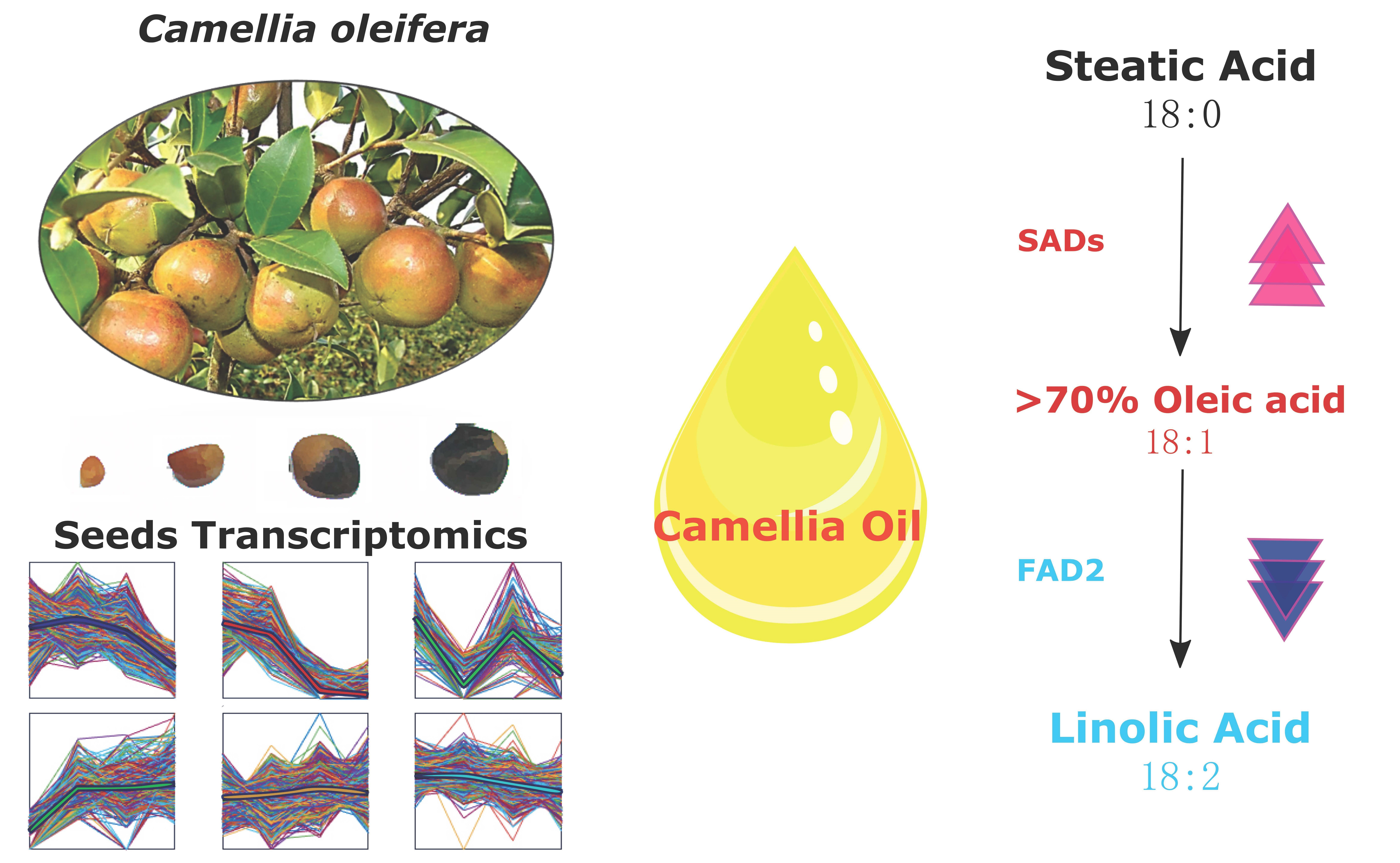
Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition


Abstract
:
1. Introduction
2. Results
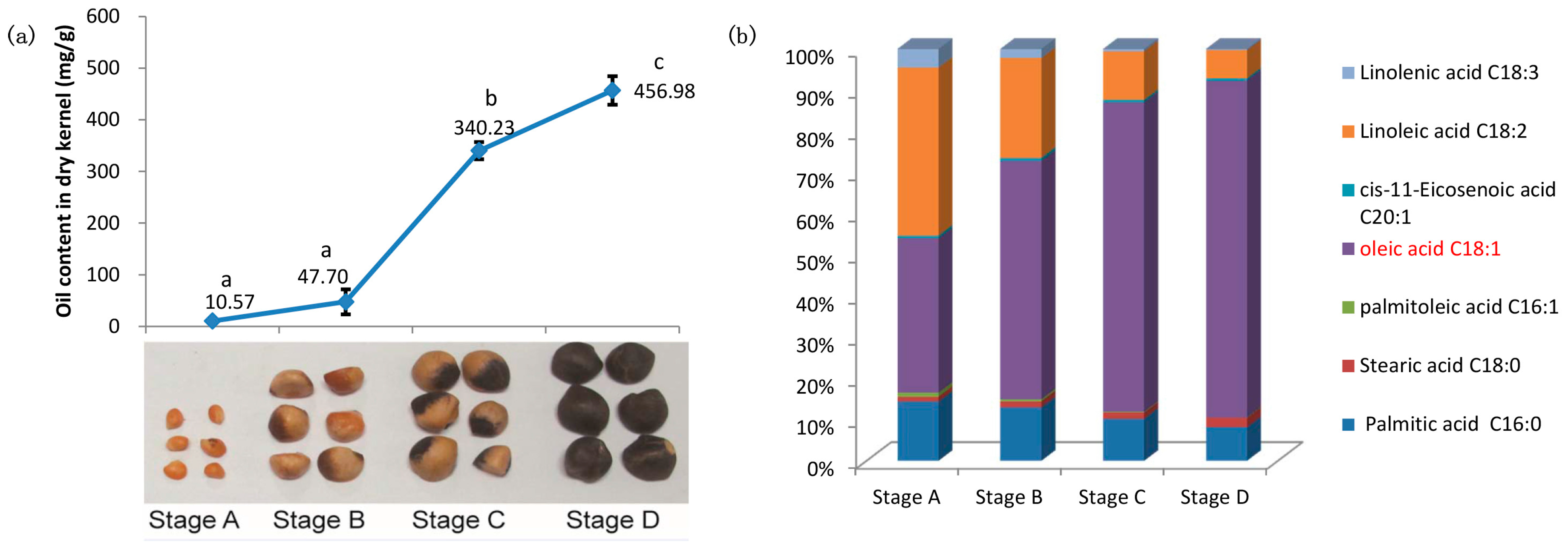
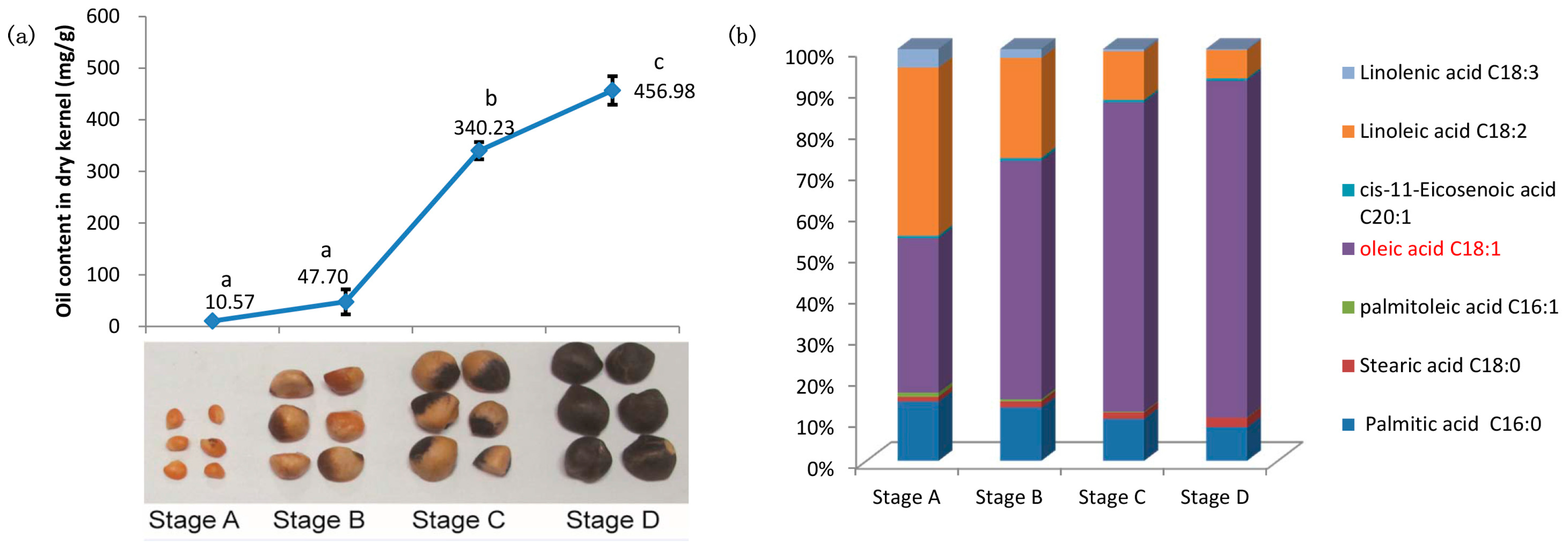
2.1. Characterizations of Oil Content and Fatty Acid (FA) Composition during Seed Development of C. oleifera
2.2. RNA-Seq, De Novo Transcriptome Assembly and Functional Annotation
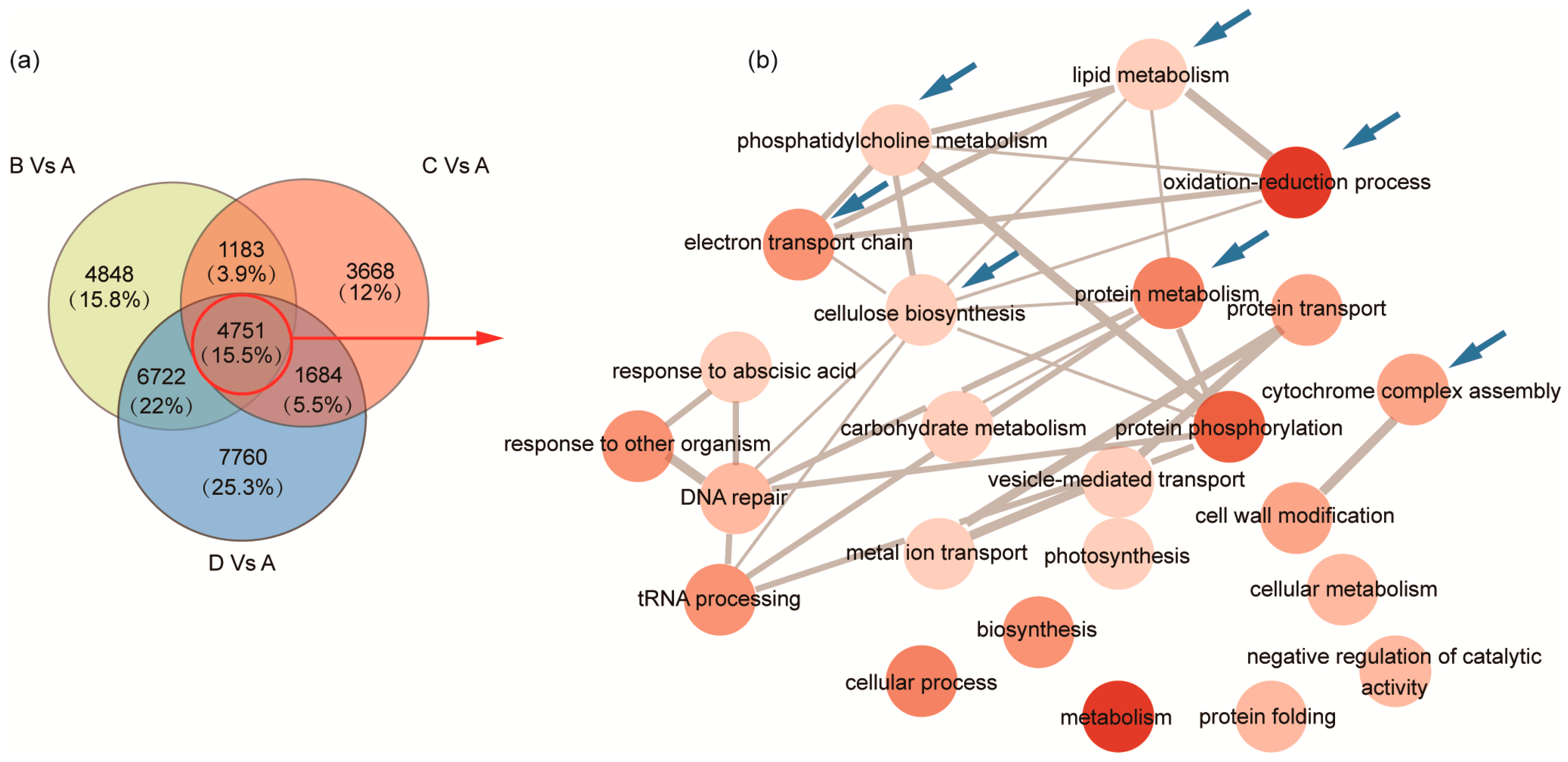
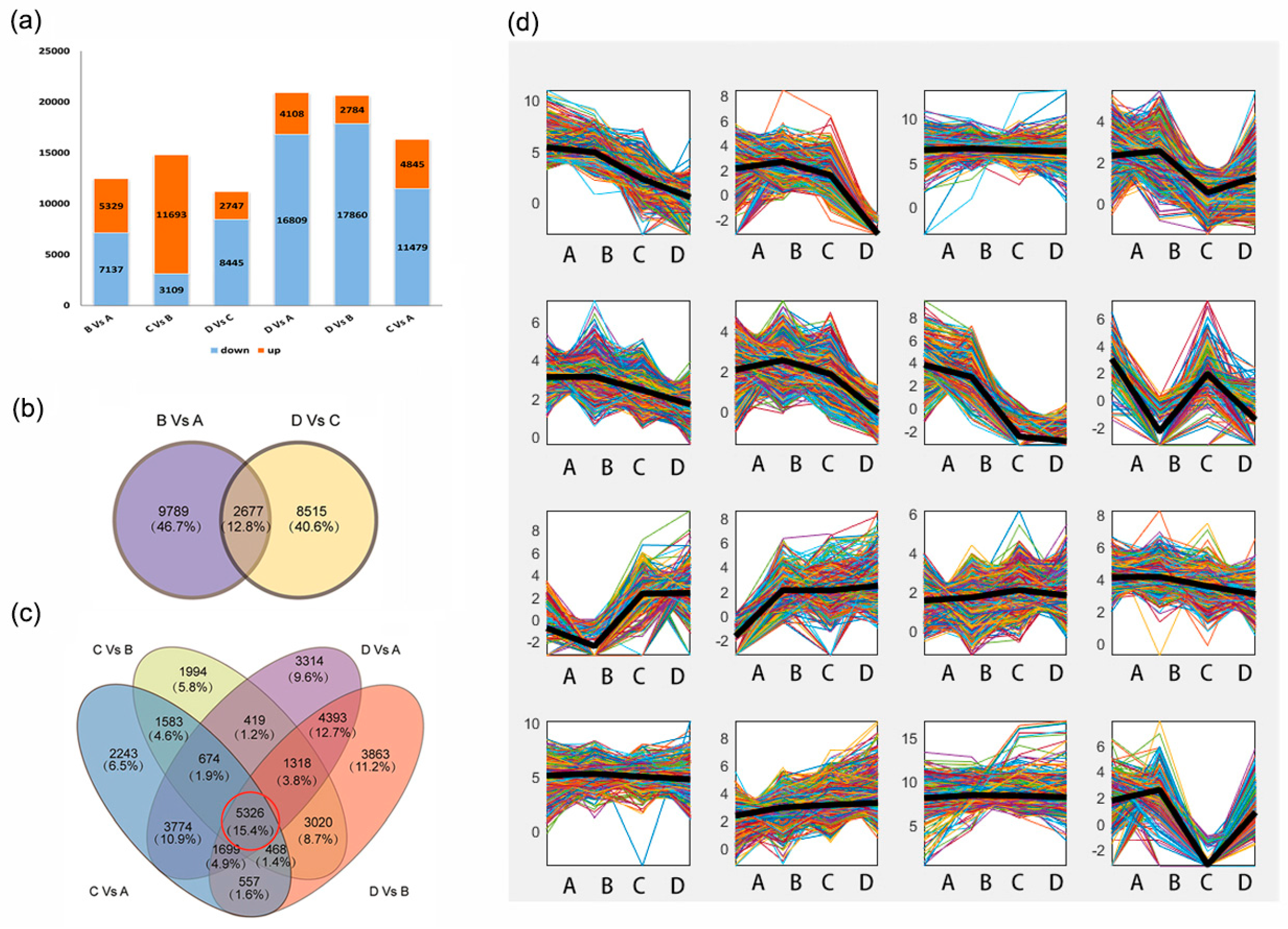
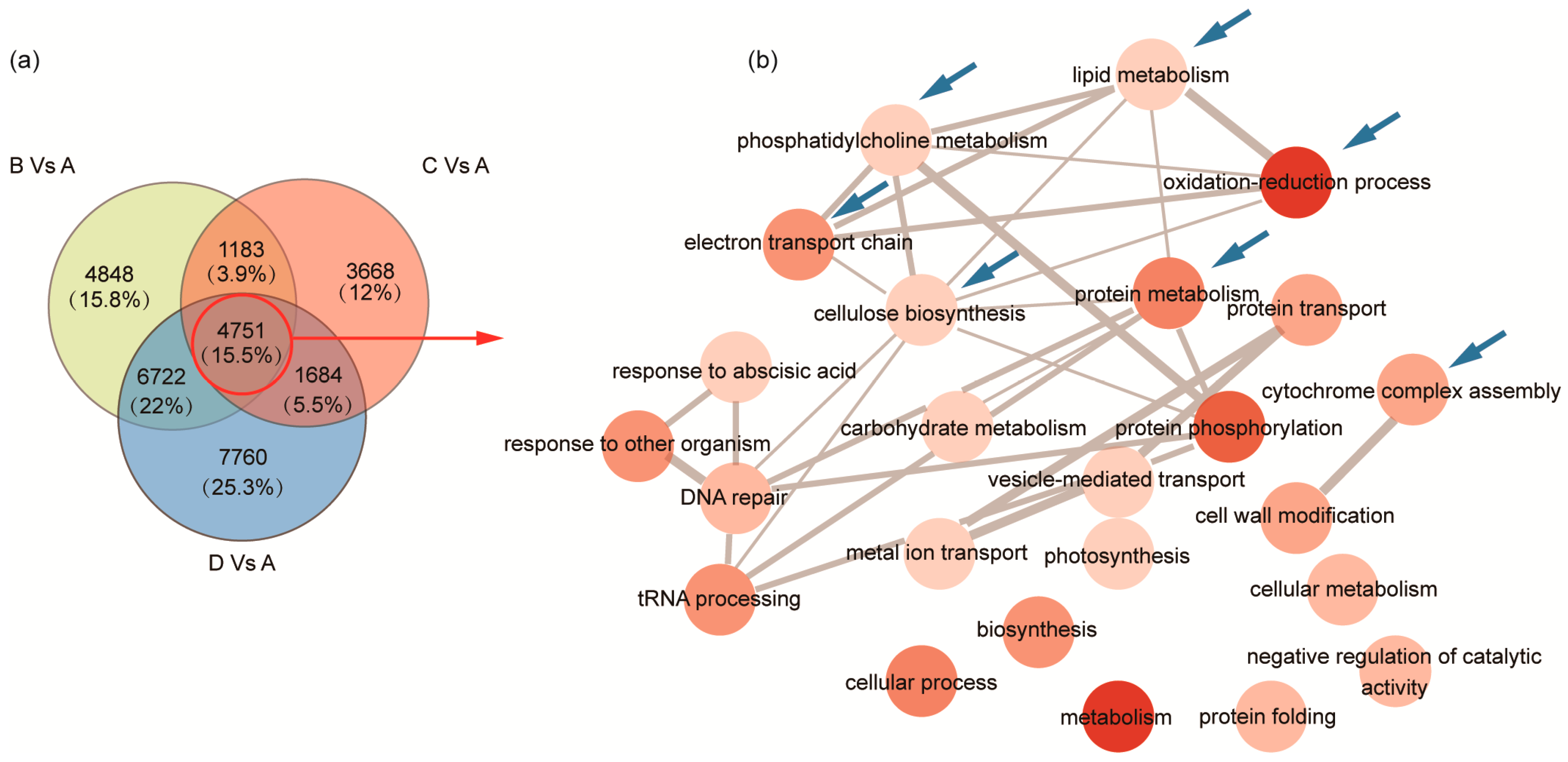
2.3. Identification of DEGs and GO Enrichment Analysis during Seed Development
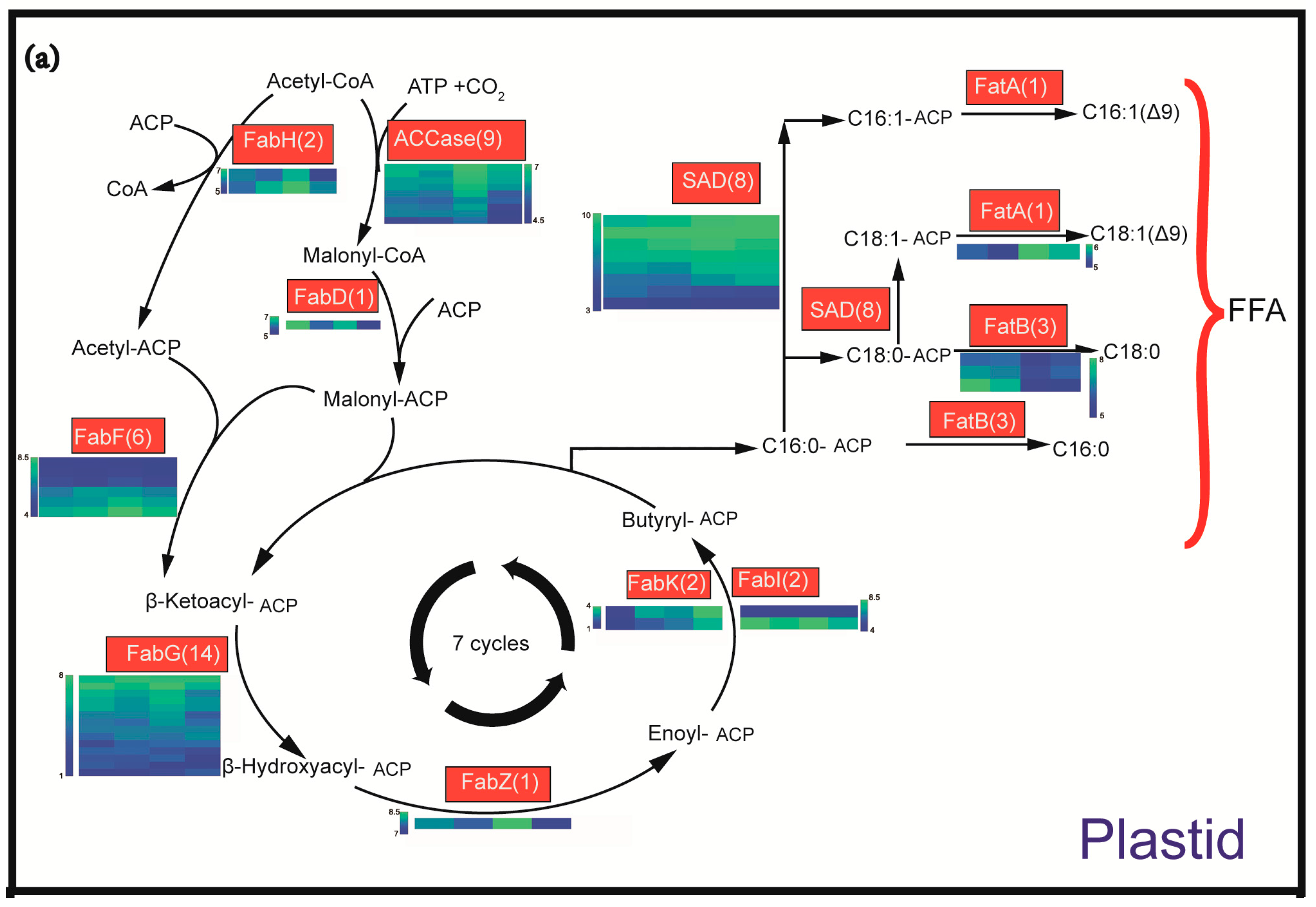
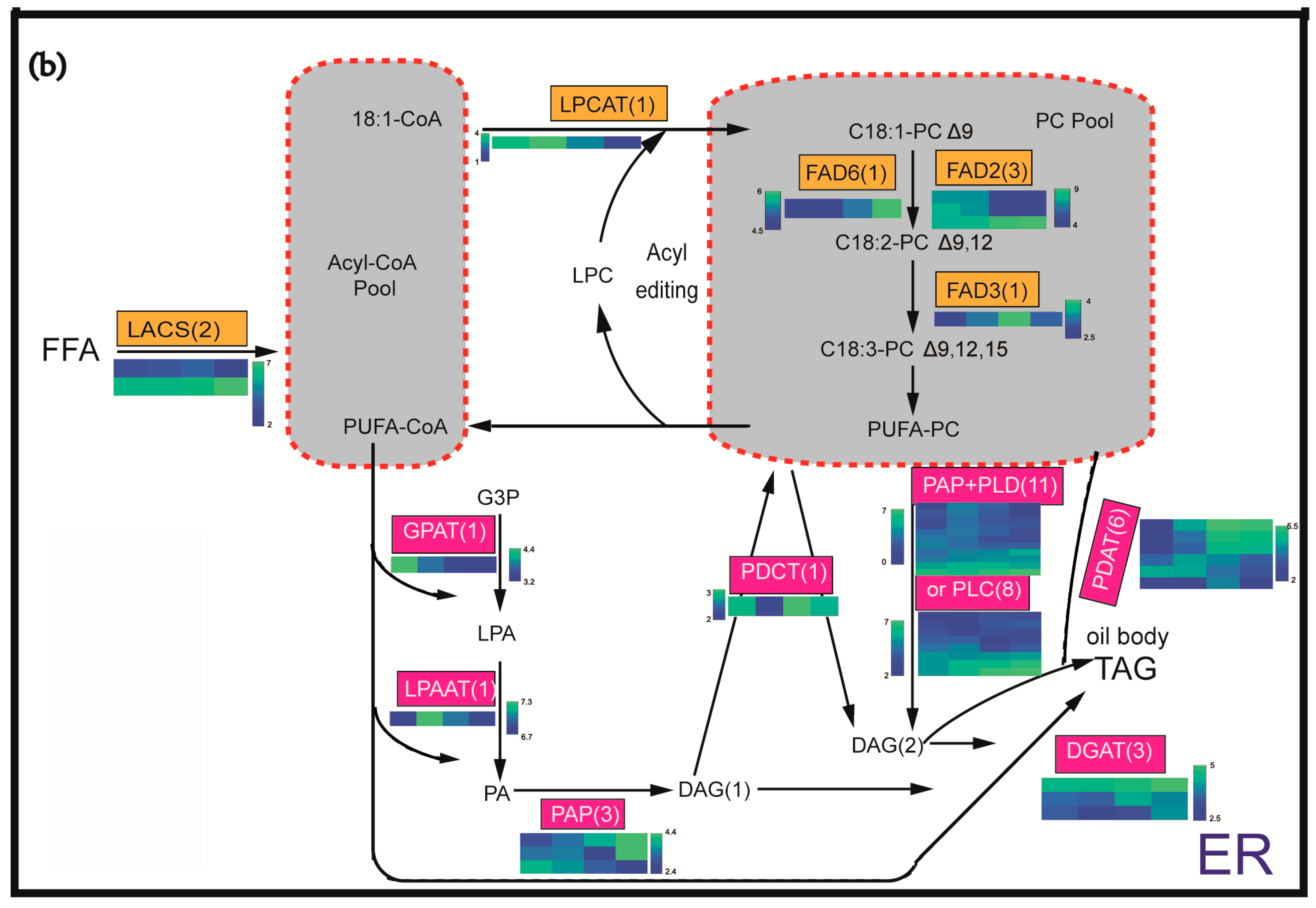
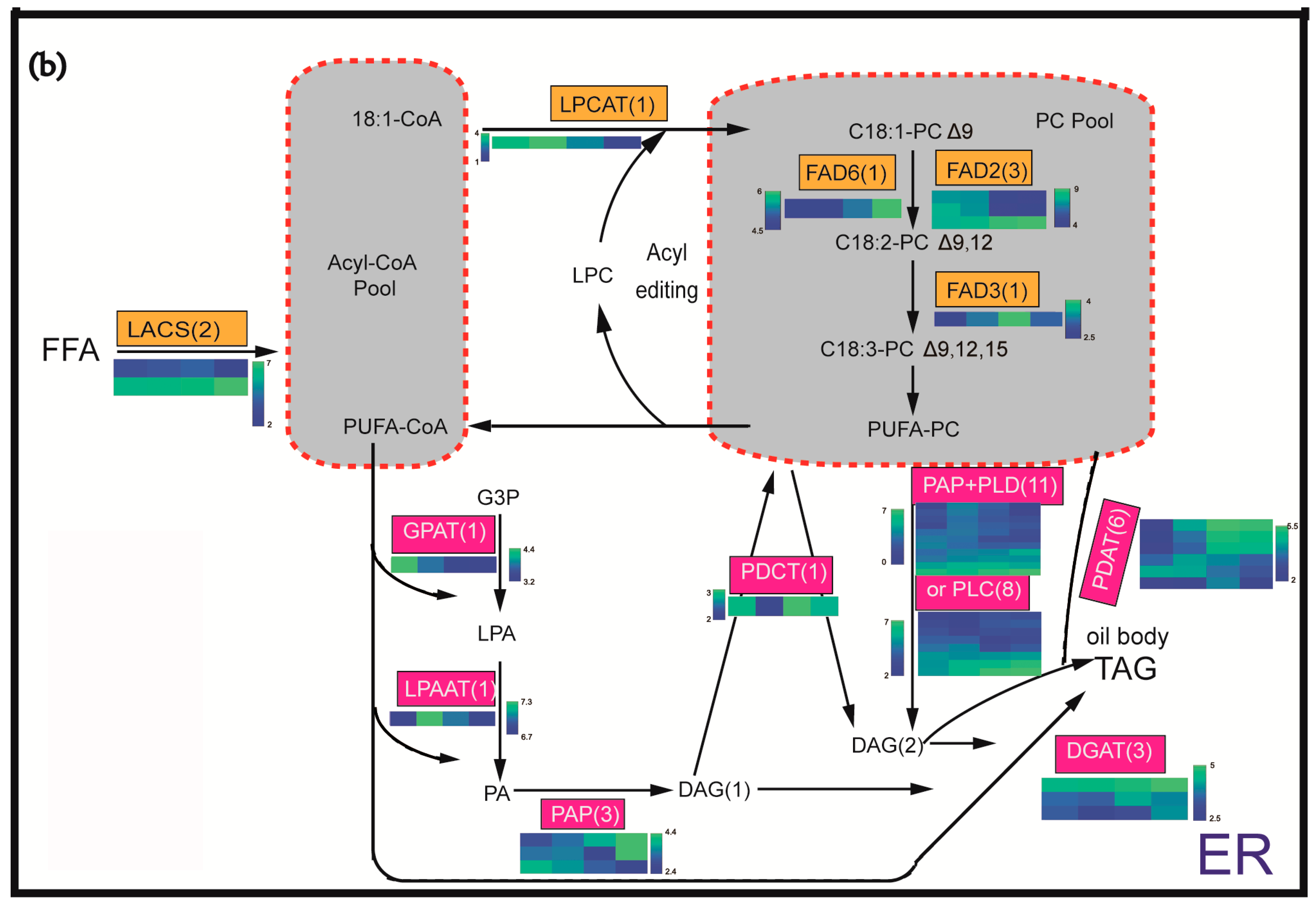
2.4. Identification and Expression Profiling of Enzymes Involved in Lipid Biosynthesis
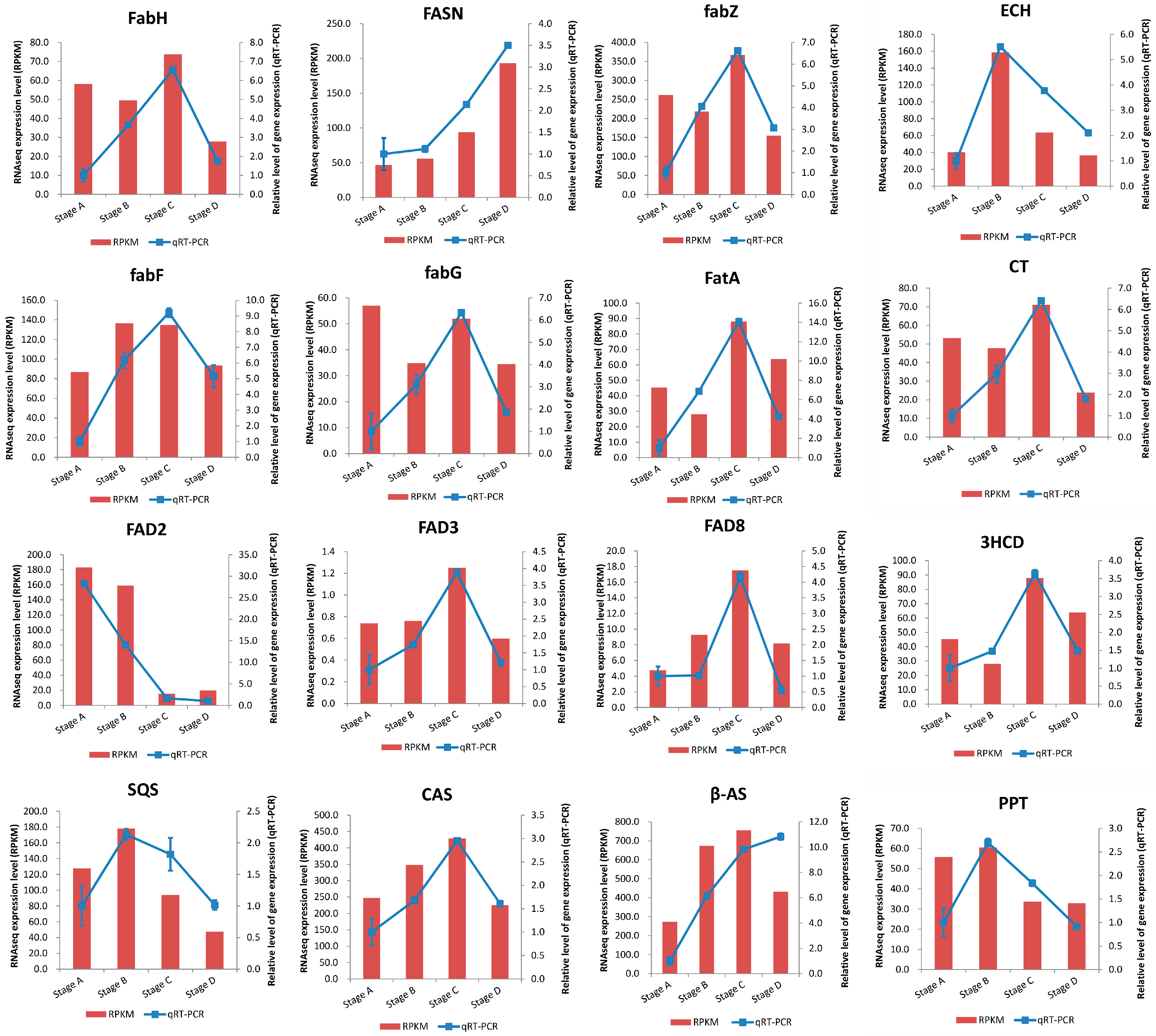
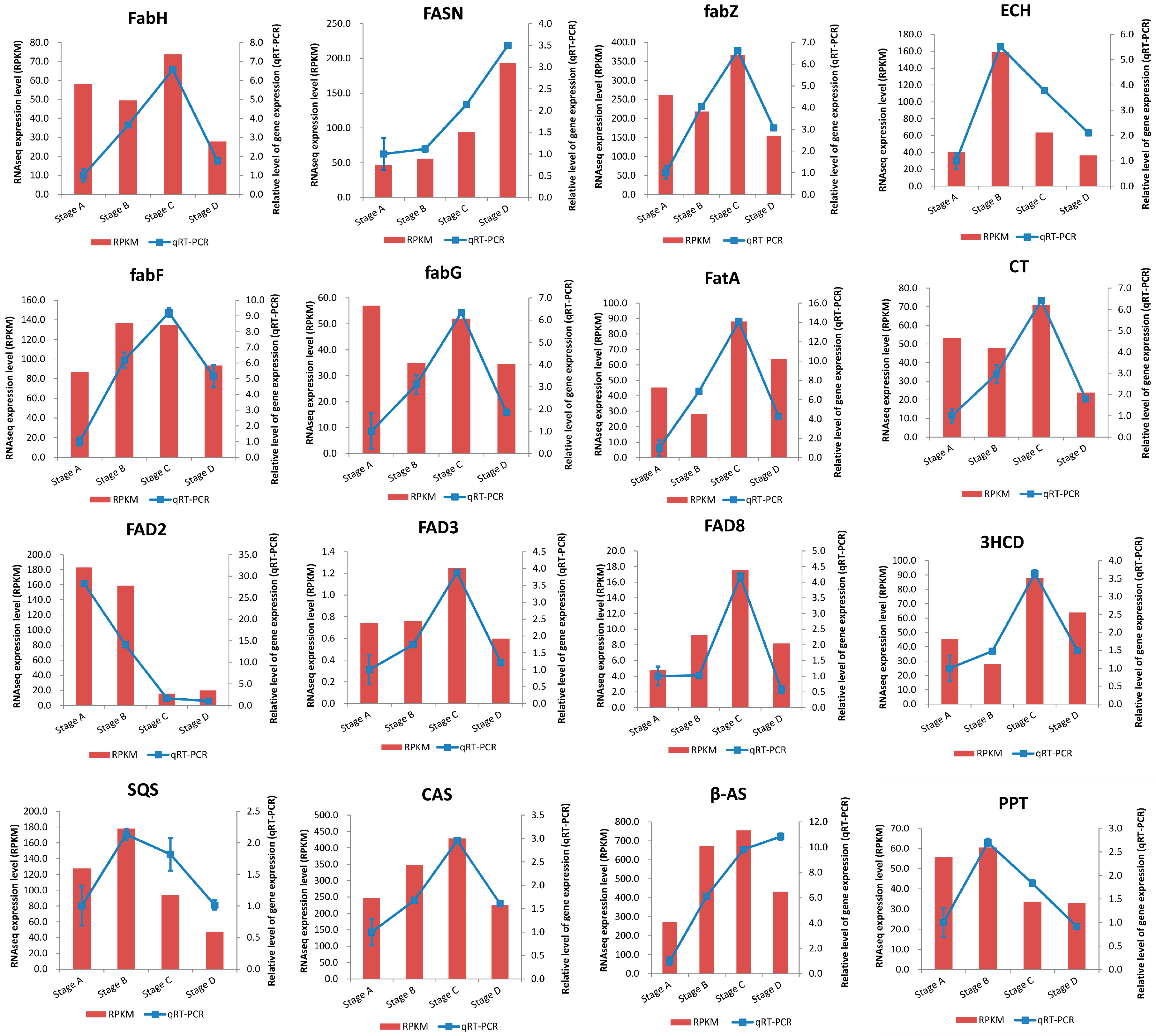
2.5. Validation of RNA-Seq Results by qRT-PCR
3. Discussion
3.1. Oil Content and FA Composition Correlated with Expression Profiles of Lipid Biosynthesis Genes during the Seed Development of C. oleifera
3.2. A Synergistic Regulation of SADs and FAD2 Contributing to the High 18:1 FA Content
3.3. Utilization of DAG Production by PLD or PLC Plus PAP as the Substrate for TAG Synthesis May Be the Important Pathway of the Incorporation of FA PC-Derived in TAG in C. oleifera
3.4. DGATs May Be Rate-Limiting Genes in the Control of the TAG Flux and Oil Production
4. Materials and Methods
4.1. Plant Materials
4.2. Oil Content and FA Component Analysis
4.3. Total RNA Extraction
4.4. Transcriptome Sequencing, Assembly and Functional Annotation
4.5. Clustering, Identification of DEGs and GO Enrichment
4.6. Quantitative RT-PCR Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| COG | Clusters of Orthologous Groups |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| PDAT | Phospholipid:diacylglycerol acyltransferase |
| FatA and FatB | Acyl-ACP thioesterase |
| PC | Phosphatidylcholine |
