Computational Modeling of the Staphylococcal Enterotoxins and Their Interaction with Natural Antitoxin Compounds

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
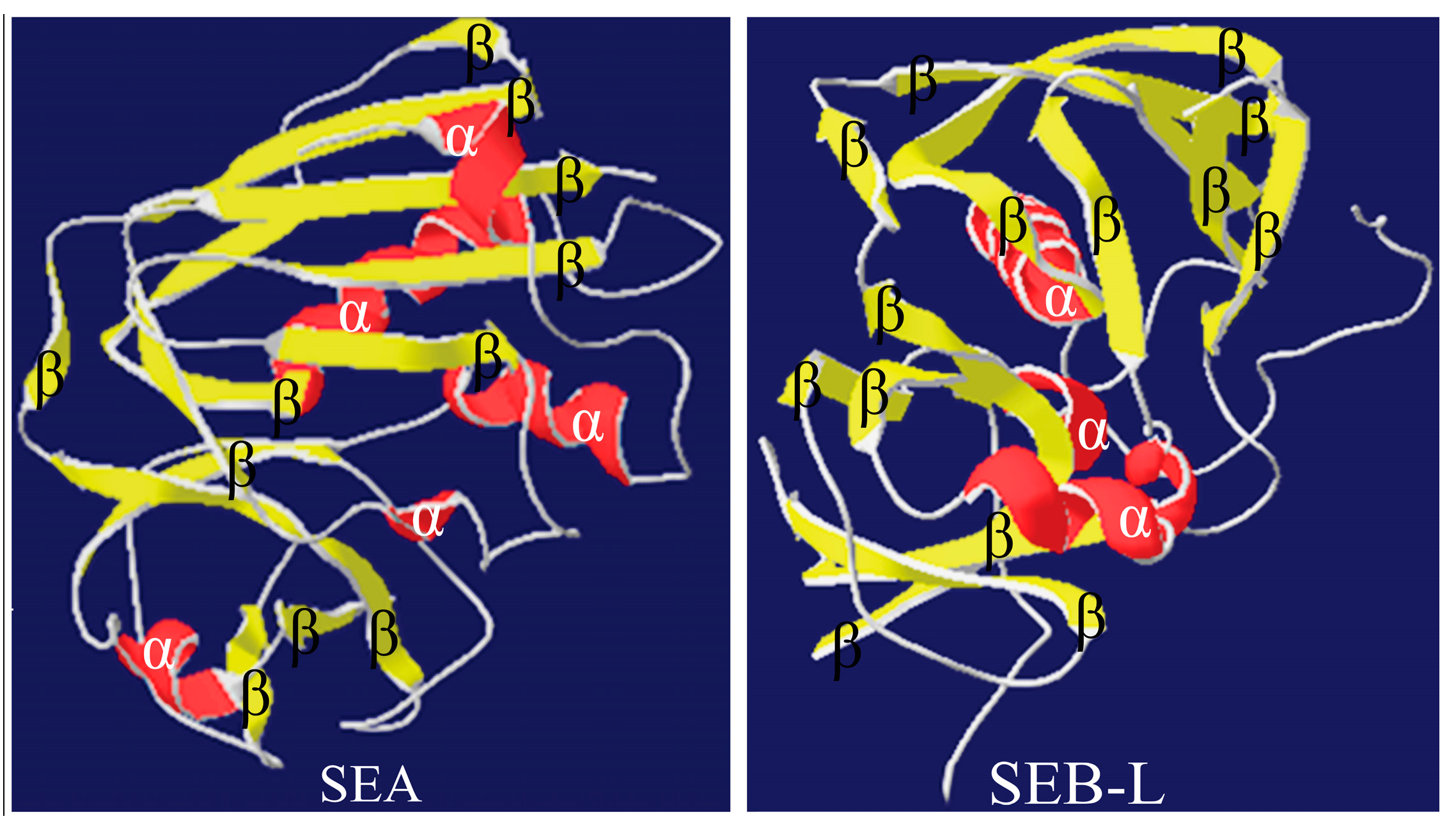
2.1. Homology Modeling of SEA and SEB-Like Proteins
2.2. Homology Modeling of the 3-D Structures
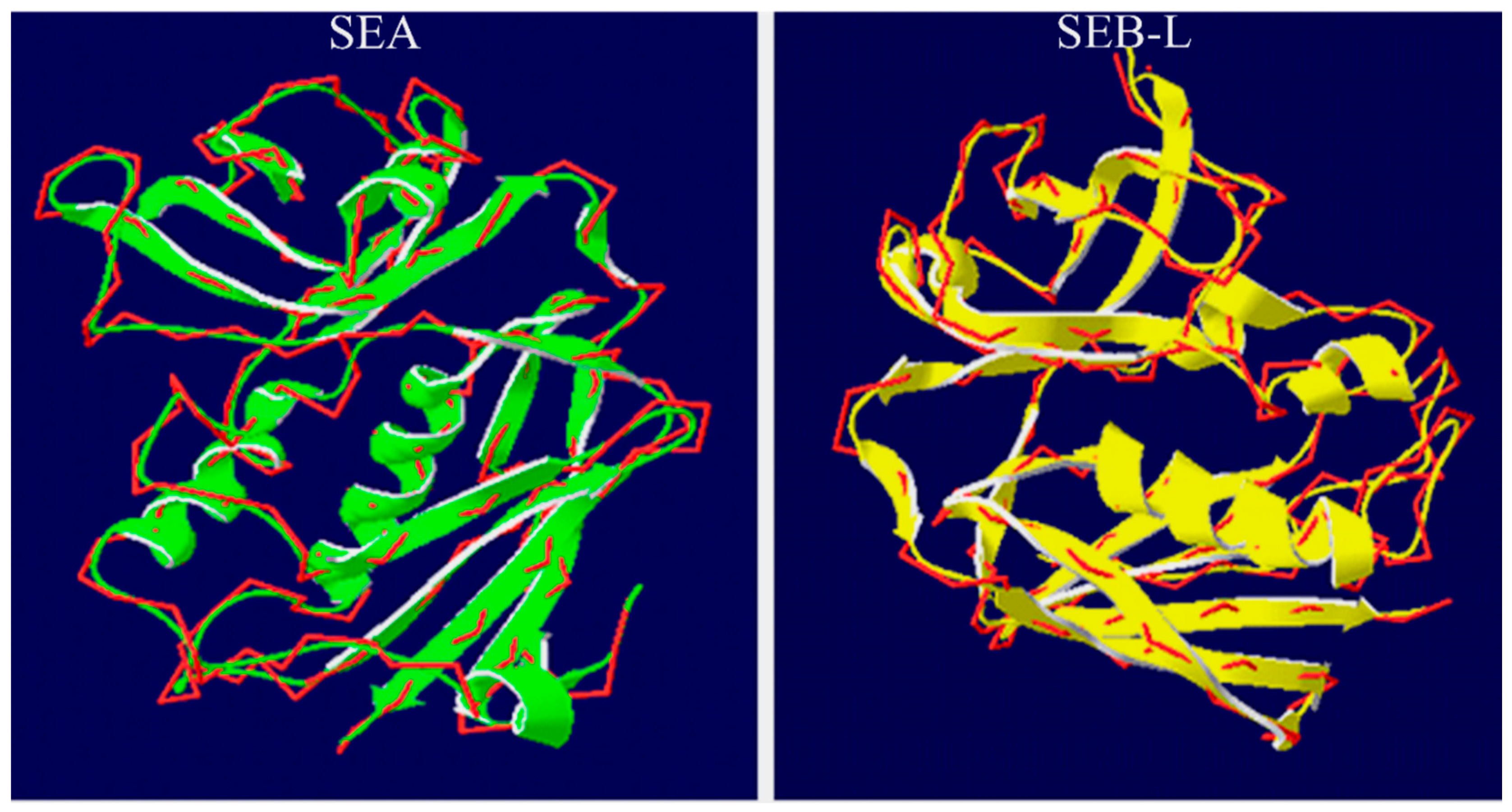
2.3. Validation of the 3-D Structures
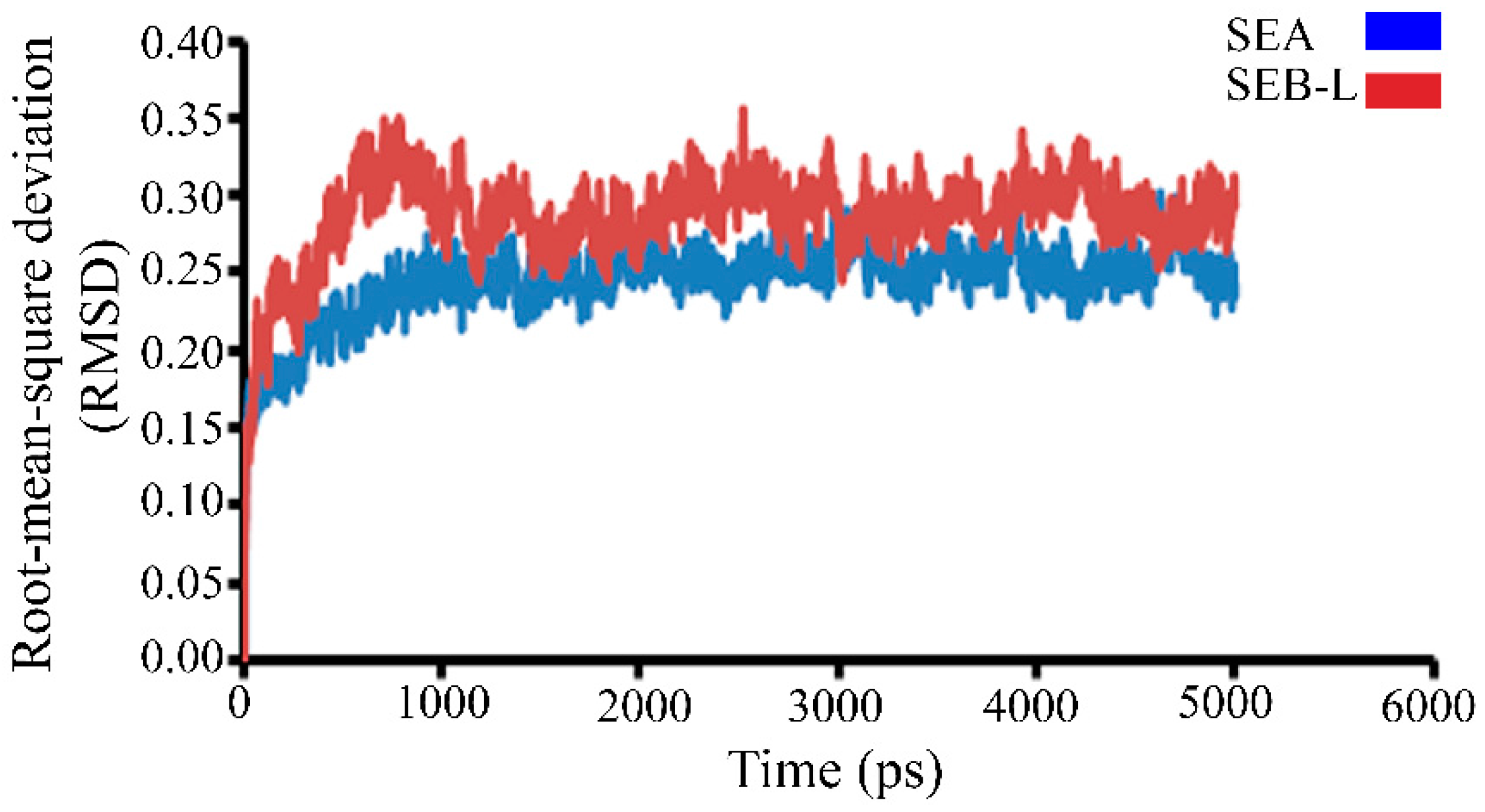
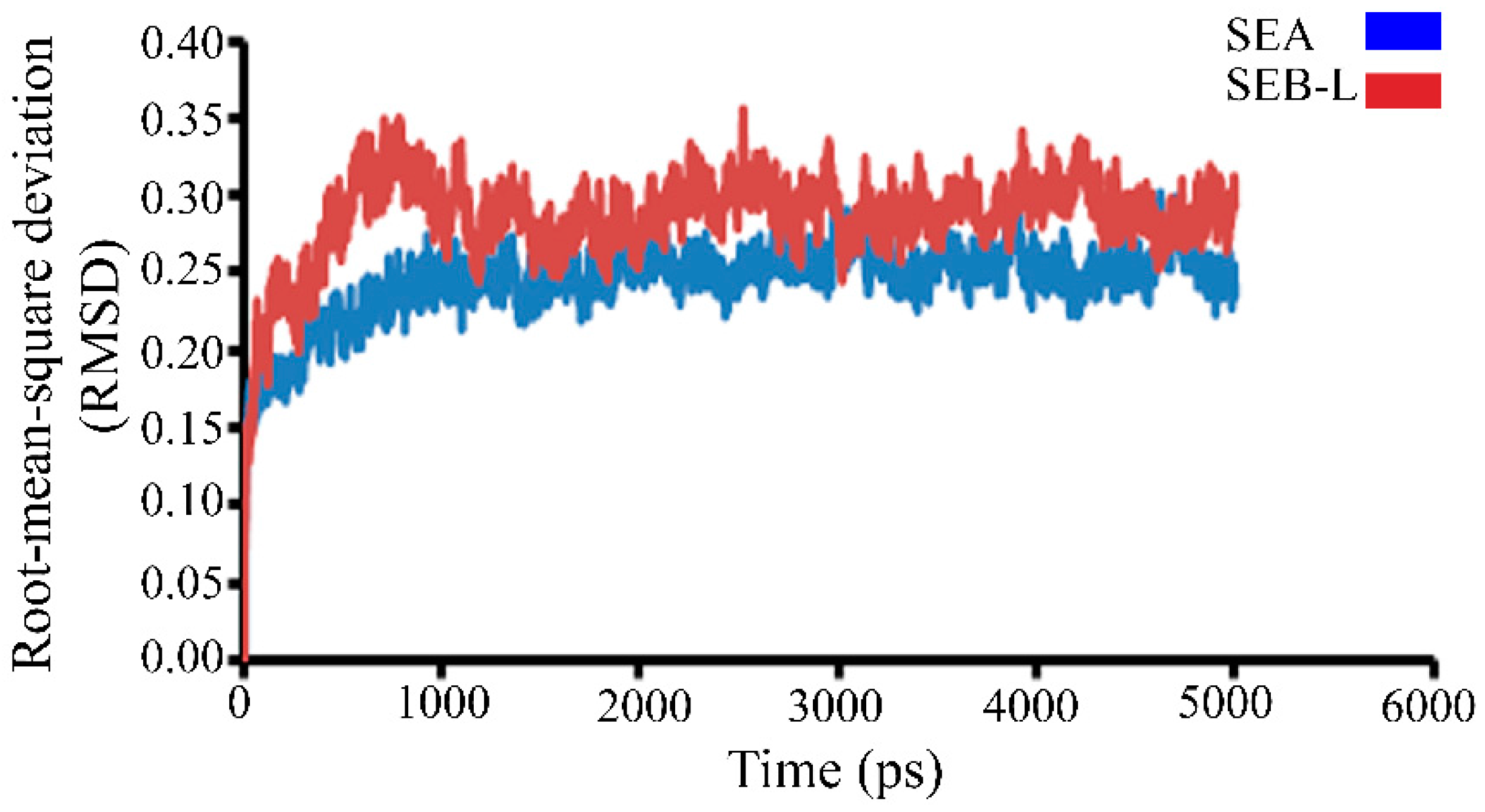
2.3.1. Molecular Dynamics Studies
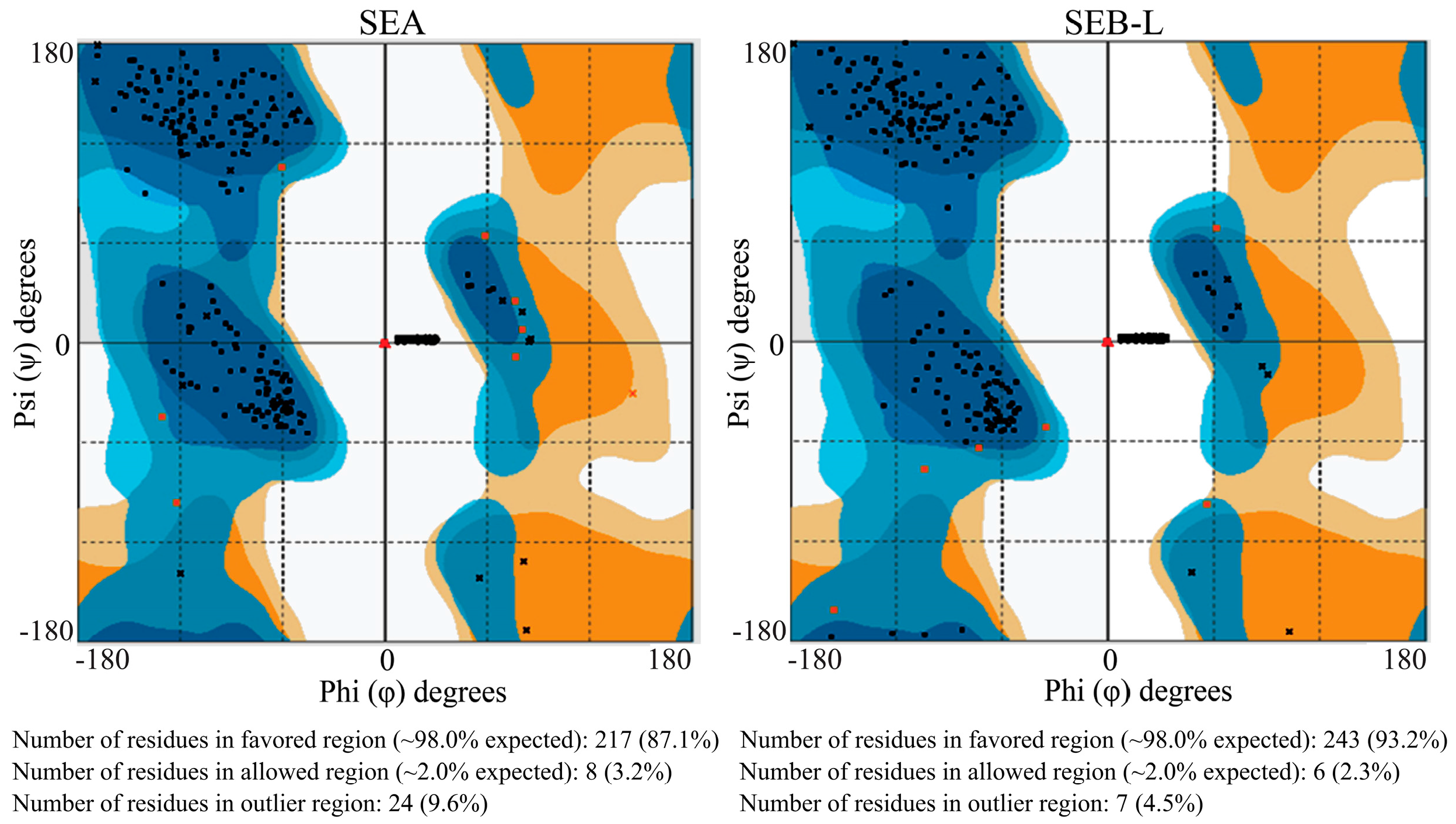
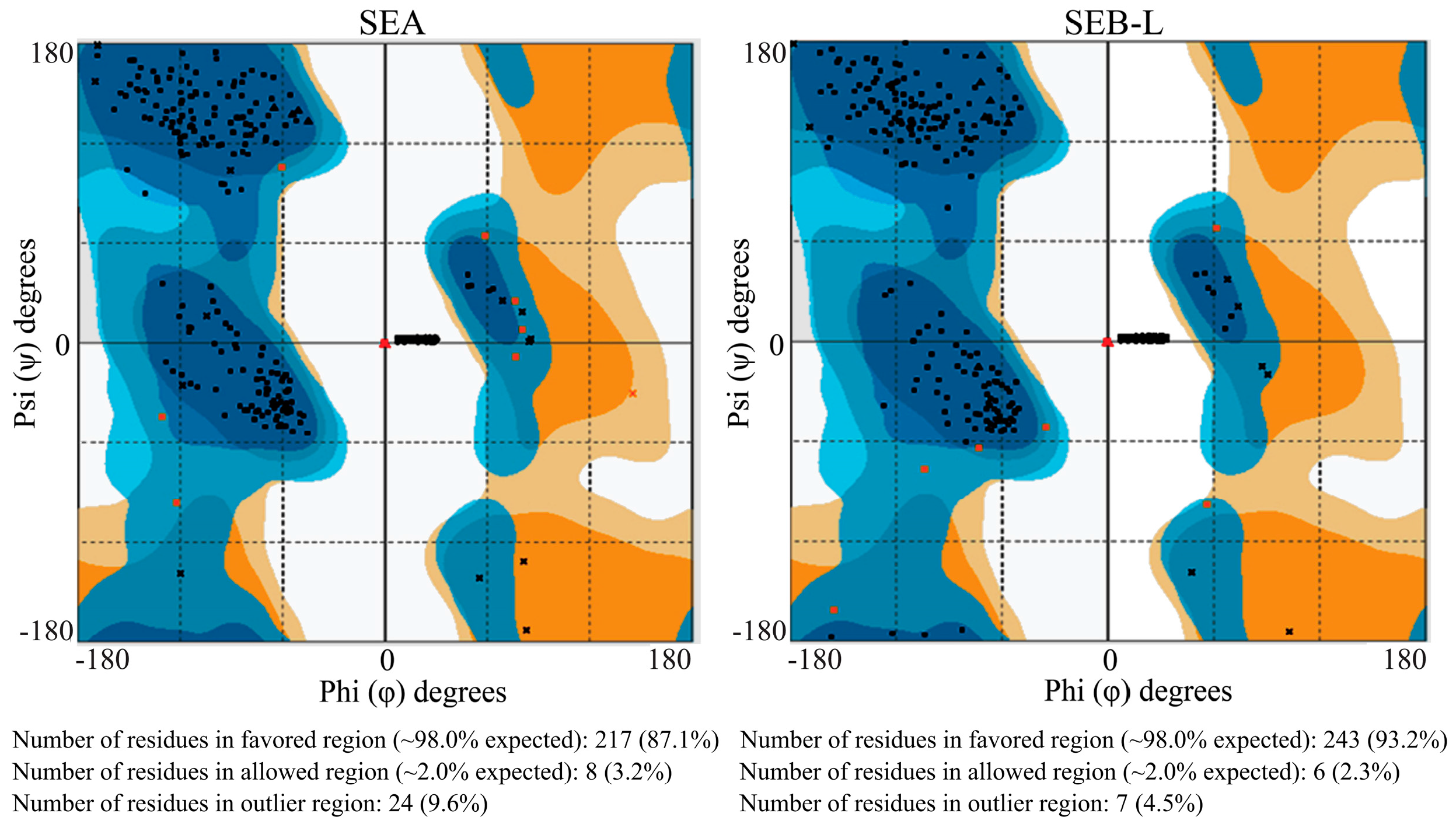
2.3.2. Ramachandran Plot Analysis
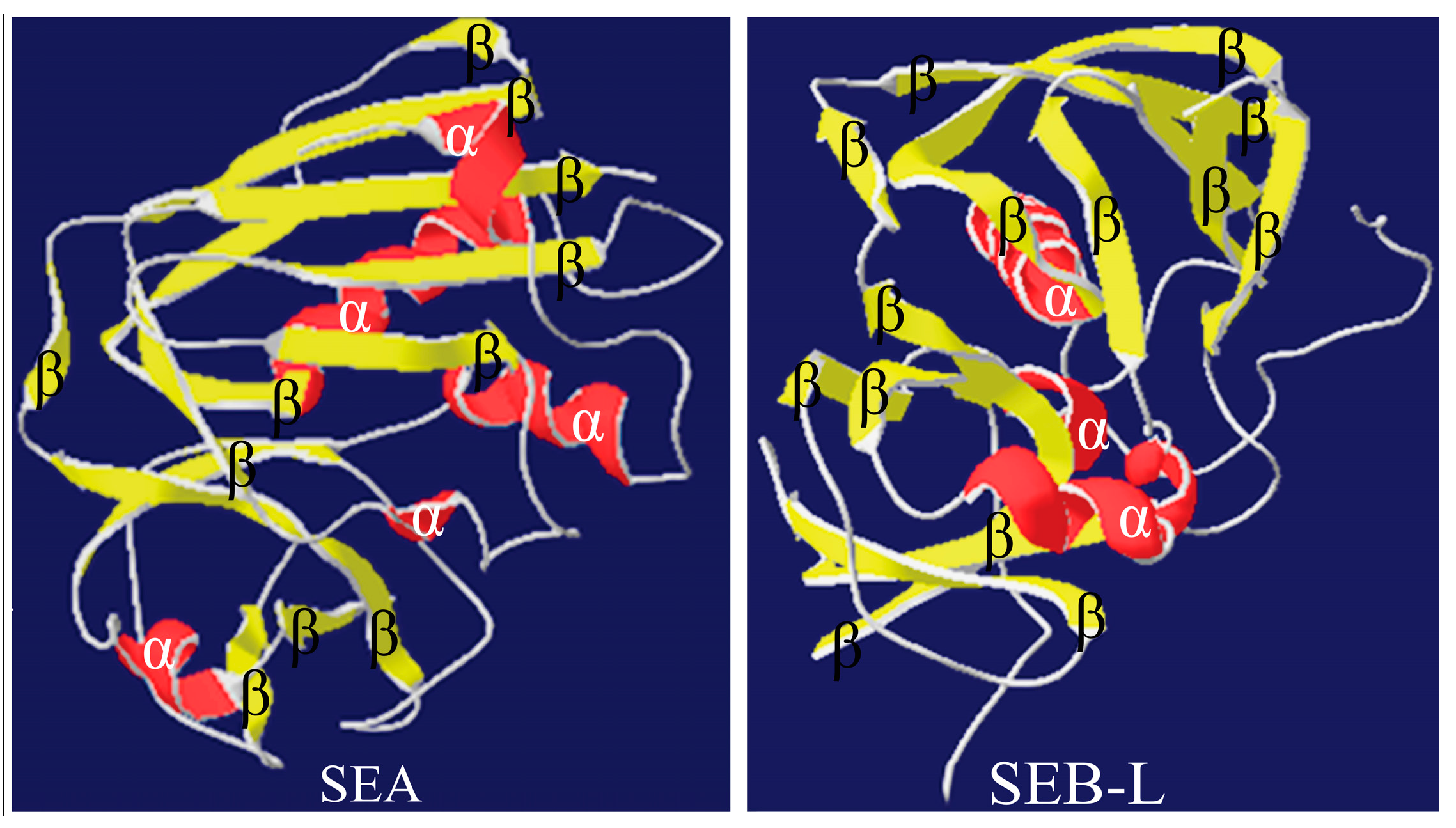
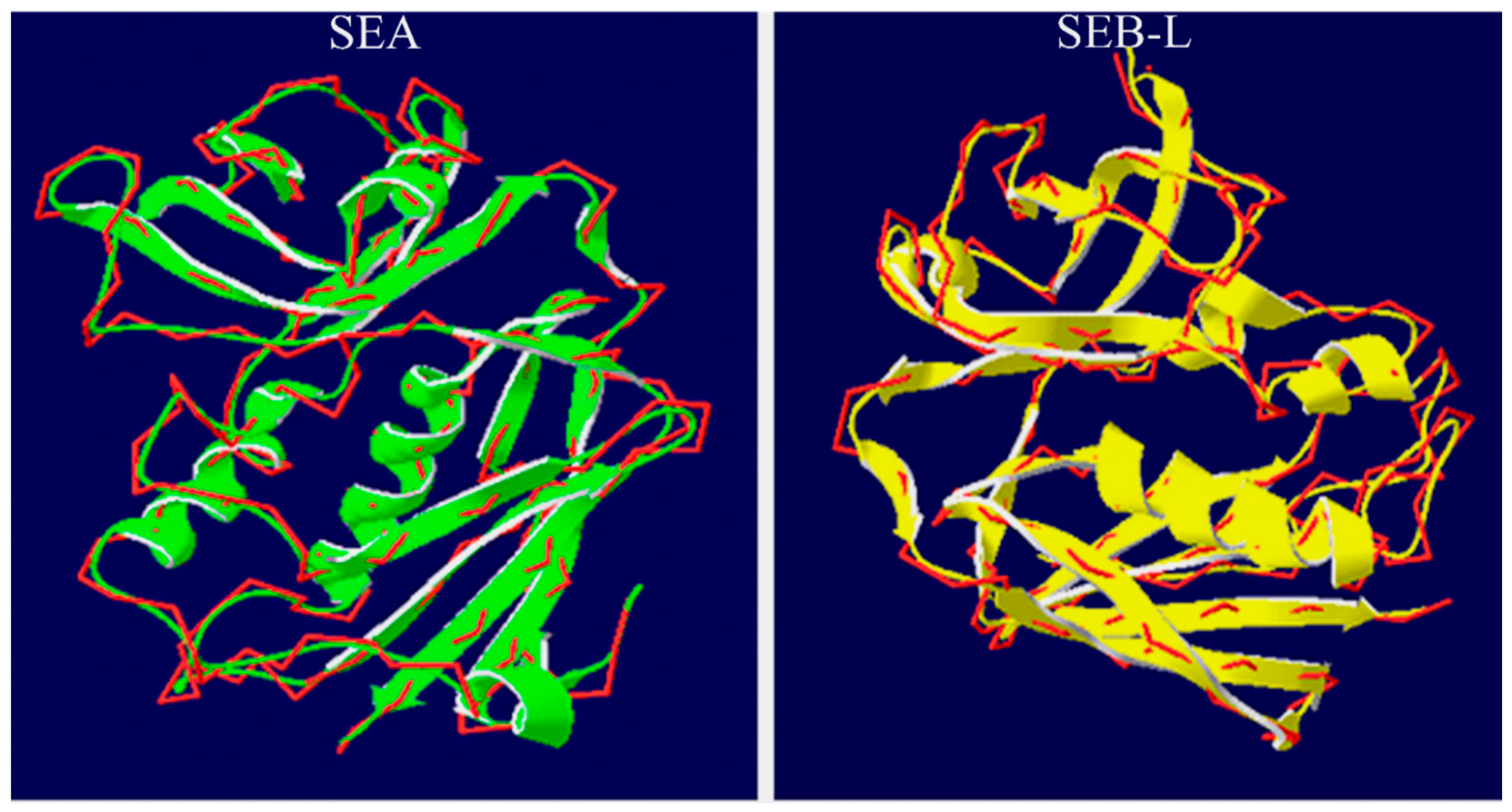
2.3.3. Analysis of the SEA and SEB-Like 3-D Structures
2.4. Docking Study
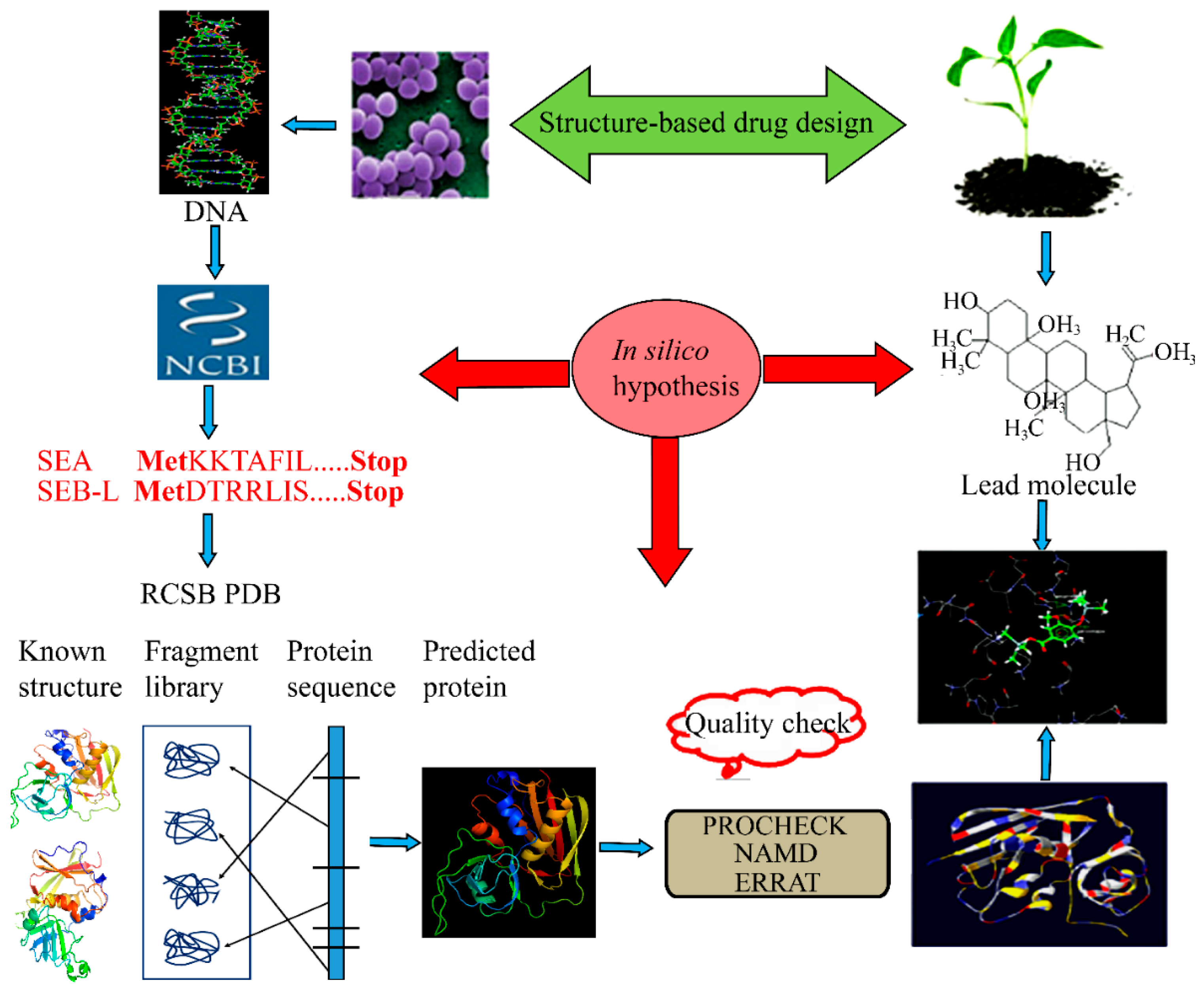
3. Materials and Methods
3.1. Source of Data
3.2. Generation of the 3-D Structure Using Homology Modeling
3.3. Molecular Dynamics
3.4. Binding Site Identification
3.5. Substrate Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3-D | Three-Dimensional |
| atm | Atmospheric Pressure |
| CASTp | Computer Atlas of Surface Topology of Proteins |
| CHARMM | Chemistry at Harvard Macromolecular Mechanics |
| FRED | Fast Rigid Exhaustive Docking |
| K | Kelvin |
| MS | Molecular Surface |
| NAMD | NAnoscale Molecular Dynamics |
| NCBI | National Center for Biotechnology Information |
| PDB | Protein Data Bank |
| PDFs | Probability Functions |
| ps and fs | Pico- and Femtoseconds |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| RMSD | Root-Mean-Square Deviation |
| SEA | Staphylococcal Enterotoxin A |
| SEB | Staphylococcal Enterotoxin B |
| SEs | Staphylococcal Enterotoxins |
References
- Mustapha, M.; Bukar-Kolo, Y.M.; Geidam, Y.A.; Gulani, I.A. Review on Methicillin-resistant Staphylococcus aureus (MRSA) in Dogs and Cats. Int. J. Anim. Vet. Adv. 2014, 6, 61–73. [Google Scholar]
