Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
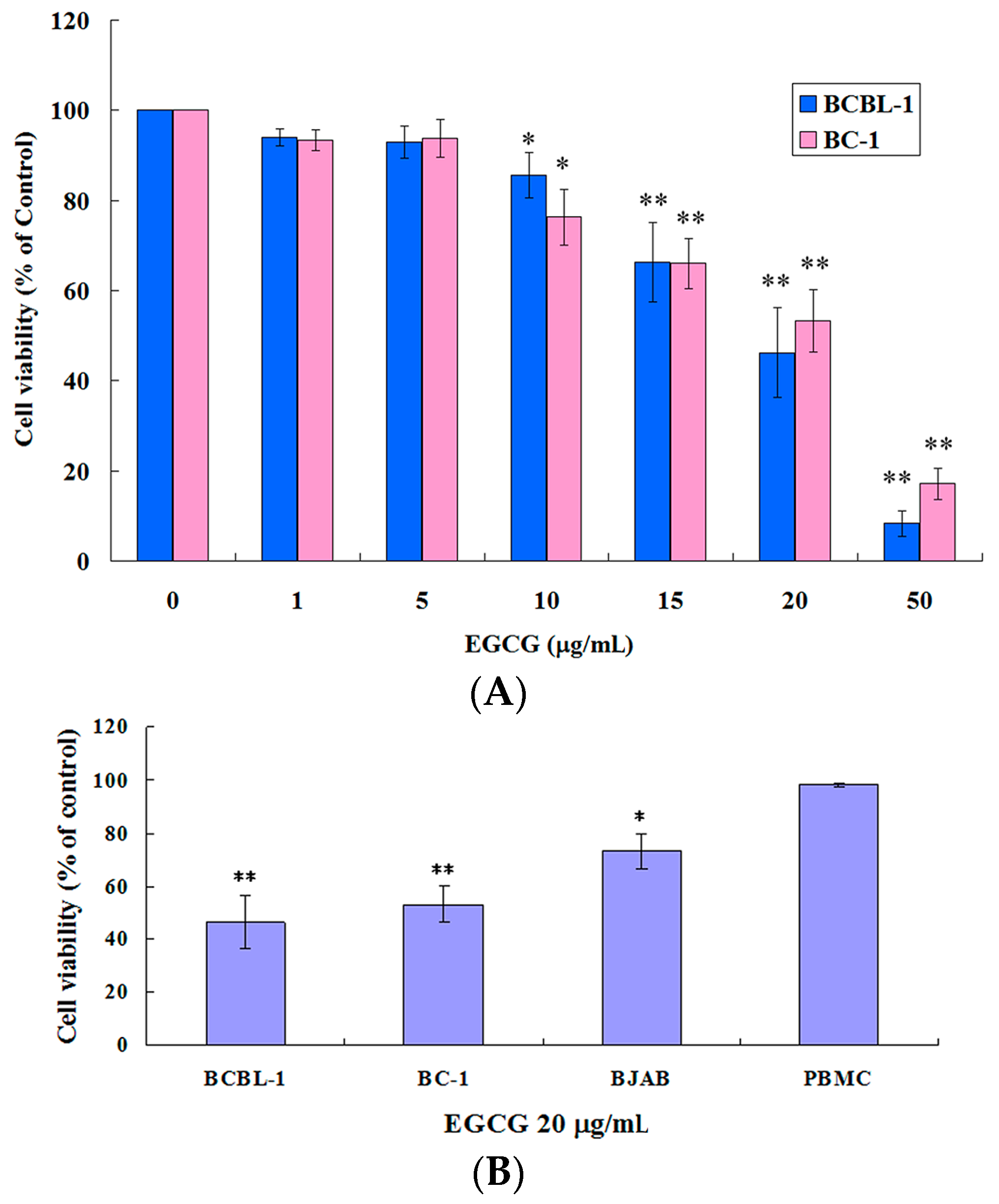
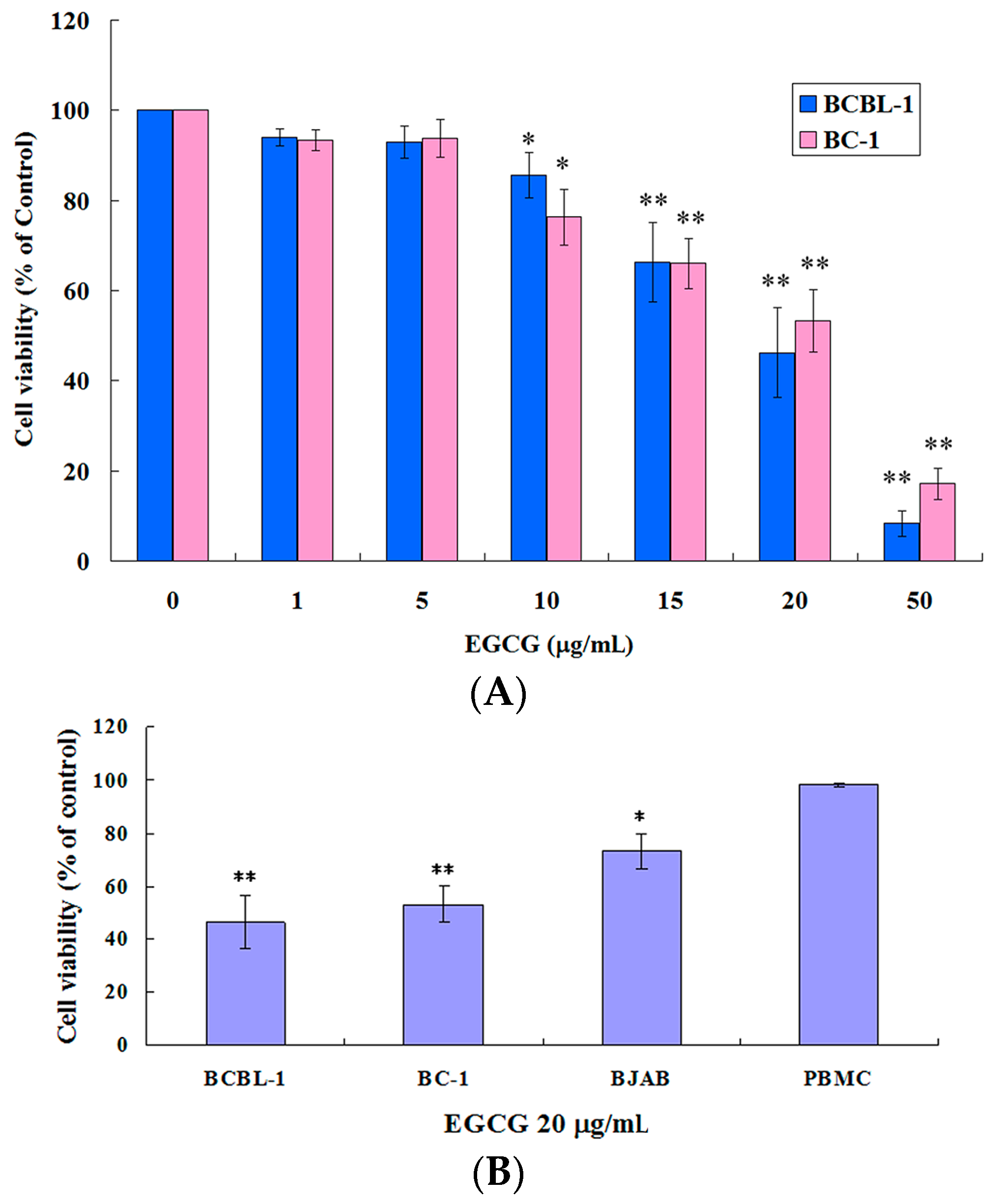
2.1. EGCG Inhibited the Growth of HHV8-Harboring PEL Cells
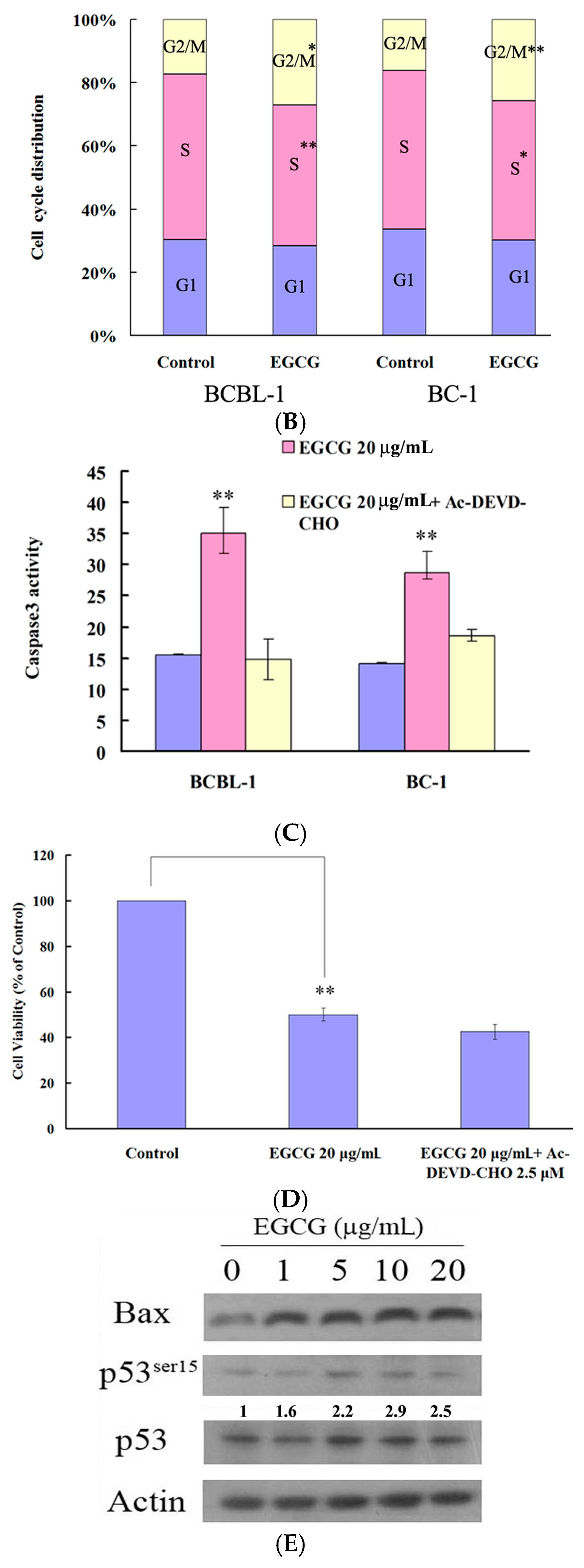
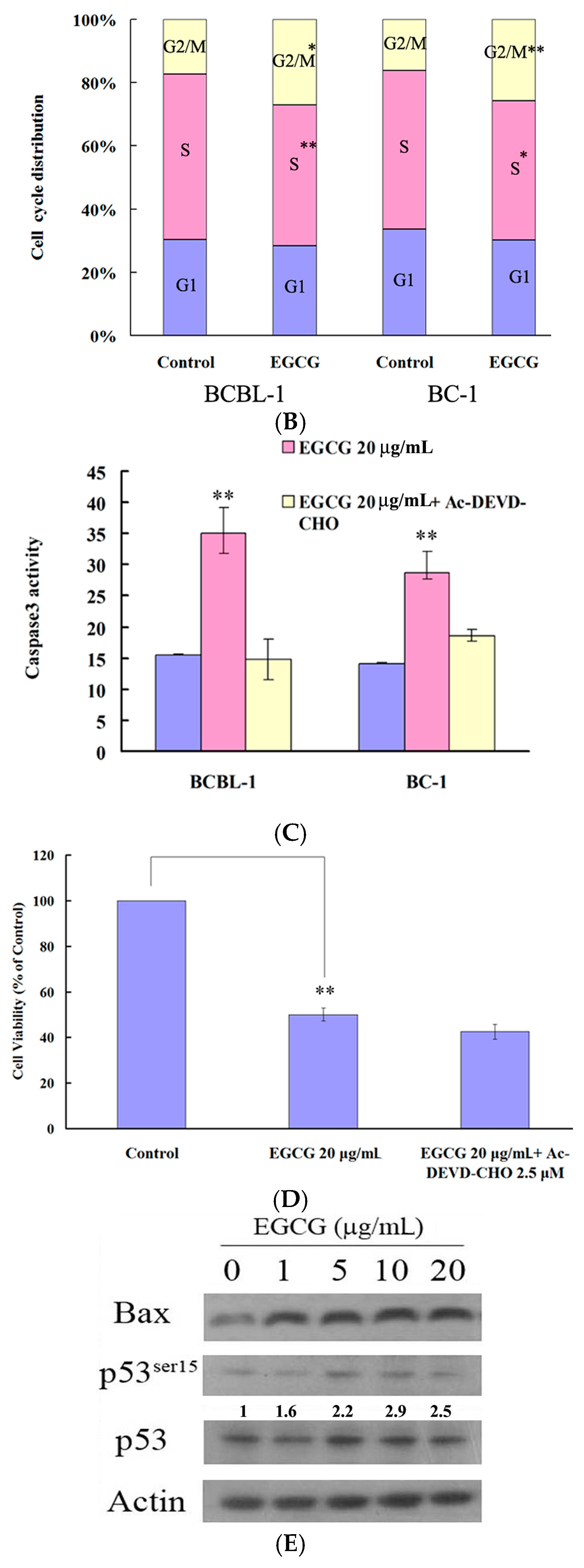
2.2. EGCG Induced G2-M Arrest and Apoptosis in PEL Cells
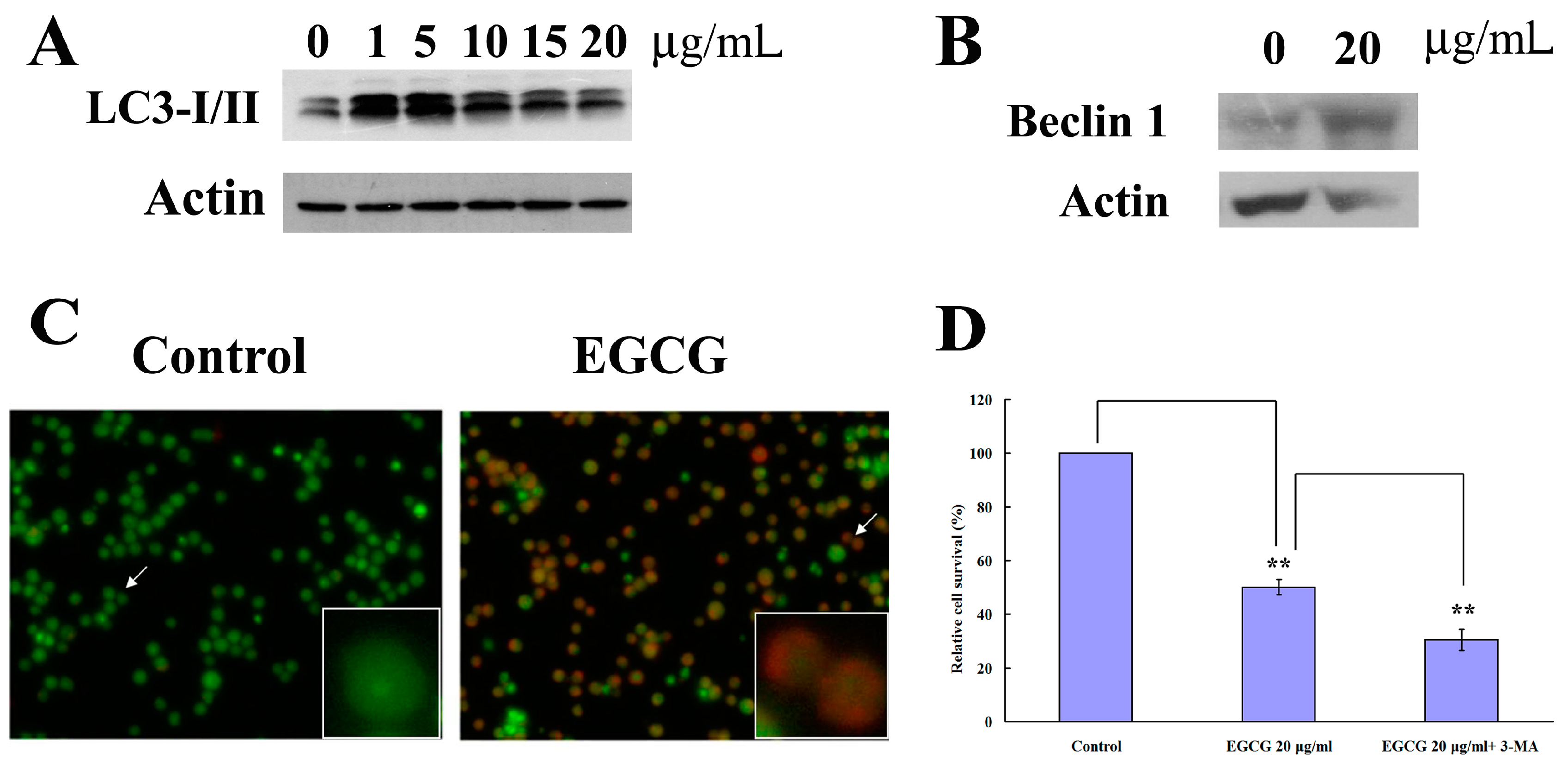
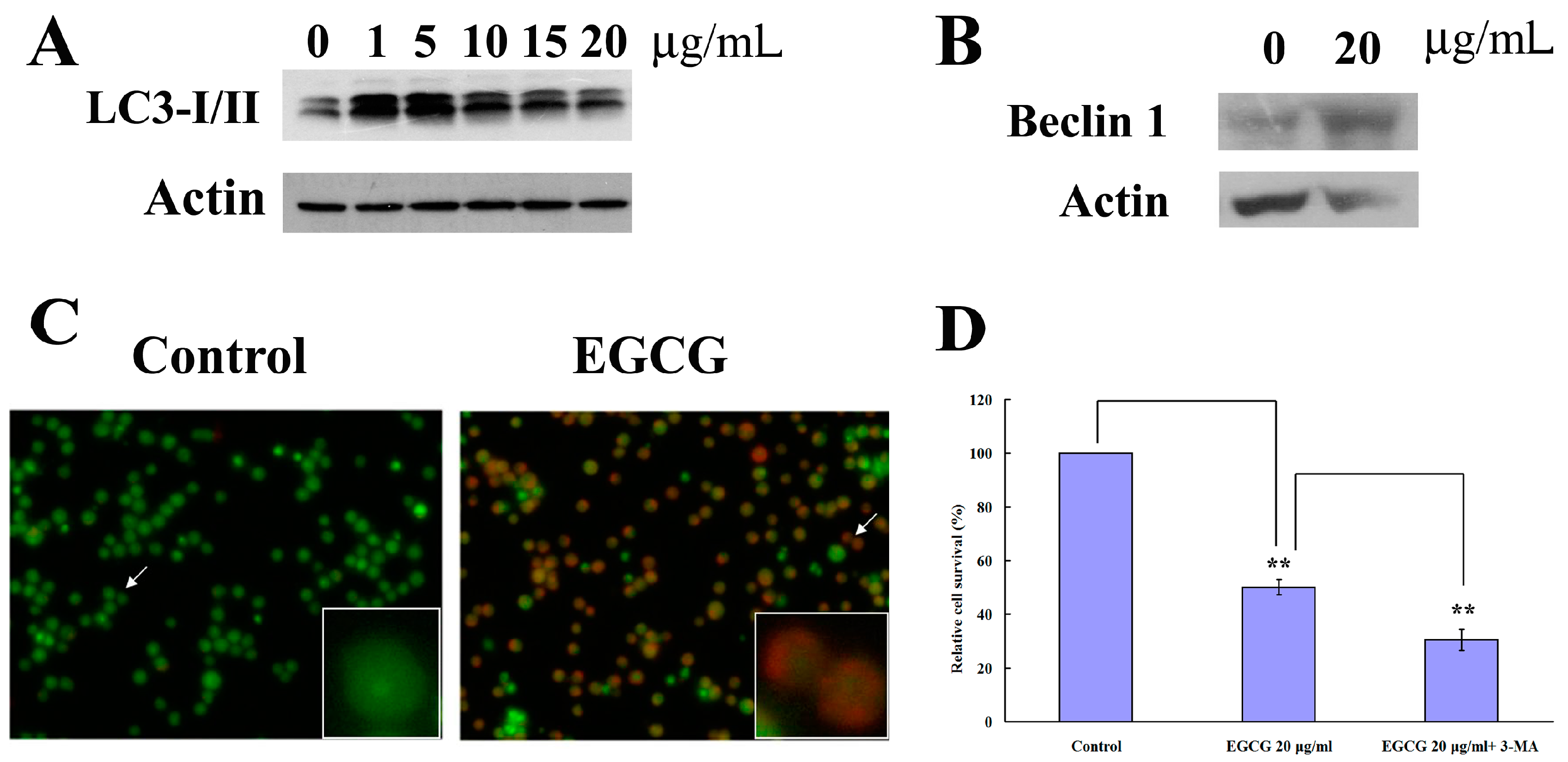
2.3. EGCG Induced Autophagy in PEL Cells
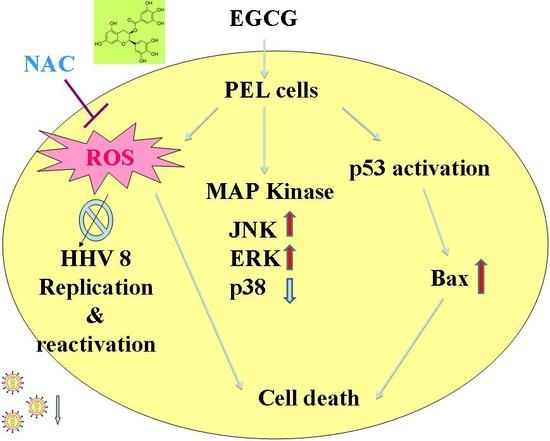
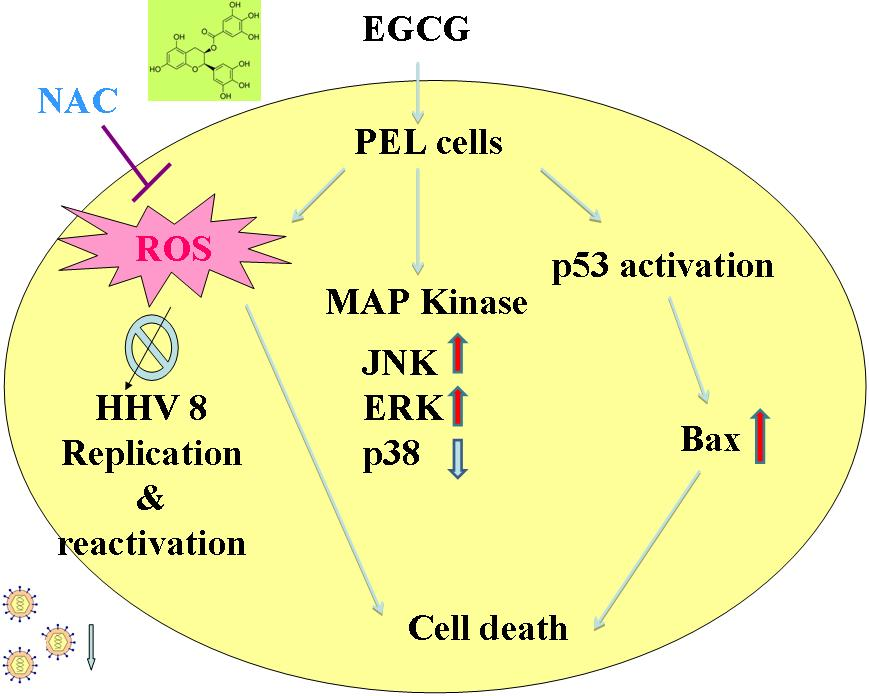
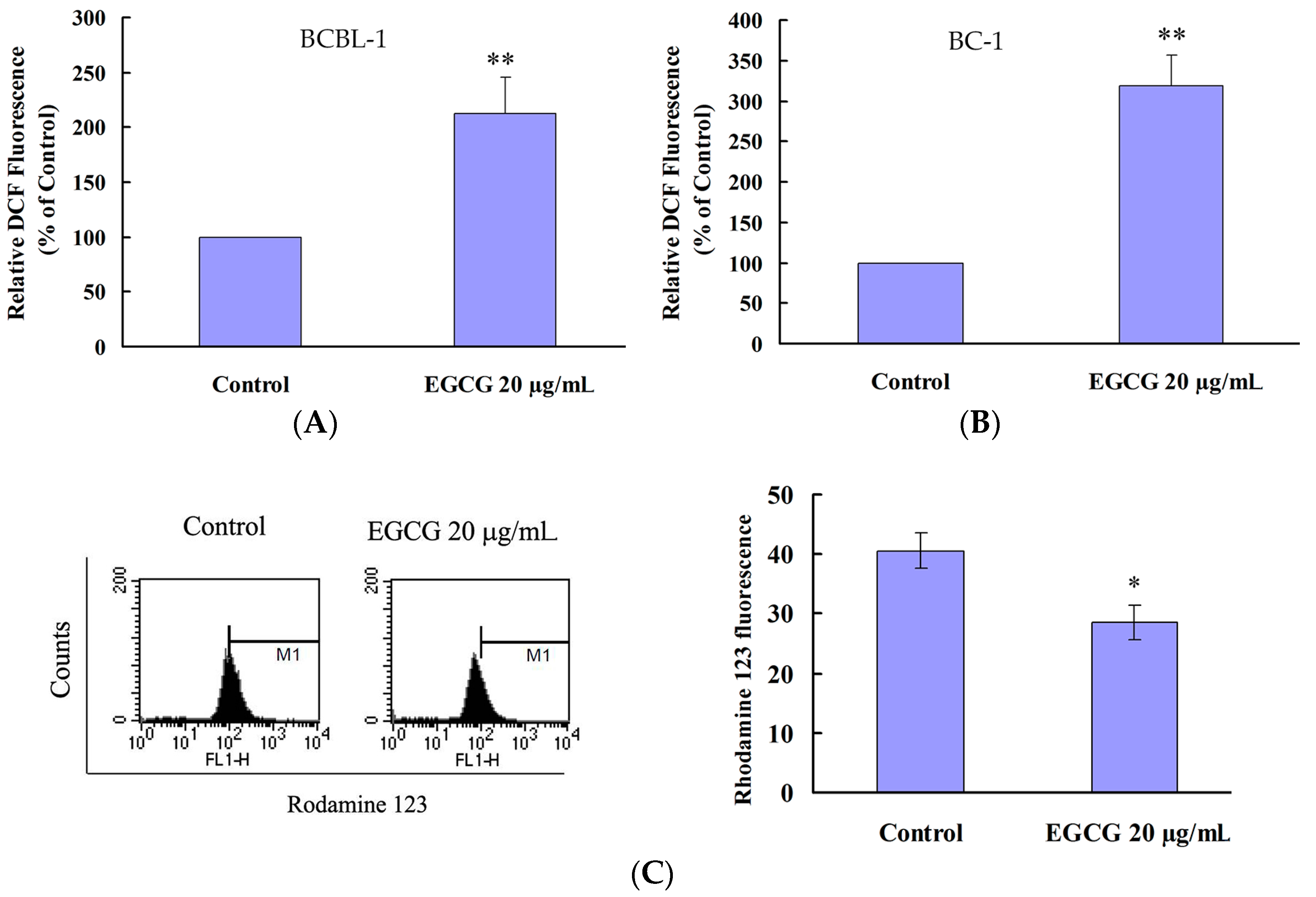
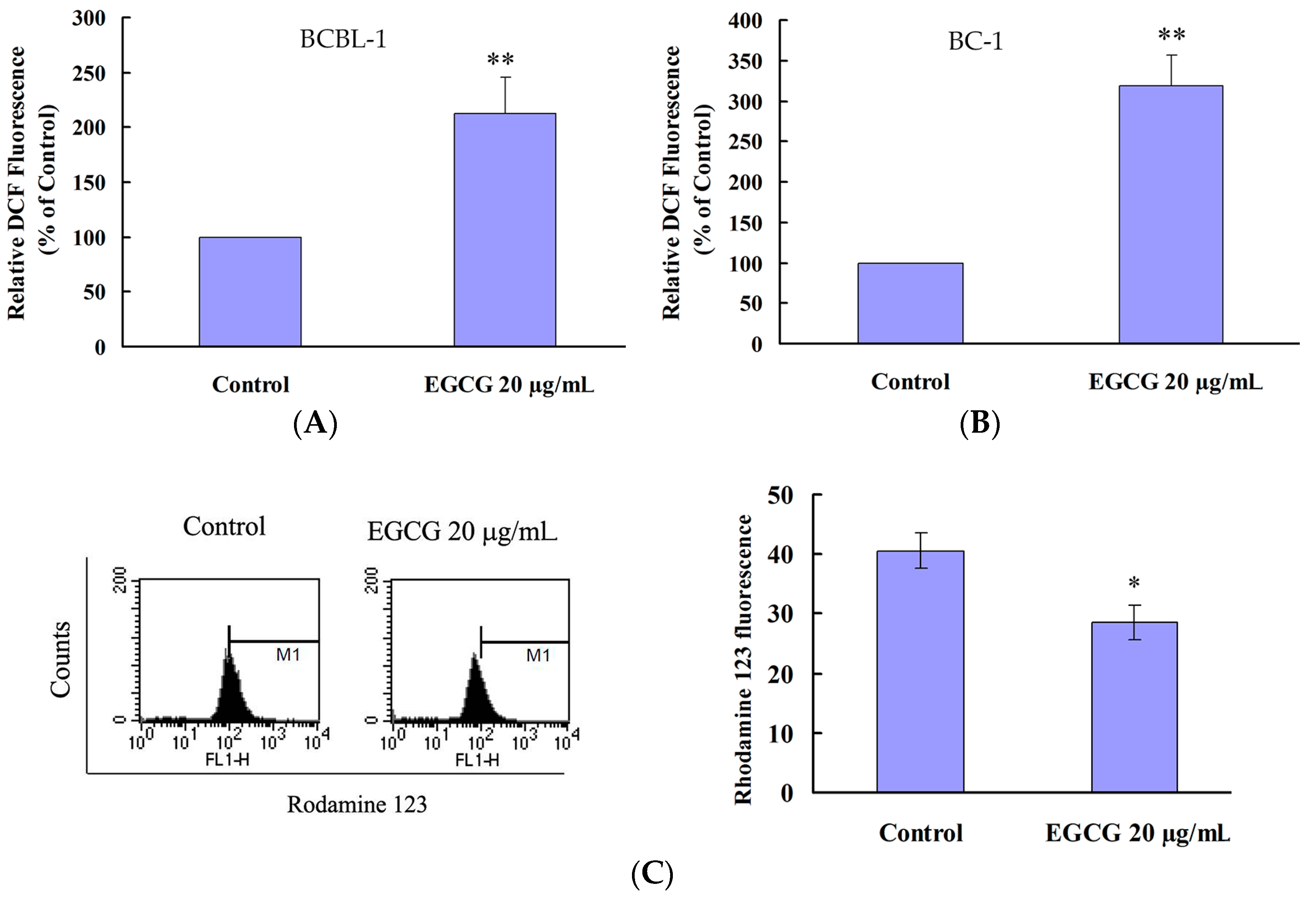
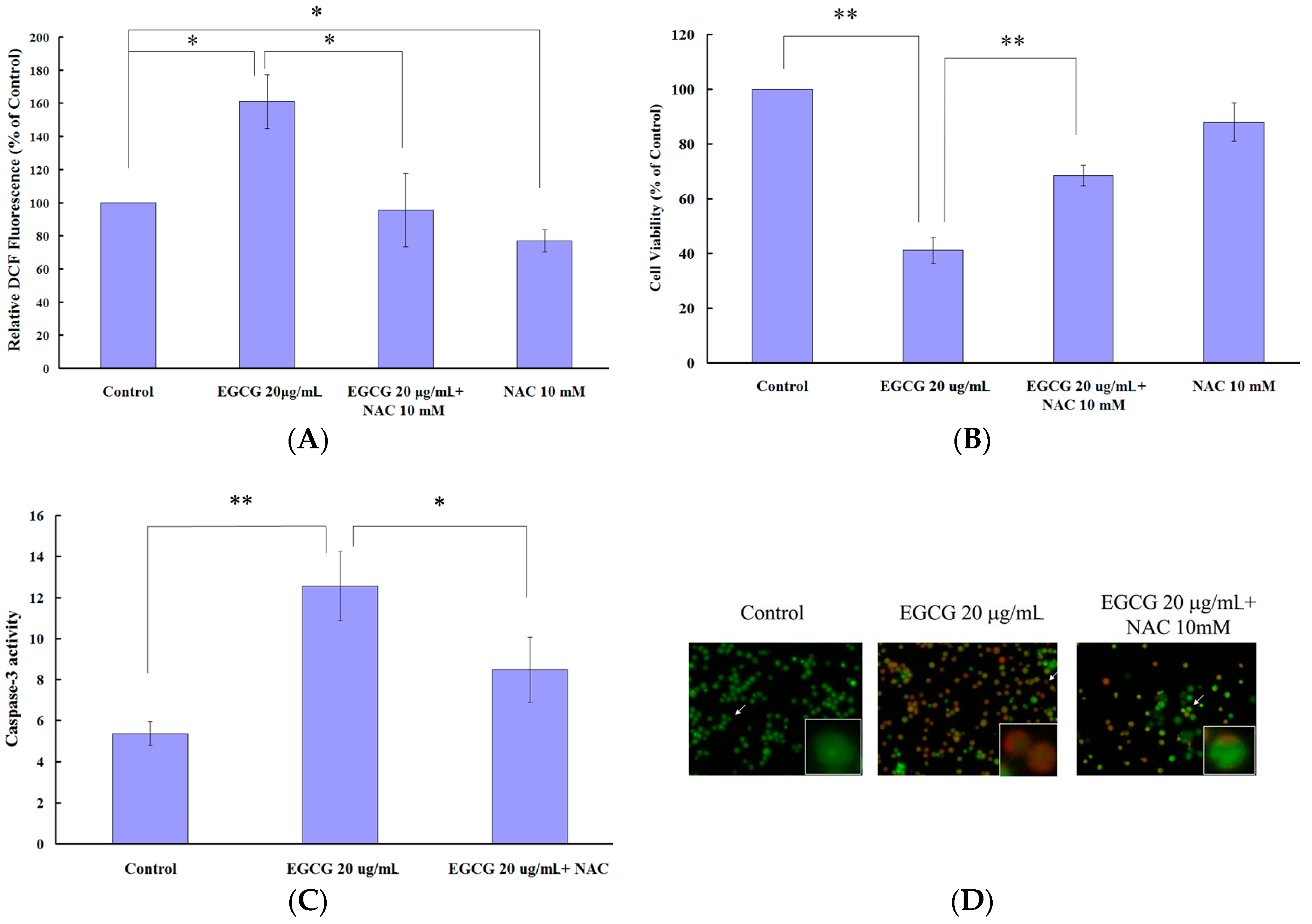
2.4. EGCG Induced Apoptosis and Autophagy through ROS Generation
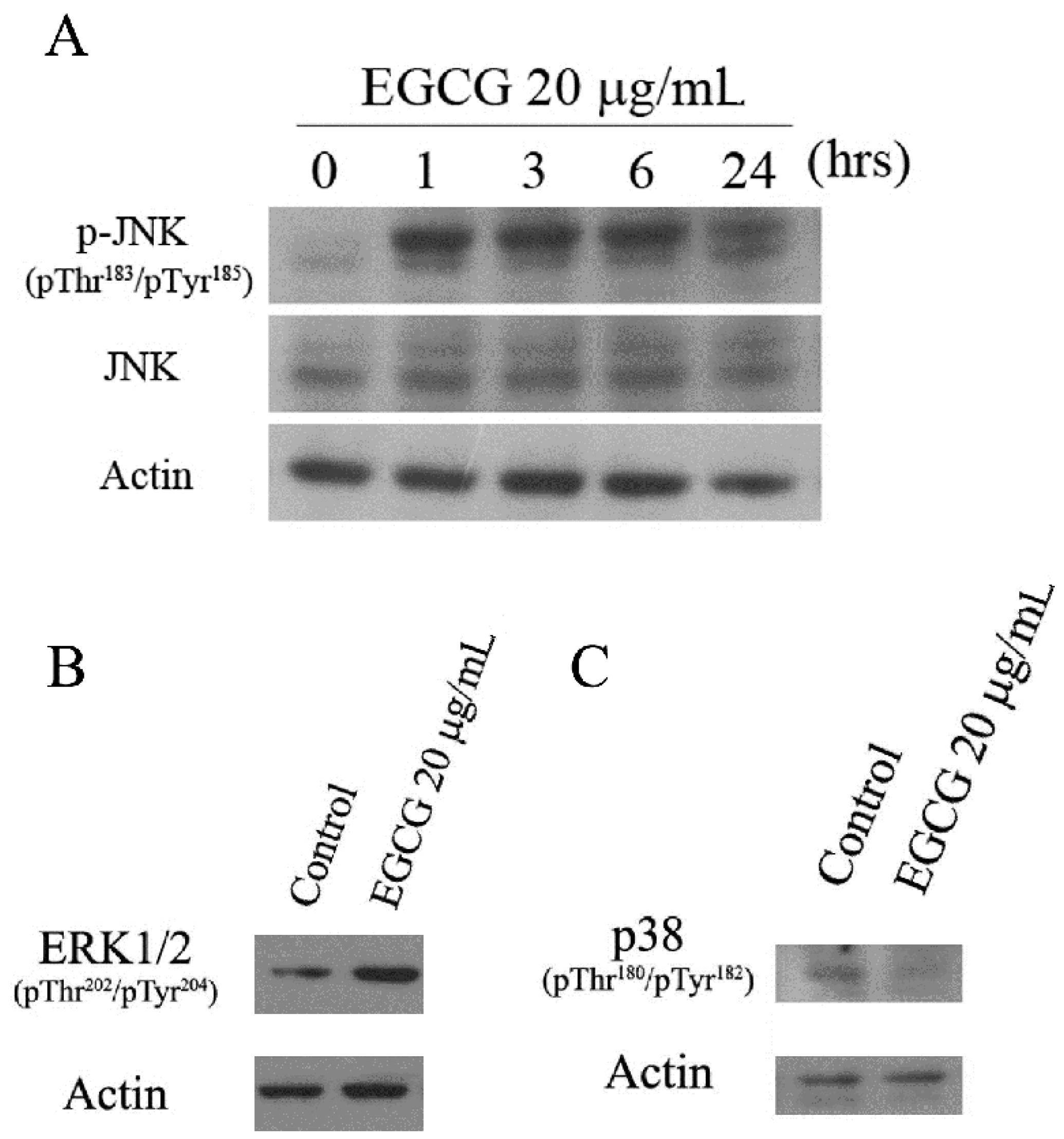
2.5. EGCG Induced Mitogen-Activated Protein Kinase Activation in PEL Cells
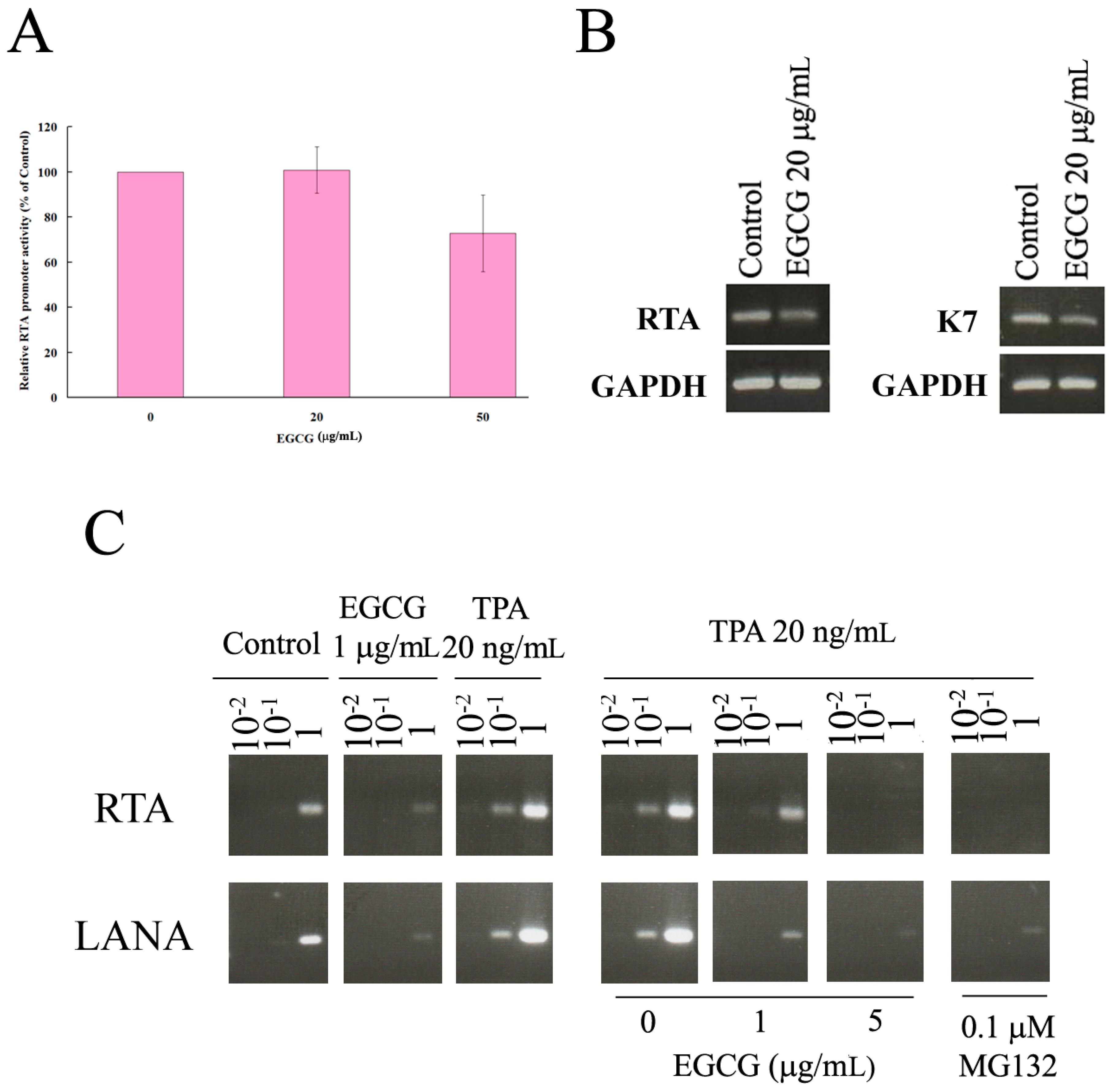
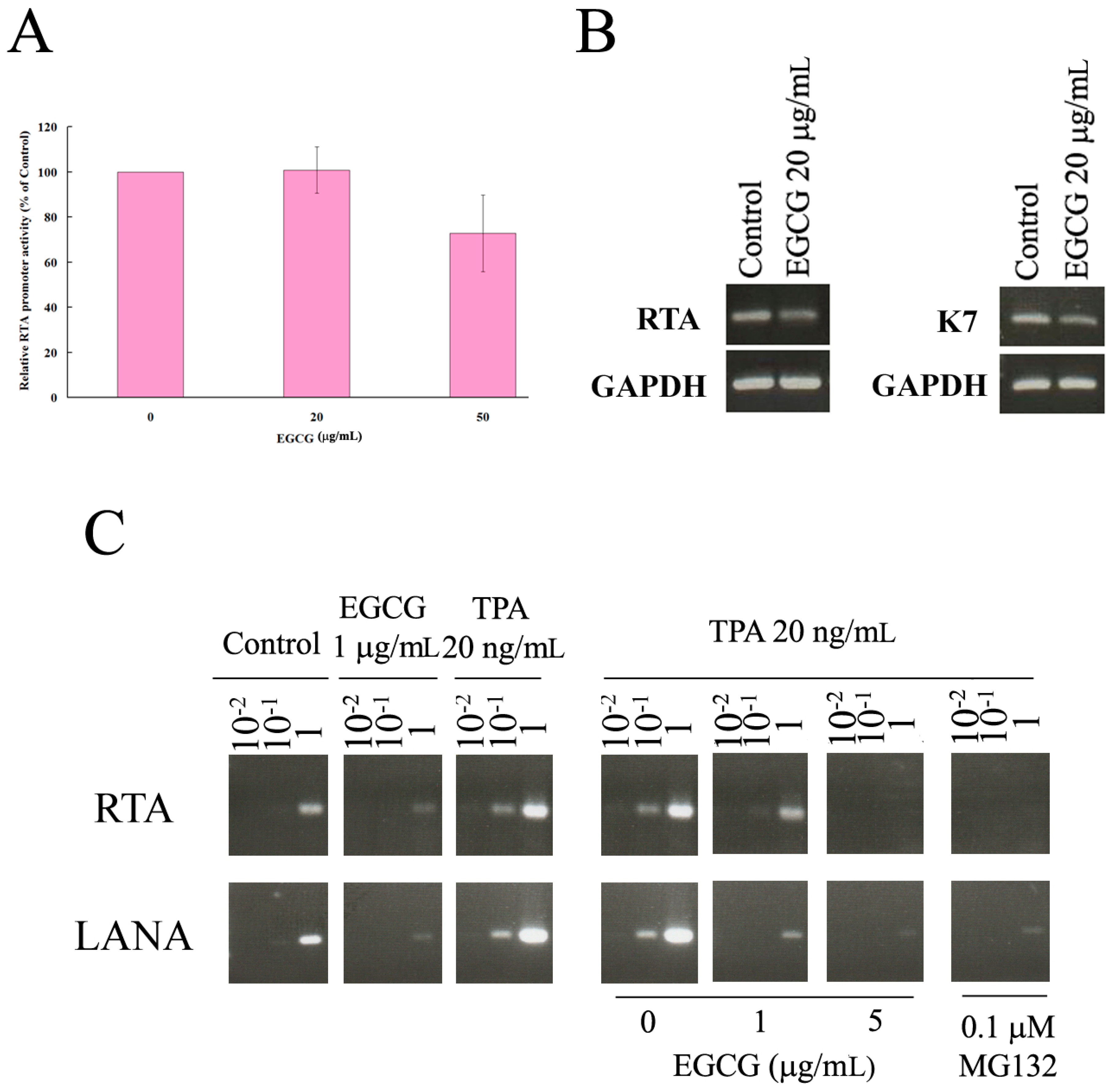
2.6. EGCG Inhibited HHV8 Replication in PEL Cells
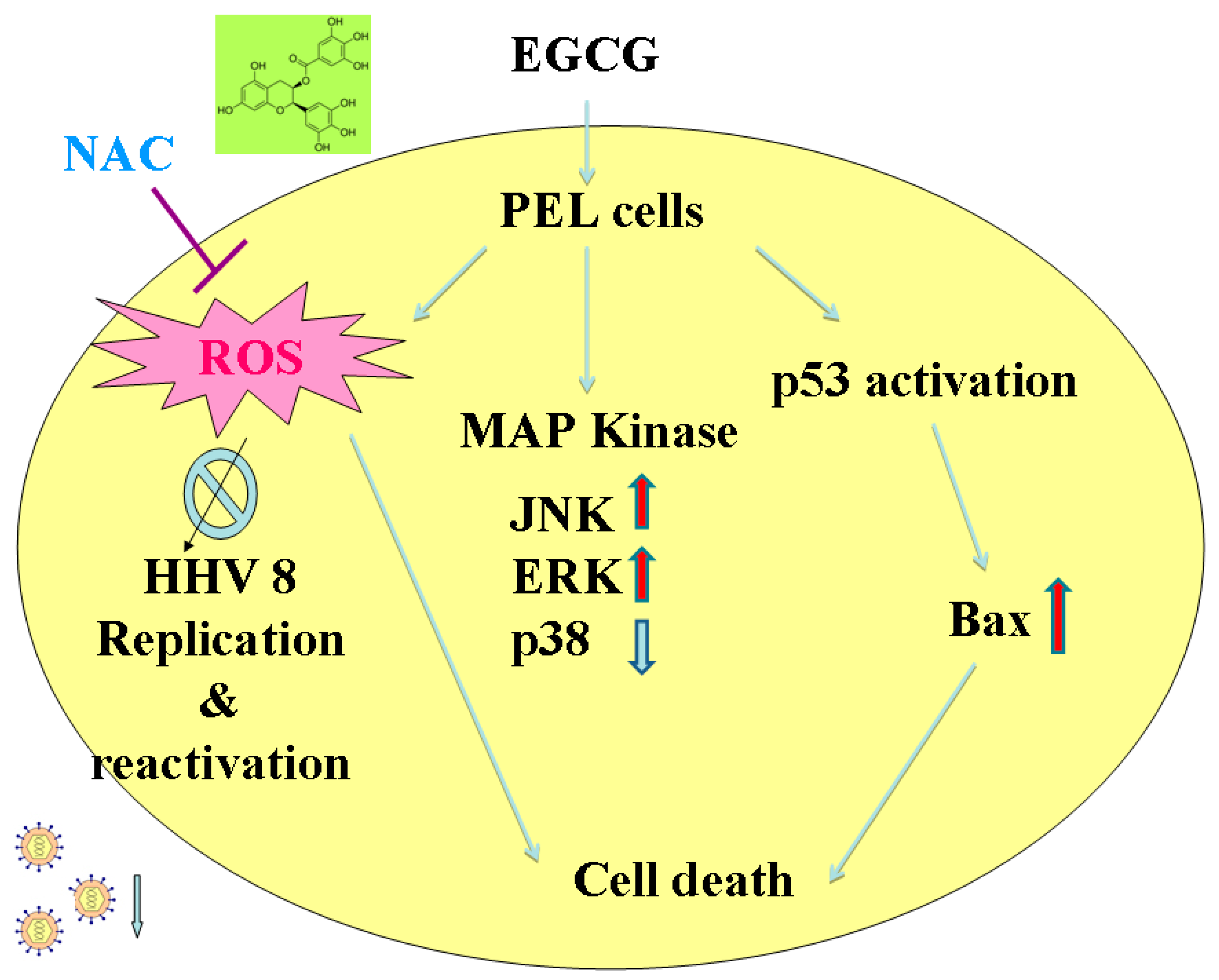
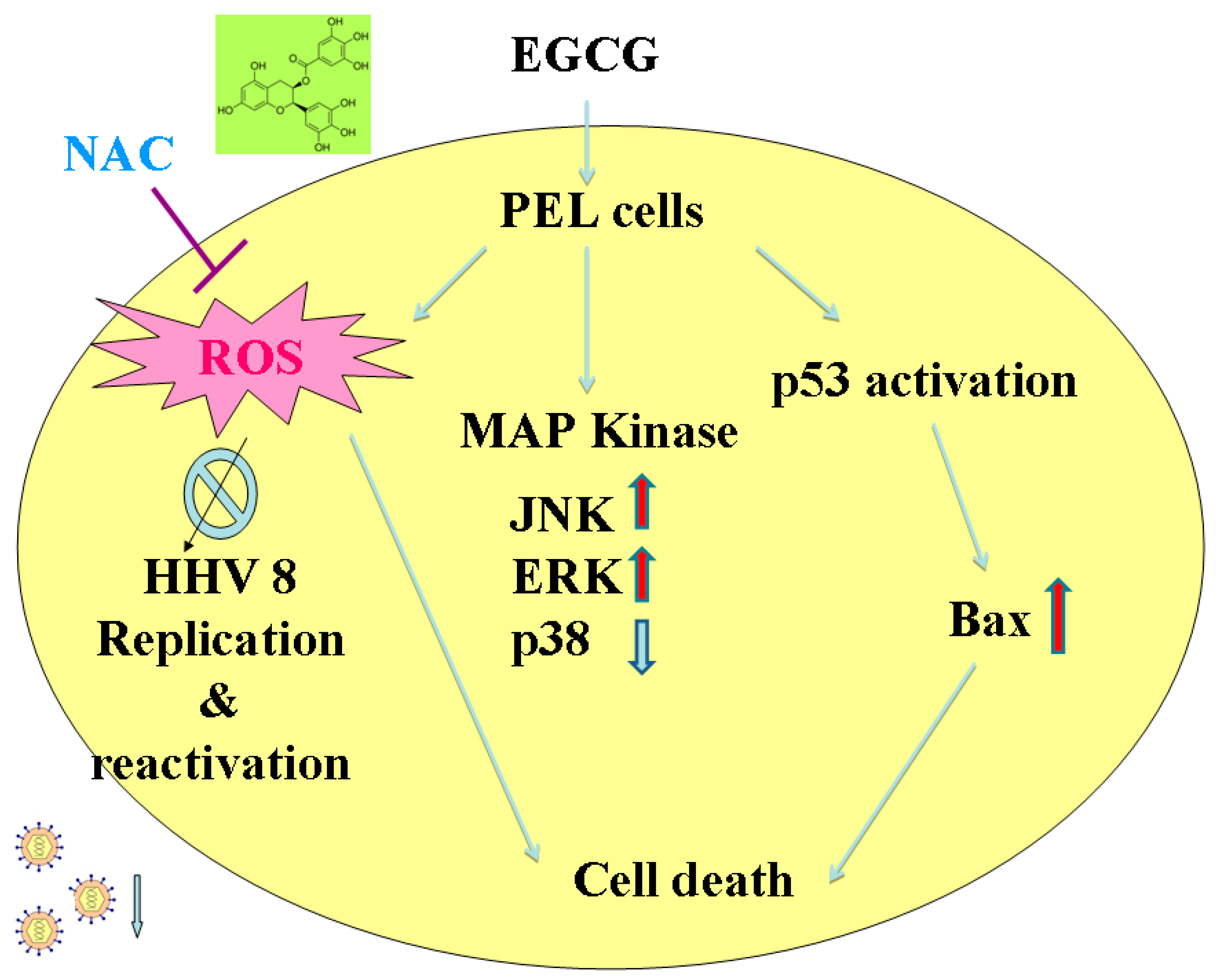
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Cell Viability Assays
4.4. Cell Cycle and Sub-G1 Analysis
