Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions?


Abstract
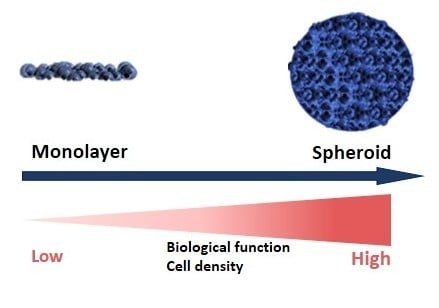
:
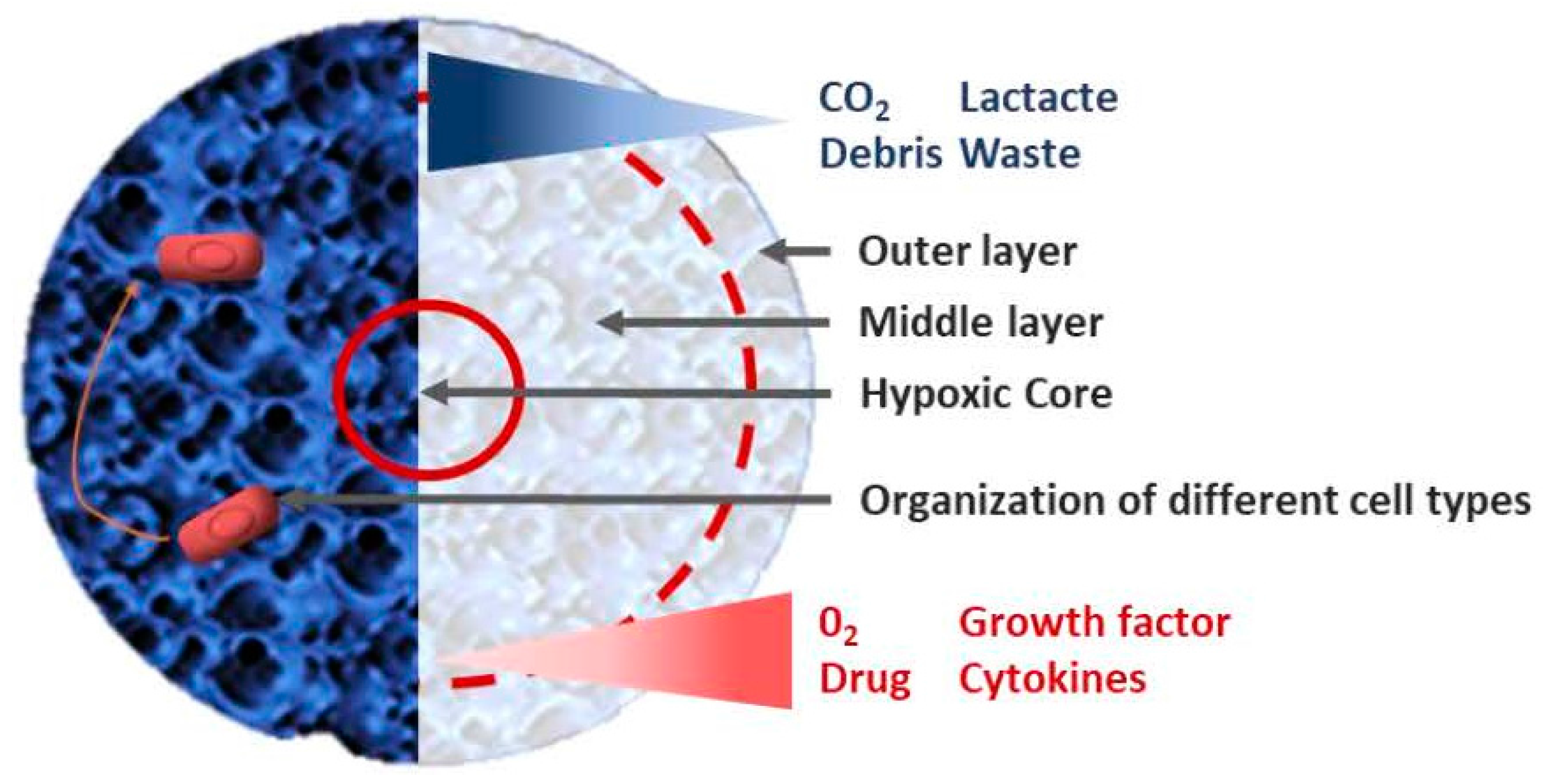
1. Introduction
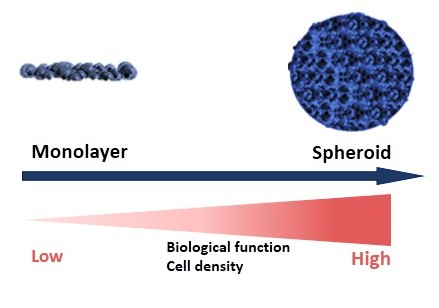
2. Different Models of 3D Culture
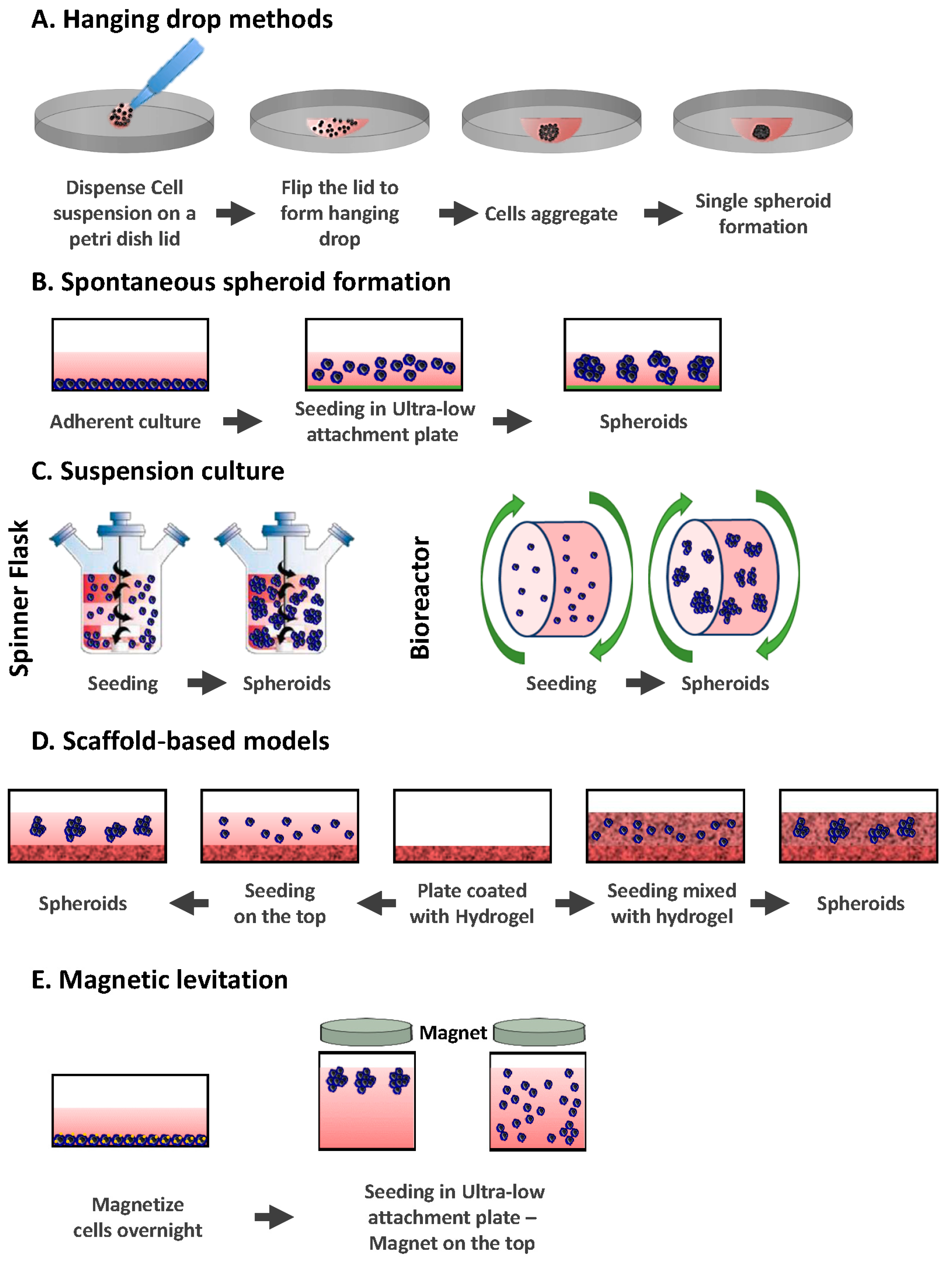
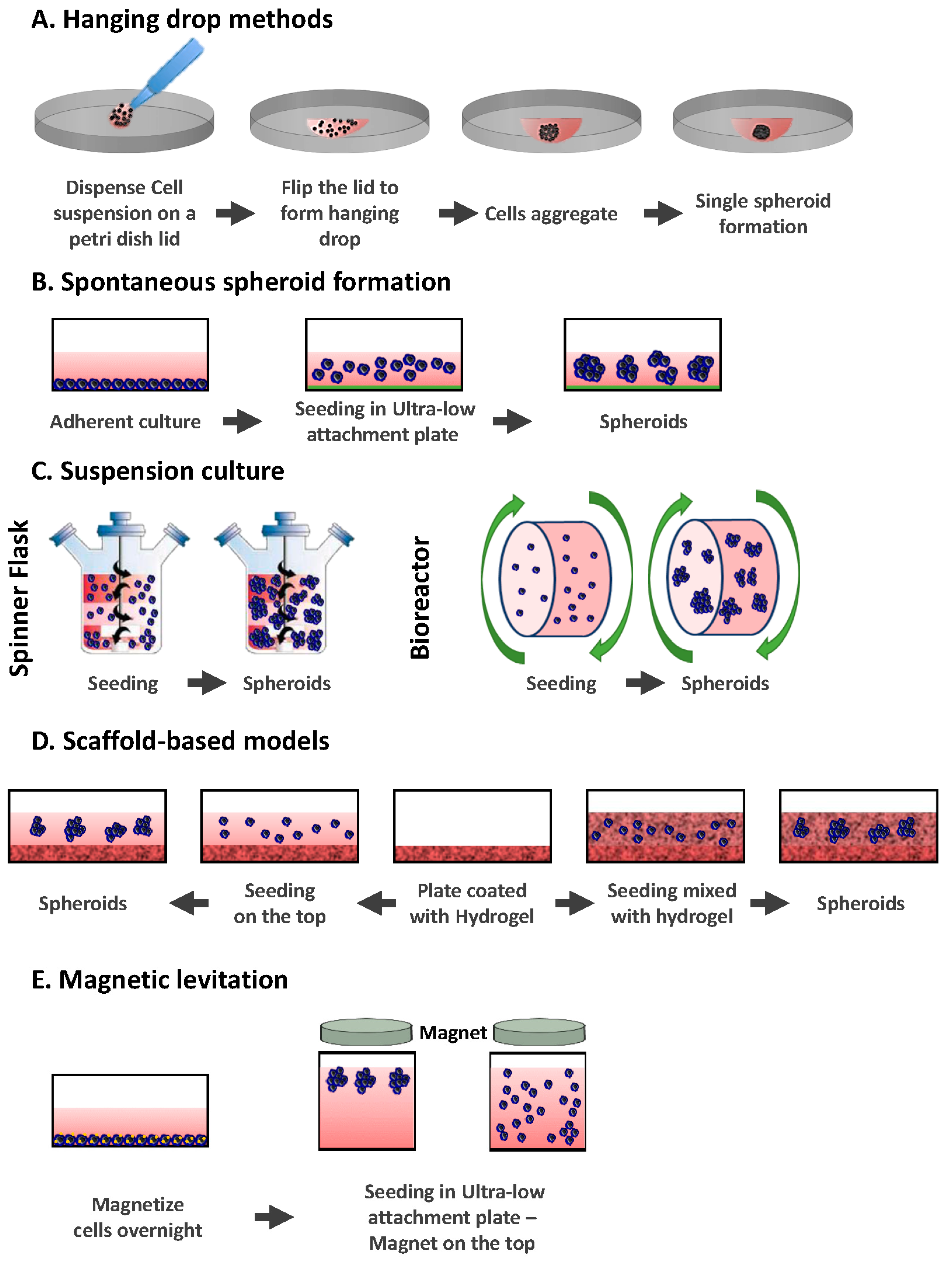
2.1. Hanging Drop Methods