- Kurjogi, M.M.; Kaliwal, B.B. Prevalence and antimicrobial susceptibility of bacteria isolated from bovine mastitis. Adv. Appl. Sci. Res. 2011, 2, 229–235. [Google Scholar]
- Julio, C.; Franco, G.; Gonzalez, L.V.; Sandra, C.; Gomez, M.; Juan, M.; Carrillo, G.; Jose, J.; Ramirez, C. Virulence factors analysis of Staphylococcus aureus isolated from bovine mastitis in Mexico. eGnosis 2008, 6, 1–9. [Google Scholar]
- Hennekinne, J.A.; de Buyser, M.L.; Dragacci, S. Staphylococcus aureus and its food poisoning toxins: Characterization and outbreak investigation. FEMS Microbiol. Rev. 2012, 36, 815–836. [Google Scholar] [CrossRef] [PubMed]
- Thanert, R.; Goldmann, O.; Beineke, A.; Medina, E. Host-inherent variability influences the transcriptional response of Staphylococcus aureus during in vivo infection. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Kurjogi, M.M.; Sanakal, R.D.; Kaliwal, B.B. Antibiotic susceptibility and antioxidant activity of Staphylococcus aureus pigment staphyloxanthin on carbon tetrachloride (ccl4) induced stress in Swiss albino mice. Int. J. Biotech. Appl. 2010, 2, 33–40. [Google Scholar]
- Dunyach-Remy, C.; Essebe, C.N.; Sotto, A.; Levigne, J.P. Staphylococcus aureus toxins and diabetic foot ulcers: Role in pathogenesis and interest in diagnosis. Toxins 2016, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Essmann, F.; Bantel, H.; Totzke, G.; Engels, I.H.; Sinha, B.; Schulze, O.K.; Janicke, R.U. Staphylococcus aureus α-toxin-induced cell death: Predominant necrosis despite apoptotic caspase activation. Cell. Death. Differ. 