4.5. Caspase-3 Activity Assay
4.6. Acidic Vesicular Organelle (AVO) Staining
4.7. Intracellular Reactive Oxygen Species (ROS) Determination
4.8. Measurement of Mitochondrial Membrane Potential
4.9. Western Blot Analysis
4.10. Luciferase Reporter Assay
4.11. Reverse Transcription-PCR
4.12. Polymerase Chain Reaction (PCR) for Viral Load
4.13. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [PubMed]
- Cai, Q.; Verma, S.C.; Choi, J.Y.; Ma, M.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus inhibits interleukin-4-mediated STAT6 phosphorylation to regulate apoptosis and maintain latency. J. Virol. 2010, 84, 11134–11144. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Cesarman, E.; Spina, M.; Gloghini, A.; Schulz, T.F. HIV-associated lymphomas and gamma-herpesviruses. Blood 2009, 113, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Gerard, L.; Gabarre, J.; Molina, J.M.; Rapp, C.; Abino, J.F.; Cadranel, J.; Chevret, S.; Oksenhendler, E. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J. Clin. Oncol. 2005, 23, 4372–4380. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.; Taskeen, M.; Mohammad, I.; Huo, C.; Chan, T.H.; Dou, Q.P. Recent advances on tea polyphenols. Front. Biosci. 2012, 4, 111–131. [Google Scholar] [CrossRef]