2.2. Spontaneous Spheroid Formation: Non-Adherent Surface Methods/Ultra Low Attachment Plates
2.3. Suspension Culture
2.3.1. Spinner Flasks
2.3.2. Bioreactors
2.4. Scaffold-Based Models: Hydrogels
2.5. Magnetic Levitation
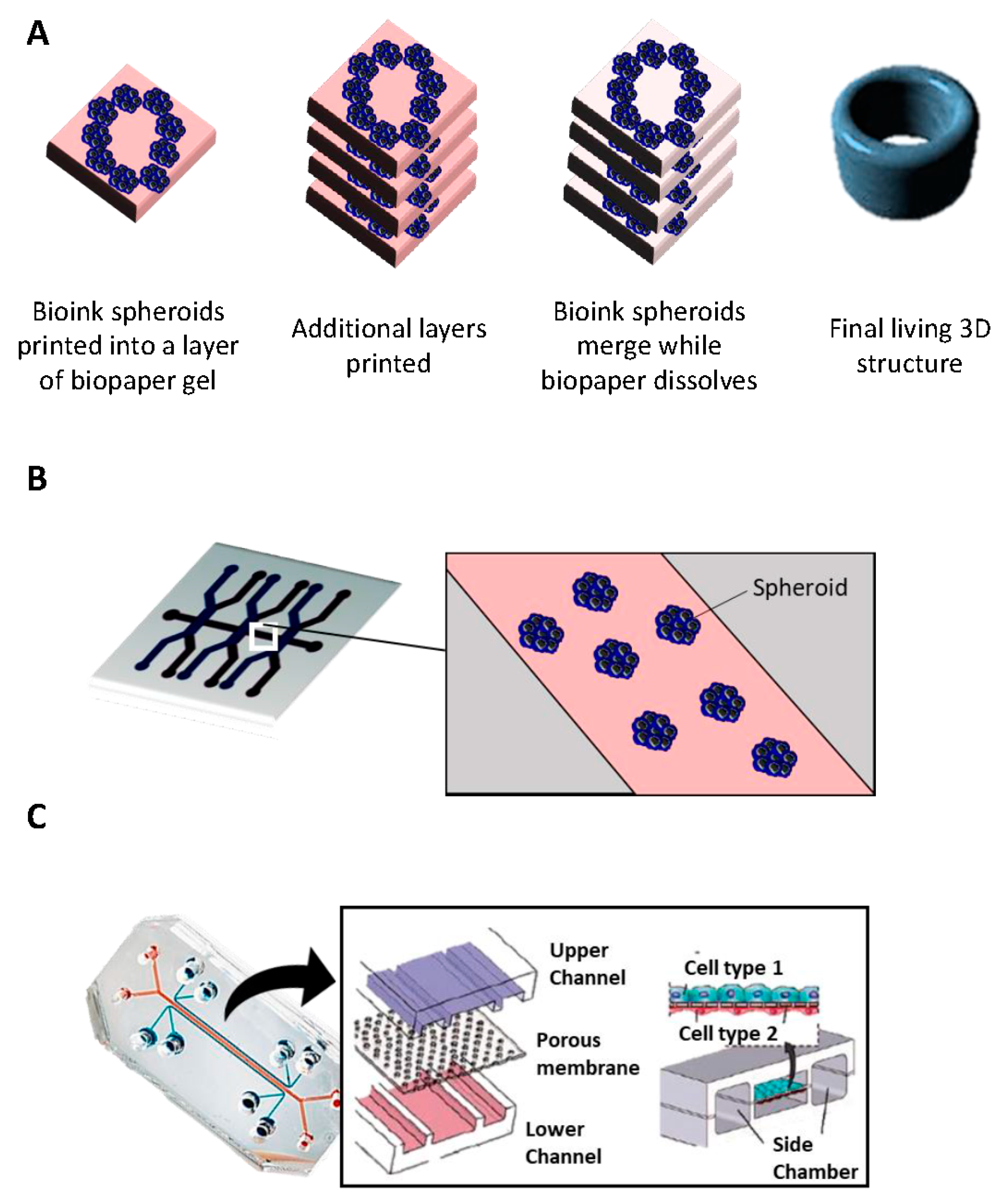
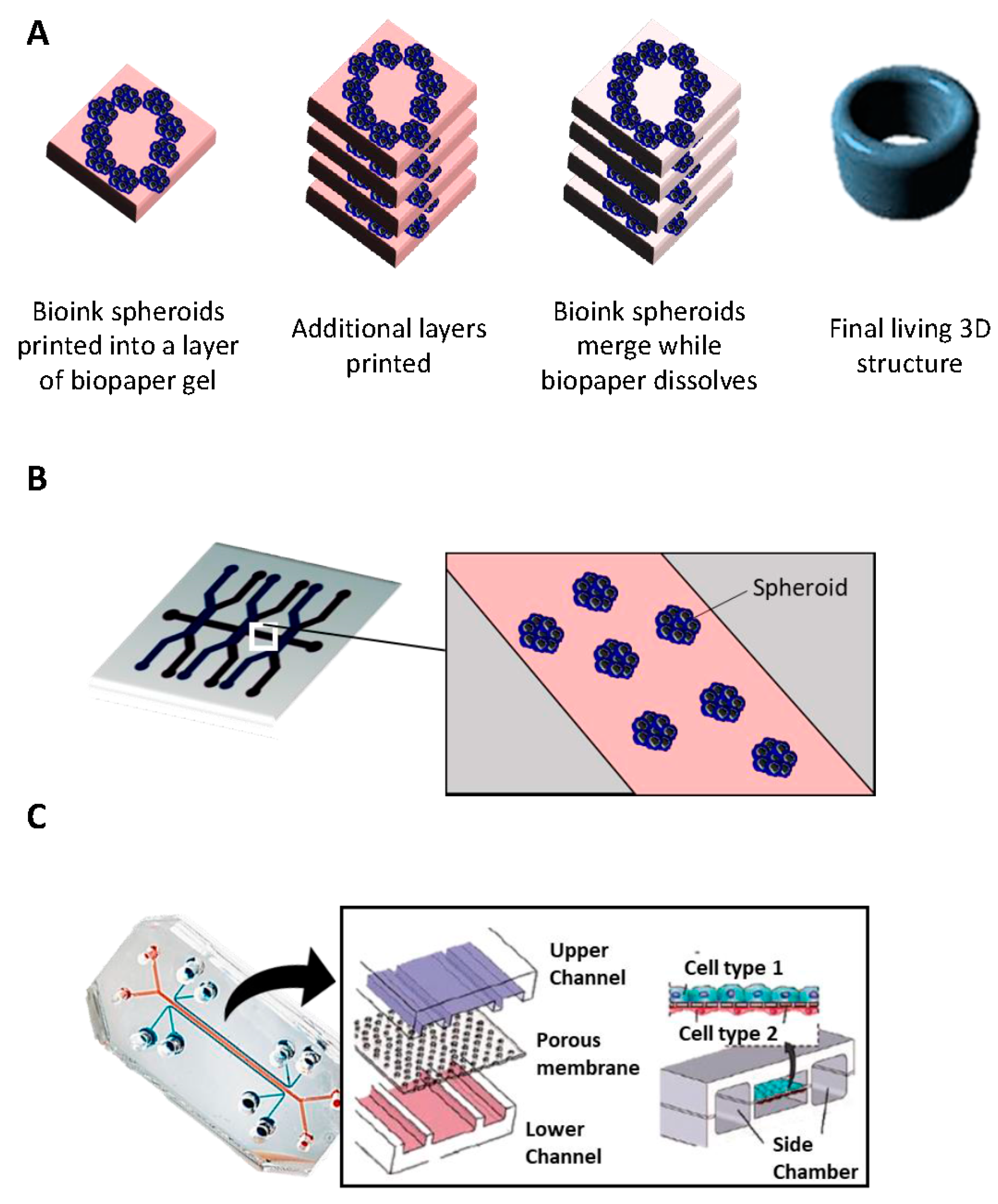
2.6. Bioprinting
- i
- Inkjet printing uses the assistance of a digital computer to control a narrow nozzle to generate droplets that are deposited on a scaffold in a precise and controlled manner [54]. There are four main current techniques used to create these droplets: thermal, piezoelectric, acoustic, and electrostatic.
- ii
- Extrusion printing is another version of inkjet printing. It differs from inkjet printing by the fact that it can print on viscous materials. This is achieved through the use of an air-force pump or a mechanical screw plunger to dispense bioinks. A continuous force is applied, allowing the constant printing of cylindrical lines instead of a single bioink droplet [55].
- iii
- Laser-assisted printing system uses a donor layer composed of a metal type energy-absorbing layer (e.g., titatium or gold) on the top and a layer of bioink solution at the bottom [56]. During the printing, a small part of the energy-absorbing layer is exposed to a focused laser beam, which generates a bubble in the bioink solution that is pushed onto a receiving substrate (where printing is achieved) [51].