2003, 10, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Johler, S.; Weder, D.; Bridy, C.; Huguenin, M.C.; Robert, L.; Hummerjohann, J.; Stephan, R. Outbreak of staphylococcal food poisoning among children and staff at a Swiss boarding school due to soft cheese made from raw milk. J. Dairy Sci. 2015, 98, 2944–2948. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.J. Recognition and management of Staphylococcus aureus toxin-mediated disease. Intern. Med. J. 2005, 35, S106–S119. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.; Rasooly, A. Staphylococcal enterotoxins. Int. J. Food Microbiol. 2000, 61, 1–10. [Google Scholar] [CrossRef]
- Argudin, M.A.; Mendoza, M.C.; Rodicio, M.R. Food poisoning and Staphylococcus aureus enterotoxins. Toxins 2010, 2, 1751–1773. [Google Scholar] [CrossRef] [PubMed]
- Derzelle, S.; Dilasser, F.; Duquenne, M.; Deperrois, V. Differential temporal expression of the staphylococcal enterotoxins genes during cell growth. Food. Microbiol. 2009, 26, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Larkin, E.A.; Carman, R.J.; Krakauer, T.; Stiles, B.G. Staphylococcus aureus: The toxic presence of a pathogen extraordinaire. Curr. Med. Chem. 2009, 16, 4003–4019. [Google Scholar] [CrossRef] [PubMed]
- Evenson, M.L.; Hinds, M.W.; Bernstein, R.S.; Bergdoll, M.S. Estimation of human dose of staphylococcal enterotoxin A from a large outbreak of staphylococcal food poisoning involving chocolate milk. Int. J. Food Microbiol. 1988, 7, 311–316. [Google Scholar] [CrossRef]
- Bergdoll, M.S. Enterotoxins. In Staphylococci and Staphylococcal Infections; Easman, C.S.F., Adlam, C., Eds.; Academic Press Inc.: London, UK, 1983; Volume 2, pp. 559–598. [Google Scholar]
- Schantz, E.J.; Roessler, W.G.; Wagman, J.; Spero, L.; Dunnery, D.A.; Bergdoll, M.S. Purification of staphylococcal enterotoxin B. Biochemistry 1965, 4, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal Enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed]