| GPAT | Acyl-CoA:glycerol-3-phosphate acyltransferase |
| LPAAT | Acyl-CoA:lyso-phosphatidic acid acyltransferase |
| PUFA | Polyunsaturated fatty acid |
| ER | Endoplasmic reticulum |
| FabH | Acetyl CoA:ACP transacylase |
| FabD | Malonyl CoA:ACP transacylase |
| FabF | β-Ketoacyl-ACP synthase |
| FabG | β-Ketoacyl-ACP reductase |
| FabZ | β-Hydroxyacyl ACP dehydrase |
| FabI and FabK | Enoyl-ACP reductase |
| FFA | Free fatty acid |
| LACS | Long chain acyl-CoA synthetase |
| LPCAT | Acyl-CoA:LPC acyltransferase |
| LPC | Lysophosphatidylcholine |
| FAD | Fatty acid desaturase |
| G3P | Glycerol-3-phosphate |
| GPAT | Acyl-CoA:G3P acyltransferase |
| LPA | Lyso-phosphatidic acid |
| PA | Phosphatidic acid |
| PAP | PA phosphatase |
| PLC | Phospholipase C |
| PLD | Phospholipase D |
References
- Li, H.; Zhou, G.Y.; Zhang, H.Y.; Liu, J.A. Research progress on the health function of tea oil. J. Med. Plants Res. 2011, 5, 485–489. [Google Scholar]
- Yao, X.; Wang, Y.; Wang, K.; Ren, H. Effects of geographic latitude and longitude on fat and its fatty acid composition of oil-tea Camellia seeds. China Oils Fats 2011, 36, 31–34. [Google Scholar]
- Yang, C.; Liu, X.; Chen, Z.; Lin, Y.; Wang, S. Comparison of Oil Content and Fatty Acid Profile of Ten New Camellia oleifera Cultivars. J. Lipids 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ohlrogge, J.B. Design of New Plant Products: Engineering of Fatty Acid Metabolism. Plant Physiol. 1994, 104, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ren, S.; Tan, K. Study on climatic regionalization and layer and belt distribution of oiltea Camellia quality in China. J. Nat. Res. 1999, 14, 123–127. [Google Scholar]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Ohlrogge, J.B.; Jaworski, J.G. Regulation of Fatty Acid Synthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 109–136. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.D.; Ohlrogge, J.B. Compartmentation of triacylglycerol accumulation in plants. J. Biol. Chem. 2012, 287, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Bafor, M.; Jonsson, L.; Stobart, A.K.; Stymne, S. Regulation of triacylglycerol biosynthesis in embryos and microsomal preparations from the developing seeds of Cuphea lanceolata. Biochem. J. 1990, 272, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Ohlrogge, J. Supply of fatty acid is one limiting factor in the accumulation of triacylglycerol in developing embryos. Plant Physiol. 1999, 120, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Lepiniec, L. Physiological and developmental regulation of seed oil production. Prog. Lipid Res. 2010, 49, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Wuilleme, S.; To, A.; Rochat, C.; Lepiniec, L. Role of WRINKLED1 in the transcriptional regulation of glycolytic and fatty acid biosynthetic genes in Arabidopsis. Plant J. Cell Mol. Biol. 2009, 60, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Ruuska, S.A.; Girke, T.; Benning, C.; Ohlrogge, J.B. Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 2002, 14, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.H.; Cha, T.S. A mesocarp-and species-specific cDNA clone from oil palm encodes for sesquiterpene synthase. Plant Sci. Int. J. Exp. Plant Biol. 2000, 154, 153–160. [Google Scholar] [CrossRef]
- Thompson, G.A.; Scherer, D.E.; Foxall-Van Aken, S.; Kenny, J.W.; Young, H.L.; Shintani, D.K.; Kridl, J.C.; Knauf, V.C. Primary structures of the precursor and mature forms of stearoyl-acyl carrier protein desaturase from safflower embryos and requirement of ferredoxin for enzyme activity. Proc. Natl. Acad. Sci. USA 1991, 88, 2578–2582. [Google Scholar] [CrossRef] [PubMed]
- Fofana, B.; Cloutier, S.; Duguid, S.; Ching, J.; Rampitsch, C. Gene expression of stearoyl-ACP desaturase and Δ12 fatty acid desaturase 2 is modulated during seed development of flax (Linum usitatissimum). Lipids 2006, 41, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Zhao, Y.; Prakash, C.S.; He, G.; Yin, D. Insights into the novel members of the FAD2 gene family involved in high-oleate fluxes in peanut. Genome 2015, 58, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Guschina, I.A.; O’Hara, P.; Slabas, A.R.; Quant, P.A.; Fawcett, T.; Harwood, J.L. Metabolic control analysis of developing oilseed rape (Brassica napus cv Westar) embryos shows that lipid assembly exerts significant control over oil accumulation. New Phytol. 2012, 196, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Lardizabal, K.; Effertz, R.; Levering, C.; Mai, J.; Pedroso, M.C.; Jury, T.; Aasen, E.; Gruys, K.; Bennett, K. Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol. 2008, 148, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Shockey, J.M.; Gidda, S.K.; Chapital, D.C.; Kuan, J.C.; Dhanoa, P.K.; Bland, J.M.; Rothstein, S.J.; Mullen, R.T.; Dyer, J.M. Tung tree DGAT1 and DGAT2 have nonredundant functions in triacylglycerol biosynthesis and are localized to different subdomains of the endoplasmic reticulum. Plant Cell 2006, 18, 2294–2313. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.C.; Zhang, Y.; Kumar, A.; Francis, T.; Giblin, E.M.; Barton, D.L.; Ferrie, J.R.; Laroche, A.; Shah, S.; Zhu, W.; et al. Molecular modification of triacylglycerol accumulation by over-expression of DGAT1 to produce canola with increased seed oil content under field conditions. Botany 2009, 87, 533–543. [Google Scholar] [CrossRef]
- Clemente, T.E.; Cahoon, E.B. Soybean Oil: Genetic Approaches for Modification of Functionality and Total Content. Plant Physiol. 2009, 151, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Zhang, T.; Zhang, Q.X.; Chen, M.; Wang, Z.J.; Zheng, B.S.; Xia, G.H.; Yang, X.Y.; Huang, C.Y.; Huang, Y.J. The mechanism of high contents of oil and oleic acid revealed by transcriptomic and lipidomic analysis during embryogenesis in Carya cathayensis Sarg. BMC Genom. 