2.7. Microfluidic Platforms
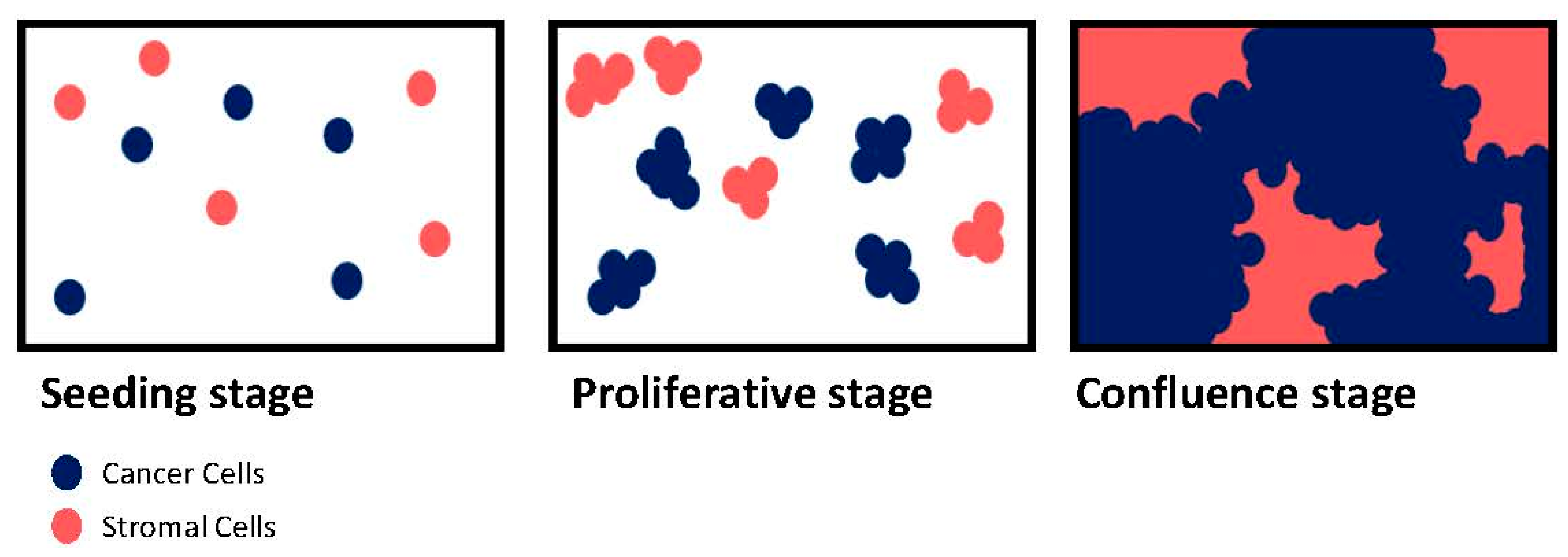
3. Tumor-Microenvironment Interactions: 2D vs. 3D
3.1. Proliferation
3.2. Migration and Invasion
3.3. Resistance to Treatment
3.3.1. Cytotoxic Agents
3.3.2. Targeted Therapy
4. Conclusions
Conflicts of Interest
References
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour-stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hall, H.G.; Parry, G. How does the extracellular matrix direct gene expression? J. Theor. Biol. 1982, 99, 31–68. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.M.; Khauv, D.; Bissell, M.J.; Radisky, D.C. Change in cell shape is required for matrix metalloproteinase-induced epithelial-mesenchymal transition of mammary epithelial cells. J. Cell. Biochem. 2008, 105, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Boudreau, A.; Bissell, M.J. Tissue architecture and function: Dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009, 28, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Cukierman, E. Modeling tissue morphogenesis and cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des. Dev. Ther. 2014, 8, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Kelm, J.M.; Ehler, E.; Nielsen, L.K.; Schlatter, S.; Perriard, J.C.; Fussenegger, M. Design of artificial myocardial microtissues. Tissue Eng. 2004, 10, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, A.; Young, J.; Nielsen, J.E.; Joensen, U.N.; Toft, B.G.; Rajpert-De Meyts, E.; Loveland, K.L. Hanging drop cultures of human testis and testis cancer samples: A model used to investigate activin treatment effects in a preserved niche. Br. J. Cancer 2014, 110, 2604–2614. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, H. Methods for inducing embryoid body formation: In vitro differentiation system of embryonic stem cells. J. Biosci. Bioeng. 2007, 103, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.C.; Hsiao, A.Y.; Allen, S.G.; Torisawa, Y.S.; Ho, M.; Takayama, S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst 2011, 136, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, A.Y.; Tung, Y.C.; Kuo, C.H.; Mosadegh, B.; Bedenis, R.; Pienta, K.J.; Takayama, S. Micro-ring structures stabilize microdroplets to enable long term spheroid culture in 384 hanging drop array plates. Biomed. Microdevices 2012, 14, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, A.Y.; Tung, Y.C.; Qu, X.; Patel, L.R.; Pienta, K.J.; Takayama, S. 384 hanging drop arrays give excellent Z-factors and allow versatile formation of co-culture spheroids. Biotechnol. Bioeng. 2012, 109, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Ward, M.R.; Rowley, K.R.; Wold, R.M.; Takayama, S.; Buckanovich, R.J.; Mehta, G. Formation of stable small cell number three-dimensional ovarian cancer spheroids using hanging drop arrays for preclinical drug sensitivity assays. Gynecol. Oncol. 2015, 138, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [PubMed]
- Sutherland, R.M.; Inch, W.R.; McCredie, J.A.; Kruuv, J. A multi-component radiation survival curve using an in vitro tumour model. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1970, 18, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Yuhas, J.M.; Li, A.P.; Martinez, A.O.; Ladman, A.J. A simplified method for production and growth of multicellular tumor spheroids. Cancer Res. 1977, 37, 3639–3643. [Google Scholar] [PubMed]