- Kurjogi, M.M.; Kaliwal, R.B.; Shivasharana, C.T.; Kaliwal, B.B. Characterization of toxin genes in Staphylococcus aureus isolated from milk of cows with mastitis. Int. J. Recent Sci. Res. 2012, 3, 841–846. [Google Scholar]
- Baba, T.; Bae, T.; Schneewind, O.; Takeuchi, F.; Hiramatsu, K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: Polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008, 190, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nabuurs, S.B.; Gert, V. Homology modeling. In Structural Bioinformatics; Bourne, P.E., Helge Weissig, H., Eds.; Wiley: Hoboken, NJ, USA, 2003; pp. 507–521. [Google Scholar]
- Singh, N.K.; Britto, C.; Pakkkianathan, M.K.; Kumar, M.; Daddam, J.R.; Sridhar, J.S.; Kannan, M.; Pillai, G.G.; Krishnan, M. Computational studies on molecular interactions of 6-thioguanosine analogs with Anthrax toxin receptor 1. Interdiscip. Sci. Comput. Life Sci. 2012, 4, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Misura, K.M.; Baker, D. Progress and challenges in high resolution refinement of protein structure models. Proteins 2005, 59, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.; Word, J.M.; Prisant, M.C.; Richardson, J.S.; Richardson, D.C. Structure validation by C-alpha geometry: phi, psi and beta deviation. Proteins 2003, 50, 437–450. [Google Scholar]
- Hooft, R.W.; Sander, C.; Vriend, G.; Abola, E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Levitt, M. Comprehensive assessment of automatic structural alignment against a manual standard, the scop classification of proteins. Protein Sci. 1998, 7, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [PubMed]
- Baumann, G.; Froemmel, C.; Sander, C. Polarity as a criterion in protein design. Protein Eng. 1989, 2, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Evaluation of protein models by atomic solvation preference. J. Mol. Biol. 1992, 225, 93–105. [Google Scholar] [CrossRef]