2016, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, E.; Banas, A.; Stymne, S. Fatty acid distribution and lipid metabolism in developing seeds of laurate-producing rape (Brassica napus L.). Planta 1997, 203, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Bourgis, F.; Kilaru, A.; Cao, X.; Ngando-Ebongue, G.F.; Drira, N.; Ohlrogge, J.B.; Arondel, V. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc. Natl. Acad. Sci. USA 2011, 108, 12527–12532. [Google Scholar] [CrossRef] [PubMed]
- Unver, T.; Wu, Z.; Sterck, L.; Turktas, M.; Lohaus, R.; Li, Z.; Yang, M.; He, L.; Deng, T.; Escalante, F.J.; et al. Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E9413–E9422. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Yang, H.; Wei, C.; Yu, O.; Zhang, Z.; Jiang, C.; Sun, J.; Li, Y.; Chen, Q.; Xia, T.; et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, X.; Huang, X.; Duan, S.; Long, C.; Chen, J.; Rong, J. Leaf transcriptome analysis of a subtropical evergreen broadleaf plant, wild oil-tea Camellia (Camellia oleifera), revealing candidate genes for cold acclimation. BMC Genom. 2017, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yang, Z.; Bai, W.; Chen, S.; Xu, W.; El-Kassaby, Y.A.; Chen, H. Transcriptome comparative analysis of two Camellia species reveals lipid metabolism during mature seed natural drying. Trees 2017, 31, 1827–1848. [Google Scholar] [CrossRef]
- Dong, B.; Wu, B.; Hong, W.; Li, X.; Li, Z.; Xue, L.; Huang, Y. Transcriptome analysis of the tea oil Camellia (Camellia oleifera) reveals candidate drought stress genes. PLoS ONE 2017, 12, e0181835. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.L.; Tan, X.F.; Zhang, L.; Jiang, N.; Cao, H.P. Identification and expression of fructose-1,6-bisphosphate aldolase genes and their relations to oil content in developing seeds of tea oil tree (Camellia oleifera). PLoS ONE 2014, 9, e107422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Q.; Tan, X.F.; Xie, L.S.; Chen, H.P.; Qiu, J.; Hu, F.M. The cDNA cloning and characteristic of stearoyl-ACP desaturase gene of Camellia oleifera. Sci. Silvae Sin. 2008, 44, 155–159. [Google Scholar]