- Ivascu, A.; Kubbies, M. Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J. Biomol. Screen. 2006, 11, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Z.; Chang, H.Y. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol. J. 2008, 3, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B. Three-dimensional tissue culture models in cancer biology. Semin. Cancer Biol. 2005, 15, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Leidig, T.; Rodday, B.; Lindemann, C.; Mueller-Klieser, W. Test system for trifunctional antibodies in 3D MCTS culture. J. Biomol. Screen. 2009, 14, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.P.; Goodwin, T.J.; Wolf, D.A. Cell culture for three-dimensional modeling in rotating-wall vessels: An application of simulated microgravity. J. Tissue Cult. Methods 1992, 14, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Eglen, R.M. Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [PubMed]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Aggeler, J.; Ram, T.G.; Bissell, M.J. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development 1989, 105, 223–235. [Google Scholar] [PubMed]
- Aggeler, J.; Ward, J.; Blackie, L.M.; Barcellos-Hoff, M.H.; Streuli, C.H.; Bissell, M.J. Cytodifferentiation of mouse mammary epithelial cells cultured on a reconstituted basement membrane reveals striking similarities to development in vivo. J. Cell Sci. 1991, 99, 407–417. [Google Scholar] [PubMed]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Guo, T.W.; Wu, A.P.; Wells, A.; Gertler, F.B.; Lauffenburger, D.A. Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3D arises from an intrinsic increase in speed but an extrinsic matrix- and proteolysis-dependent increase in persistence. Mol. Biol. Cell 2008, 19, 4249–4259. [Google Scholar] [CrossRef] [PubMed]
- Walters, B.D.; Stegemann, J.P. Strategies for directing the structure and function of three-dimensional collagen biomaterials across length scales. Acta Biomater. 2014, 10, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.D.; Carvajal, N.; Jin, A.; Matsumoto, K.; Yamada, K.M. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 2015, 6, 8720. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.Y.; Jones, C.E.; Anseth, K.S. PEG-peptide hydrogels reveal differential effects of matrix microenvironmental cues on melanoma drug sensitivity. Integr. Biol. 2017, 9, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Mukundan, S.; Jaramillo, M.; Oesterreich, S.; Sant, S. Three-Dimensional Breast Cancer Models Mimic Hallmarks of Size-Induced Tumor Progression. Cancer Res. 2016, 76, 3732–3743. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.J.; Martino, M.M.; de Laporte, L.; Tortelli, F.; Briquez, P.S.; Hubbell, J.A. Engineering the regenerative microenvironment with biomaterials. Adv. Healthc. Mater. 2013, 2, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Yoon, H.Y.; Koo, H.; Ko, S.H.; Shim, J.S.; Lee, J.H.; Kim, K.; Kwon, I.C.; Kim, D.D. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic® for tumor-targeted delivery of docetaxel. Biomaterials 2011, 32, 7181–7190. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jambhrunkar, S.; Thorn, P.; Chen, J.; Gu, W.; Yu, C. Hyaluronic acid modified mesoporous silica nanoparticles for targeted drug delivery to CD44-overexpressing cancer cells. Nanoscale 2013, 5, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Highley, C.B.; Prestwich, G.D.; Burdick, J.A. Recent advances in hyaluronic acid hydrogels for biomedical applications. Curr. Opin. Biotechnol. 2016, 40, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Souza, G.R.; Molina, J.R.; Raphael, R.M.; Ozawa, M.G.; Stark, D.J.; Levin, C.S.; Bronk, L.F.; Ananta, J.S.; Mandelin, J.; Georgescu, M.M.; et al. Three-dimensional tissue culture based on magnetic cell levitation. Nat. Nanotechnol. 2010, 5, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.; Gage, J.A.; Raphael, R.M.; Moore, R.H.; Killian, T.C.; Grande-Allen, K.J.; Souza, G.R. Assembly of a three-dimensional multitype bronchiole coculture model using magnetic levitation. Tissue Eng. Part C Methods 2013, 19, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiee, A.; Atala, A. Printing Technologies for Medical Applications. Trends Mol. Med. 2016, 22, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Duchamp, M.; Oklu, R.; Ellisen, L.W.; Langer, R.; Khademhosseini, A. Bioprinting the Cancer Microenvironment. ACS Biomater. Sci. Eng. 2016, 2, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Huang, Y.; Schilling, A.F.; Hubbell, K.; Cui, X. Organ Bioprinting: Are We There Yet? Adv. Healthc. Mater. 