- Gregoret, L.M.; Cohen, F.E. Novel method for the rapid evaluation of packing in protein structures. J. Mol. Biol. 1990, 211, 959–974. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Purohit, R.; Sethumadhavan, R. Structural basis for the resilience of darunavir (TMC114) resistance major mutation of HIV-1 protease. Interdiscip. Sci. Comput. Life Sci. 2009, 1, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Shinde, M.G.; Modi, S.J.; Kulkarni, V.M. Synthesis, pharmacological evaluation, molecular docking and in silico ADMET prediction of nitric oxide releasing biphenyls as anti-inflammatory agents. J. Appl. Pharm. Sci. 2017, 7, 37–47. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Elokely, K.M.; Doerksen, R.J. Docking challenge: Protein sampling and molecular docking performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.S.; Abbasi, S.W. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor. Biol. Med. Model. 2013, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Damayanthi, D.I. Molecular docking analyses of cynodon dactylon derived phytochemicals against white spot syndrome virus (wssv) structural protein vp26. Int. J. Appl. Biol. Pharm. Tech. 2015, 6, 182–188. [Google Scholar]
- Renee, M.K.; Ervin, P.; Kazuo, K.; Ken, H.; Aleksandra, R.; Boguslaw, S.; Stieglitz, K.A. Shape matters: Improving docking results by prior analysis of geometric attributes of binding sites. JSM Chem. 2016, 4, 1020. [Google Scholar]
- Sathish, K.; Paramashivam, K.E.; Boopala, B.N.; Ramamoorthy, M.D.; Suganthana, B.; Kannan, N.D. In silico pharmacokinetic and molecular docking studies of small molecules derived from Indigofera aspalathoides Vahl targeting receptor tyrosine kinases. Bioinformation 2015, 11, 73–84. [Google Scholar]
- Cammisa, M.; Correra, A.; Andreotti, G.; Maria, V.C. Identification and analysis of conserved pockets on protein surfaces. BMC Bioinform. 2013, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, S.; Makela, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Flekhter, O.B.; Karachurina, L.T.; Nigmatullina, L.R.; Sapozhnikova, T.A.; Baltina, L.A.; Zarudiĭ, F.S.; Galin, F.Z.; Spirikhin, L.V.; Tolstikov, G.A.; Pliasunova, O.A. Synthesis and pharmacological activity of Betulin dinicotinate. Bioorganicheskaia Khimiia 2002, 28, 543–550. [Google Scholar] [PubMed]
- Pisha, E.; Chai, H.; Lee, I.S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, E.; Bebenek, E.; Kadela-Tomanek, M.; Latocha, M.; Jelsch, C.; Wenger, E.; Boryczka, S. Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity. Molecules 2016, 21, 1123. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic acid: A natural product with anticancer activity. Mol. Nutr. Food Res. 2009, 53, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Kommera, H.; Kaluđerović, G.N.; Kalbitz, J.; Paschke, R. Synthesis and Anticancer Activity of Novel Betulinic acid and Betulin Derivatives. Arch. Pharm. 2010, 343, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.; Saleh, M. Bioactive Betulin produced by marine Paecilomyces WE3-F. J. Appl. Pharm. Sci. 2016, 6, 34–40. [Google Scholar] [CrossRef]
- Bringmann, G.; Saeb, W.; Assi, L.A.; Francois, G.; Sankara-Narayanan, A.S.; Peters, K.; Peters, E.M. Betulinic acid: Isolation from Triphyophyllum peltatum and Ancistrocladus heyneanus, antimalarial activity, and crystal structure of the benzyl ester. Planta Med. 1997, 63, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Recio, M.C.; Giner, R.M.; Manez, S.; Gueho, J.; Julien, H.R.; Hostettmann, K.; Rios, J.L. Investigations on the steroidal anti-inflammatory activity of triterpenoids from Diospyros leucomelas. Planta Med. 1995, 61, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Suresh, C.; Zhao, H.; Gumbs, A.; Chetty, C.S.; Bose, H.S. New ionic derivatives of betulinic acid as highly potent anti-cancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.S.; Lee, K.H. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994, 57, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Q.L.; Zheng, M.F.; Ren, H.; Lei, T.; Wu, P.; Zhou, Z.Y.; Wei, X.Y.; Tan, J.W. Bioactive 30-noroleanane triterpenes from the pericarps of Akebia trifoliata. Molecules 2014, 19, 4301–4312. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.; Liang, J.Y.; Feng, X. Research in nor-oleanane triterpenoids. Nat. Prod. Res. Dev. 2011, 23, 577–581. [Google Scholar]
- Zhen, Q.A.; Yang, C.R. Chemotaxonomic study on the family of Lardizabalaceae. Chin. Bull. Bot. 2001, 18, 332–339. [Google Scholar]
- Wenbing, D.; Ye, Li.; Guanhua, L.; Hualiang, H.; Zhiwen, L.; Youzhi, L. New 30-noroleanane triterpenoid saponins from Holboellia coriacea Diels. Molecules 2016, 21, 734. [Google Scholar]
- Wang, Q.-Z.; Liu, X.-F.; Shan, Y.; Guan, F.-Q.; Chen, Y.; Wang, X.-Y.; Wang, M.; Feng, X. Two new nortriterpenoid saponins from Salicornia bigelovii Torr. and their cytotoxic activity. Fitoterapia 2012, 83, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Kale, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2001, 56, 257–265. [Google Scholar] [CrossRef]
- Feller, S.E.; MacKerell, A.D., Jr. An Improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. [Google Scholar] [CrossRef]
- Grubmuller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, S.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamic studies of proteins. J. Phys. Chem. 1998, 102, 3586–3616. [Google Scholar]
- Brunger, A. X-PLOR, Version 3.1: A System for X-ray Crystallography and NMR; Yale University: New Haven, CT, USA, 1992. [Google Scholar]
- Diaz, A.; Horjales, E.; Rudino, P.E.; Arrola, R.; Hansberg, W. Unusual Cys-Tyr Covalent Bond in a Large Catalase. J. Mol. Biol. 2004, 342, 971–985. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
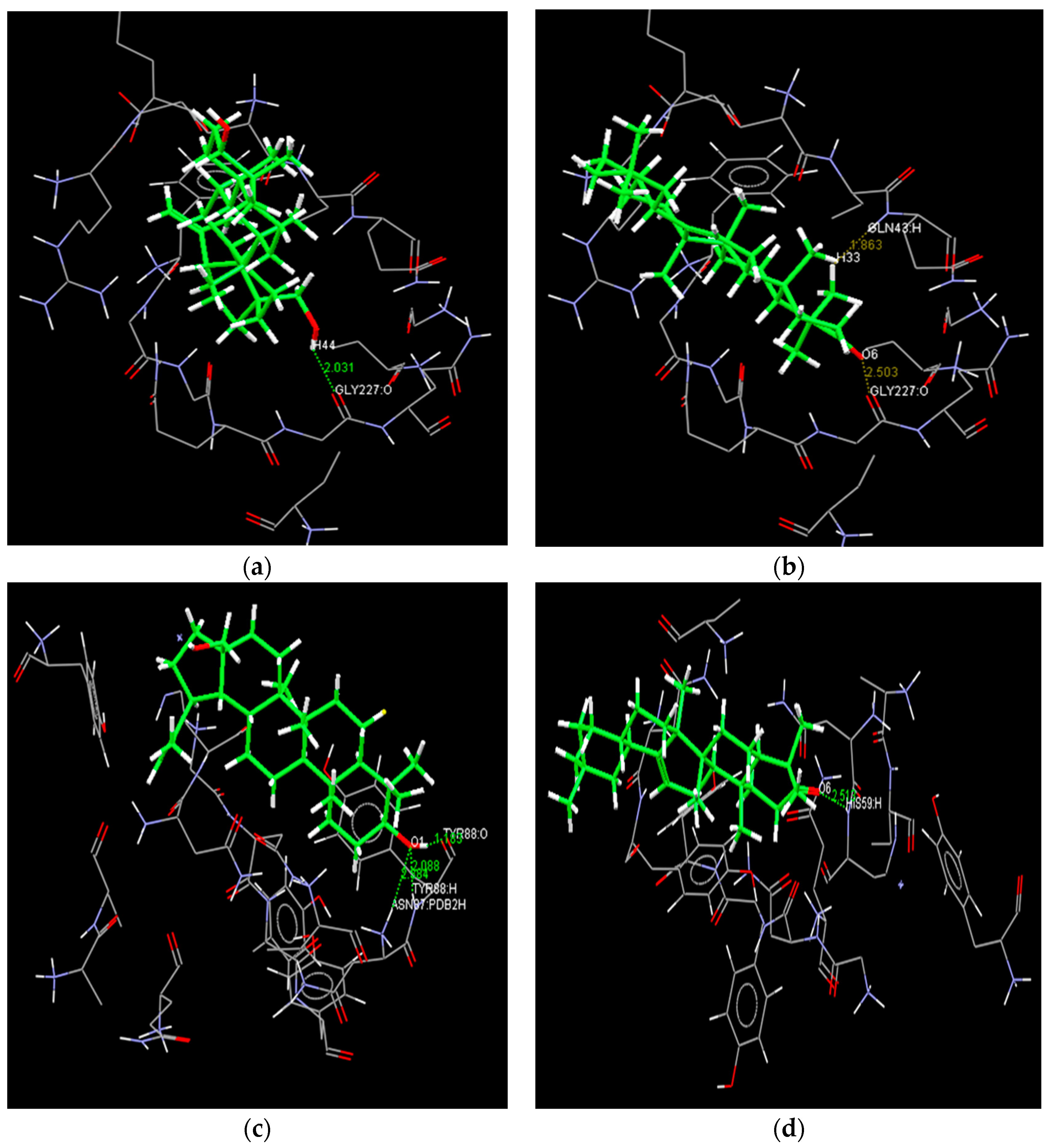{kind=link}
| Compound Name | PubChem CID | Molecular Formula | Molecular Weight (g/mol) | Compound Structure | Staphylococcal Enterotoxin (SE) in Interaction | Free Energy (kcal/mol) |
|---|---|---|---|---|---|---|
| Betulin | 72326 | C30H50O2 | 442.728 |  | SEA | −147.39 |
| SEB-like | −139.97 | |||||
| 28-Norolean-12-en-3-one | 101616676 | C29H46O | 410.686 |  | SEA | −83.97 |
| SEB-like | −88.20 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurjogi, M.; Satapute, P.; Jogaiah, S.; Abdelrahman, M.; Daddam, J.R.; Ramu, V.; Tran, L.-S.P. Computational Modeling of the Staphylococcal Enterotoxins and Their Interaction with Natural Antitoxin Compounds. Int. J. Mol. Sci. 2018, 19, 133. https://doi.org/10.3390/ijms19010133
Kurjogi M, Satapute P, Jogaiah S, Abdelrahman M, Daddam JR, Ramu V, Tran L-SP. Computational Modeling of the Staphylococcal Enterotoxins and Their Interaction with Natural Antitoxin Compounds. International Journal of Molecular Sciences. 2018; 19(1):133. https://doi.org/10.3390/ijms19010133
Chicago/Turabian StyleKurjogi, Mahantesh, Praveen Satapute, Sudisha Jogaiah, Mostafa Abdelrahman, Jayasimha Rayalu Daddam, Venkatesh Ramu, and Lam-Son Phan Tran. 2018. "Computational Modeling of the Staphylococcal Enterotoxins and Their Interaction with Natural Antitoxin Compounds" International Journal of Molecular Sciences 19, no. 1: 133. https://doi.org/10.3390/ijms19010133