- Lin, P.; Zhou, C.F.; Yao, X.H.; Cao, Y.Q. Sequence and expression characterization of two genes encoding Δ-12 fatty acid desaturases from Camellia oleifera. For. Res. 2016, 29, 743–751. [Google Scholar]
- Xia, E.H.; Jiang, J.J.; Huang, H.; Zhang, L.P.; Zhang, H.B.; Gao, L.Z. Transcriptome analysis of the oil-rich tea plant, Camellia oleifera, reveals candidate genes related to lipid metabolism. PLoS ONE 2014, 9, e104150. [Google Scholar] [CrossRef] [PubMed]
- Andre, C.; Haslam, R.P.; Shanklin, J. Feedback regulation of plastidic acetyl-CoA carboxylase by 18:1-acyl carrier protein in Brassica napus. Proc. Natl. Acad. Sci. USA 2012, 109, 10107–10112. [Google Scholar] [CrossRef] [PubMed]
- Nikolau, B.J.; Ohlrogge, J.B.; Wurtele, E.S. Plant biotin-containing carboxylases. Arch. Biochem. Biophys. 2003, 414, 211–222. [Google Scholar] [CrossRef]
- Cahoon, E.B.; Ohlrogge, J.B. Apparent Role of Phosphatidylcholine in the Metabolism of Petroselinic Acid in Developing Umbelliferae Endosperm. Plant Physiol. 1994, 104, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Voelker, T.A.; Hayes, T.R.; Cranmer, A.M.; Turner, J.C.; Davies, H.M. Genetic engineering of a quantitative trait: Metabolic and genetic parameters influencing the accumulation of laurate in rapeseed. Plant. J. 1996, 9, 229–241. [Google Scholar] [CrossRef]
- Manan, S.; Chen, B.; She, G.; Wan, X.; Zhao, J. Transport and transcriptional regulation of oil production in plants. Crit. Rev. Biotechnol. 2017, 37, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Durrett, T.P.; Ohlrogge, J.B.; Pollard, M. Analysis of Acyl Fluxes through Multiple Pathways of Triacylglycerol Synthesis in Developing Soybean Embryos. Plant Physiol. 2009, 150, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Browse, J. The significance of different diacylgycerol synthesis pathways on plant oil composition and bioengineering. Front. Plant Sci. 2012, 3, 147. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xin, Z.; Ren, Z.; Miquel, M.; Browse, J. An enzyme regulating triacylglycerol composition is encoded by the ROD1 gene of Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 18837–18842. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Fatihi, A.; Snapp, A.R.; Carlsson, A.S.; Browse, J.; Lu, C. Acyl editing and headgroup exchange are the major mechanisms that direct polyunsaturated fatty acid flux into triacylglycerols. Plant Physiol. 2012, 160, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Welti, R.; Schapaugh, W.T.; Trick, H.N. Phospholipid and triacylglycerol profiles modified by PLD suppression in soybean seed. Plant Biotechnol. J. 2011, 9, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Browse, J. The pathway of triacylglycerol synthesis through phosphatidylcholine in Arabidopsis produces a bottleneck for the accumulation of unusual fatty acids in transgenic seeds. Plant J. 2011, 68, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Katavic, V.; Reed, D.W.; Taylor, D.C.; Giblin, E.M.; Barton, D.L.; Zou, J.; Mackenzie, S.L.; Covello, P.S.; Kunst, L. Alteration of seed fatty acid composition by an ethyl methanesulfonate-induced mutation in Arabidopsis thaliana affecting diacylglycerol acyltransferase activity. Plant Physiol. 1995, 108, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Siloto, R.M.; Lehner, R.; Stone, S.J.; Weselake, R.J. Acyl-CoA:diacylglycerol acyltransferase: Molecular biology, biochemistry and biotechnology. Prog. Lipid Res. 2012, 51, 350–377. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wei, Y.; Jako, C.; Kumar, A.; Selvaraj, G.; Taylor, D.C. The Arabidopsis thaliana TAG1 mutant has a mutation in a diacylglycerol acyltransferase gene. Plant. J. Cell Mol. Biol. 1999, 19, 645–653. [Google Scholar] [CrossRef]
- Brkić, K.; Radulović, M.; Sladonja, B.; Lukić, I.; Šetić, E. Application of Soxtec apparatus for oil content determination in olive fruit. Rivista Italiana Delle Sostanze Grasse 2006, 83, 115–119. [Google Scholar]
- Lall, R.K.; Proctor, A.; Jain, V.P. A Rapid, Micro FAME Preparation Method for Vegetable Oil Fatty Acid Analysis by Gas Chromatography. J. Am. Oil Chem. Soc. 2009, 86, 309–314. [Google Scholar] [CrossRef]
- Tang, Q.Y.; Zhang, C.X. Data Processing System (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Long, Y.; Meng, J.; Bancroft, I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 2009, 7, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Patton, S.J.; Wallace, A.J.; Elles, R. Benchmark for evaluating the quality of DNA sequencing: Proposal from an international external quality assessment scheme. Clin. Chem. 2006, 52, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.F.; Lin, P.; Yao, X.H.; Wang, K.L.; Chang, J.; Han, X.J. Selection of reference genes for quantitative real-time PCR in six oil-tea Camellia based on RNA. Mol. Biol. 2013, 47, 959–975. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated Databases | Number of Unigenes | Percentage (%) | ≥300 bp | ≥1000 bp |
|---|---|---|---|---|
| Nr Annotation | 51,574 | 64.22 | 40,908 | 13,684 |
| Swiss-Prot Annotation | 33,503 | 41.72 | 28,215 | 11,410 |
| COG Annotation | 15,442 | 19.23 | 13,883 | 6721 |
| KEGG Annotation | 10,493 | 13.07 | 8551 | 3564 |
| GO Annotation | 7261 | 9.04 | 5545 | 2393 |
| Total | 51,725 | 64.41 | 40,982 | 13,689 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, P.; Wang, K.; Zhou, C.; Xie, Y.; Yao, X.; Yin, H. Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition. Int. J. Mol. Sci. 2018, 19, 118. https://doi.org/10.3390/ijms19010118
Lin P, Wang K, Zhou C, Xie Y, Yao X, Yin H. Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition. International Journal of Molecular Sciences. 2018; 19(1):118. https://doi.org/10.3390/ijms19010118
Chicago/Turabian StyleLin, Ping, Kailiang Wang, Changfu Zhou, Yunhai Xie, Xiaohua Yao, and Hengfu Yin. 2018. "Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition" International Journal of Molecular Sciences 19, no. 1: 118. https://doi.org/10.3390/ijms19010118