- Min, N.Y.; Kim, J.H.; Choi, J.H.; Liang, W.; Ko, Y.J.; Rhee, S.; Bang, H.; Ham, S.W.; Park, A.J.; Lee, K.H. Selective death of cancer cells by preferential induction of reactive oxygen species in response to (−)-epigallocatechin-3-gallate. Biochem. Biophys. Res. Commun. 2012, 421, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.K.; Wei, T.T.; Chiu, Y.F.; Tung, C.P.; Chuang, J.Y.; Hung, S.K.; Li, C.; Liu, S.T. Inhibition of Epstein-Barr virus lytic cycle by (−)-epigallocatechin gallate. Biochem. Biophys. Res. Commun. 2003, 301, 1062–1068. [Google Scholar] [CrossRef]
- Choi, K.C.; Jung, M.G.; Lee, Y.H.; Yoon, J.C.; Kwon, S.H.; Kang, H.B.; Kim, M.J.; Cha, J.H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Sin, S.H.; Dittmer, D.P. Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus lymphomas: Implications for therapy. J. Virol. 2007, 81, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Takemura, Y.; Hamada, H.; Sekido, Y.; Kubota, S. EGCG induces human mesothelioma cell death by inducing reactive oxygen species and autophagy. Cancer Cell Int. 2013, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Cakir, Y.; Ballinger, S.W. Reactive species-mediated regulation of cell signaling and the cell cycle: The role of MAPK. Antioxid. Redox Signal. 2005, 7, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Saeki, K.; Kobayashi, N.; Inazawa, Y.; Zhang, H.; Nishitoh, H.; Ichijo, H.; Isemura, M.; Yuo, A. Oxidation-triggered c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein (MAP) kinase pathways for apoptosis in human leukaemic cells stimulated by epigallocatechin-3-gallate (EGCG): A distinct pathway from those of chemically induced and receptor-mediated apoptosis. Biochem. J. 2002, 368, 705–720. [Google Scholar] [PubMed]
- Klass, C.M.; Krug, L.T.; Pozharskaya, V.P.; Offermann, M.K. The targeting of primary effusion lymphoma cells for apoptosis by inducing lytic replication of human herpesvirus 8 while blocking virus production. Blood 2005, 105, 4028–4034. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, A.; Anderson, J.; Papanastasiou, A.; Takeuchi, Y.; Boshoff, C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 2005, 105, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Saji, C.; Higashi, C.; Niinaka, Y.; Yamada, K.; Noguchi, K.; Fujimuro, M. Proteasome inhibitors induce apoptosis and reduce viral replication in primary effusion lymphoma cells. Biochem. Biophys. Res. Commun. 2011, 415, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, H. Cancer Preventive Activities of Tea Catechins. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Min, K.J.; Kwon, T.K. Anticancer effects and molecular mechanisms of epigallocatechin-3-gallate. Integr. Med. Res. 2014, 3, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Khan, H.Y.; Oves, M.; Al-Harrasi, A.; Rehmani, N.; Arif, H.; Hadi, S.M.; Ahmad, A. Cancer Therapy by Catechins Involves Redox Cycling of Copper Ions and Generation of Reactive Oxygen species. Toxins 2016, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xie, Y.; Feng, Y.; Zhang, L.; Huang, X.; Shen, X.; Luo, X. (−)-Epigallocatechingallate induces apoptosis in B lymphoma cells via caspase-dependent pathway and Bcl-2 family protein modulation. Int. J. Oncol. 2015, 46, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.I.; Braicu, C.; Zanoaga, O.; Pileczki, V.; Gherman, C.; Berindan-Neagoe, I.; Campian, R.S. Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis and autophagy in oral cancer SSC-4 cells. OncoTargets Ther. 2015, 8, 461–470. [Google Scholar] [PubMed]
- Fulda, S. Autophagy in Cancer Therapy. Front. Oncol. 2017, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F. Fooyin University: Kaohsiung, Taiwan, Unpublished Work. 2014.
- Kreuzaler, P.; Watson, C.J. Killing a cancer: What are the alternatives? Nat. Rev. Cancer 2012, 12, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, J.; Sun, R. Oxidative stress induces reactivation of Kaposi’s sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, L.; Lan, K.; Chen, X. Celecoxib Inhibits the Lytic Activation of Kaposi’s Sarcoma-Associated Herpesvirus through Down-Regulation of RTA Expression by Inhibiting the Activation of p38 MAPK. Viruses 2015, 7, 2268–2287. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, Z.; Zheng, W. A Review of the Antiviral Role of Green Tea Catechins. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Chen, Y.L.; Tsai, H.L.; Peng, C.W. EGCG debilitates the persistence of EBV latency by reducing the DNA binding potency of nuclear antigen 1. Biochem. Biophys. Res. Commun. 