2017. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, X.F.; Gao, G.; Yonezawa, T.; Cui, X. 3D bioprinting and the current applications in tissue engineering. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Boland, T.; Xu, T.; Damon, B.; Cui, X. Application of inkjet printing to tissue engineering. Biotechnol. J. 2006, 1, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Mandrycky, C.; Wang, Z.; Kim, K.; Kim, D.H. 3D bioprinting for engineering complex tissues. Biotechnol. Adv. 2016, 34, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F.; Souquet, A.; Catros, S.; Guillotin, B.; Lopez, J.; Faucon, M.; Pippenger, B.; Bareille, R.; Remy, M.; Bellance, S.; et al. High-throughput laser printing of cells and biomaterials for tissue engineering. Acta Biomater. 2010, 6, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, S.; Onal, S.; Yu, C.H.; Zhao, J.J.; Tasoglu, S. Bioprinting for cancer research. Trends Biotechnol. 2015, 33, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Torisawa, Y.S.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Microengineered physiological biomimicry: Organs-on-chips. Lab Chip 2012, 12, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, S.M.; Sylvester, D.C.; Dyer, C.E.; Stafford, N.D.; Haswell, S.J.; Greenman, J. A microfluidic system for testing the responses of head and neck squamous cell carcinoma tissue biopsies to treatment with chemotherapy drugs. Ann. Biomed. Eng. 2012, 40, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Holton, A.B.; Sinatra, F.L.; Kreahling, J.; Conway, A.J.; Landis, D.A.; Altiok, S. Microfluidic biopsy trapping device for the real-time monitoring of tumor microenvironment. PLoS ONE 2017, 12, e0169797. [Google Scholar] [CrossRef] [PubMed]
- Atac, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.J.; Domansky, K.; Kaazempur-Mofrad, M.R.; Kalezi, A.; Capitano, A.; Upadhyaya, A.; Kurzawski, P.; Wack, K.E.; Stolz, D.B.; Kamm, R.; et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol. Bioeng. 2002, 78, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Yamamoto, T.; Sakai, H.; Sakai, Y.; Fujii, T. An integrated microfluidic system for long-term perfusion culture and on-line monitoring of intestinal tissue models. Lab Chip 2008, 8, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Patra, B.; Peng, C.C.; Liao, W.H.; Lee, C.H.; Tung, Y.C. Drug testing and flow cytometry analysis on a large number of uniform sized tumor spheroids using a microfluidic device. Sci. Rep. 2016, 6, 21061. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D. Clinical and biological implications of the tumor microenvironment. Cancer Microenviron. 2012, 5, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M. Contact inhibition in tissue culture. In Vitro 1970, 6, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.M.; Jeng, D.Y.; Chang, Y. Contact inhibition, polyribosomes, and cell surface membranes in cultured mammalian cells. J. Cell. Physiol. 1974, 84, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Schmialek, P.; Geyer, A.; Miosga, V.; Nundel, M.; Zapf, B. The kinetics of contact inhibition in mammalian cells. Cell Tissue Kinet. 1977, 10, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M. Contact inhibition and malignancy. Nature 1979, 281, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiao, Z.; Meng, Y.; Zhao, Y.; Han, J.; Su, G.; Chen, B.; Dai, J. The enhancement of cancer stem cell properties of MCF-7 cells in 3D collagen scaffolds for modeling of cancer and anti-cancer drugs. Biomaterials 2012, 33, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Fato, M.; Penuela, L.; