano, L.; Caira, M.; De Carolis, E.; Fuso, L.; De Pascale, G.; Bello, G.; Antonelli, M.; et al. Diagnosis of invasive aspergillosis by a commercial real-time PCR assay for aspergillus DNA in bronchoalveolar lavage fluid samples from high-risk patients compared to a galactomannan enzyme immunoassay. J. Clin. Microbiol. 2011, 49, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, P.; Springer, B.; Vogel, U.; Meier, A.; Wrede, A.; Kiekenbeck, M.; Bange, F.C.; Böttger, E.C. Genotypic identification of mycobacteria by nucleic acid sequence determination: Report of a 2-year experience in a clinical laboratory. J. Clin. Microbiol. 1993, 31, 2882–2889. [Google Scholar] [PubMed]
- Ciardo, D.E.; Lucke, K.; Imhof, A.; Bloemberg, G.V.; Böttger, E.C. Systematic internal transcribed spacer sequence analysis for identification of clinical mold isolates in diagnostic mycology: A 5-year study. J. Clin. Microbiol. 2010, 48, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Extraction Kit | DNA Concentration (pg/μL) in the Extracts Estimated by qPCR | Input Sample Volume (μL) | DNA Extract Volume (μL) | Percentage of Bacterial DNA in Extracts | Percentage of Human DNA in Extracts | Ratio Bacterial-Human DNA | |
|---|---|---|---|---|---|---|---|
| Human | Bacterial | ||||||
| Nucleospin Soil-MN | 4197 | 5 | 600 | 60 | 0.12 | 99.88 | 0.0012 |
| Ultra-Deep Microbiome Prep-Molzym | 27 | 10 | 600 | 100 | 27.03 | 72.97 | 0.37 |
| Bacteria | Detection |
| Corynebacterium jeikeium | >1.0 × 105 CFU/mL |
| Rothia dentocariosa | 1.0 × 102 CFU/mL |
| Mycobacterium abscessus | Present |
| Fungi | Detection |
| Pupureocillium lilacinum | Present |
| Penicillium sp. | Present |
| Scopulariopsis sp. | Present |
| CLARK | Kraken | |||||||
|---|---|---|---|---|---|---|---|---|
| Viruses | HiSeq Molzym (2 × 250) | HiSeq Merged | HiSeq Trim. to 100 nt | HiSeq MN (2 × 100) | HiSeq Molzym (2 × 250) | HiSeq Merged | HiSeq Trim. to 100 nt | HiSeq MN (2 × 100) |
| Torque teno viruses | 198 | 119 | 188 | 2313 | 298 | 208 | 248 | 2992 |
| Propionibacterium phage | 35 | 15 | 33 | 3 | 50 | 26 | 45 | 3 |
| Mycobacterium phage | 26 | 12 | 17 | 0 | 27 | 12 | 17 | 0 |
| Staphylococcus phage | 26 | 12 | 23 | 0 | 24 | 17 | 12 | 0 |
| Streptococcus phage | 24 | 17 | 15 | 0 | 29 | 15 | 23 | 0 |
| Enterobacteria phage phiX174 | 0 | 0 | 0 | 27,393 | 0 | 0 | 0 | 27,405 |
| Human herpesvirus | 4 | 1 | 2 | 193 | 4 | 1 | 2 | 230 |
| Falconid herpesvirus | 0 | 0 | 0 | 133 | 0 | 0 | 0 | 144 |
| ResFinder | C. jeikeium | M. abscessus | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Identity: | 0.9 | 0.3 | 0.5 | 0.9 | 0.3 | 0.5 | 0.9 | ||
| Gene | Allele | Classes of Antibiotics | |||||||
| erm(X) | erm(X)_4_NC_005206 | Macrolide | 544 | 544 | 544 | 538 | 0 | 0 | 0 |
| erm(X) | erm(X)_2_X51472 | Macrolide | 185 | 185 | 185 | 181 | 0 | 0 | 0 |
| erm(X) | erm(X)_1_M36726 | Macrolide | 49 | 49 | 49 | 49 | 0 | 0 | 0 |
| erm(41) | erm(41)_1_EU177504 | Macrolide | 17 | 0 | 0 | 0 | 17 | 17 | 16 |
| mef(A) | mef(A)_10_AF376746 | Macrolide | 13 | 0 | 0 | 0 | 0 | 0 | 0 |
| aadD | aadD_1_AF181950 | Aminoglycoside | 10 | 0 | 0 | 0 | 0 | 0 | 0 |
| msr(D) | msr(D)_2_AF274302 | Macrolide, lincosamide and streptogramin B | 7 | 0 | 0 | 0 | 0 | 0 | 0 |
| tet(O) | tet(O)_2_M20925 | Tetracycline | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| tet(O) | tet(O)_3_Y07780 | Tetracycline | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| mph(C) | mph(C)_2_AF167161 | Macrolide | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| tet(O) | tet(O)_1_M18896 | Tetracycline | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| mecA | mecA_10_AB512767 | β-lactam | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| cfxA2 | cfxA2_1_AF504914 | β-lactam | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| cfxA | cfxA_1_U38243 | β-lactam | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| msr(A) | msr(A)_1_X52085 | Macrolide, lincosamide and streptogramin B | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| tet(M) | tet(M)_3_U08812 | Tetracycline | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total number of reads | 847 | ||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leo, S.; Gaïa, N.; Ruppé, E.; Emonet, S.; Girard, M.; Lazarevic, V.; Schrenzel, J. Detection of Bacterial Pathogens from Broncho-Alveolar Lavage by Next-Generation Sequencing. Int. J. Mol. Sci. 2017, 18, 2011. https://doi.org/10.3390/ijms18092011
Leo S, Gaïa N, Ruppé E, Emonet S, Girard M, Lazarevic V, Schrenzel J. Detection of Bacterial Pathogens from Broncho-Alveolar Lavage by Next-Generation Sequencing. International Journal of Molecular Sciences. 2017; 18(9):2011. https://doi.org/10.3390/ijms18092011
Chicago/Turabian StyleLeo, Stefano, Nadia Gaïa, Etienne Ruppé, Stephane Emonet, Myriam Girard, Vladimir Lazarevic, and Jacques Schrenzel. 2017. "Detection of Bacterial Pathogens from Broncho-Alveolar Lavage by Next-Generation Sequencing" International Journal of Molecular Sciences 18, no. 9: 2011. https://doi.org/10.3390/ijms18092011