2012, 417, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, H.; Chen, L.; Yang, L.; Li, L.; Tao, Y.; Li, W.; Li, Z.; Liu, H.; Tang, M.; et al. (−)-Epigallocatechin-3-gallate inhibition of Epstein-Barr virus spontaneous lytic infection involves ERK1/2 and PI3-K/Akt signaling in EBV-positive cells. Carcinogenesis 2013, 34, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Nance, C.L.; Siwak, E.B.; Shearer, W.T. Preclinical development of the green tea catechin, epigallocatechin gallate, as an HIV-1 therapy. J. Allergy Clin. Immunol. 2009, 123, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Traganos, F.; Darzynkiewicz, Z. Lysosomal proton pump activity: Supravital cell staining with acridine orange differentiates leukocyte subpopulations. Methods Cell Biol. 1994, 41, 185–194. [Google Scholar] [PubMed]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Chiou, Y.H.; Wang, L.Y.; Chang, S.T.; Shyu, H.W.; Chen, C.Y.; Lin, K.H.; Chou, M.C. Cisplatin disrupts the latency of human herpesvirus 8 and induces apoptosis in primary effusion lymphoma cells. Cancer Investig. 2012, 30, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.Y.; Chen, C.Y.; Shyu, H.W.; Hong, S.; Chen, H.M.; Chiou, Y.H.; Lin, K.H.; Chou, M.C.; Wang, L.Y.; Wang, Y.F. Resveratrol induces cell death and inhibits human herpesvirus 8 replication in primary effusion lymphoma cells. Chem. Biol. Interact. 2015, 242, 372–379. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-Y.; Chen, C.-Y.; Chiou, Y.-H.; Shyu, H.-W.; Lin, K.-H.; Chou, M.-C.; Huang, M.-H.; Wang, Y.-F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2018, 19, 16. https://doi.org/10.3390/ijms19010016
Tsai C-Y, Chen C-Y, Chiou Y-H, Shyu H-W, Lin K-H, Chou M-C, Huang M-H, Wang Y-F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. International Journal of Molecular Sciences. 2018; 19(1):16. https://doi.org/10.3390/ijms19010016
Chicago/Turabian StyleTsai, Ching-Yi, Chang-Yu Chen, Yee-Hsuan Chiou, Huey-Wen Shyu, Kuan-Hua Lin, Miao-Chen Chou, Mei-Han Huang, and Yi-Fen Wang. 2018. "Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells" International Journal of Molecular Sciences 19, no. 1: 16. https://doi.org/10.3390/ijms19010016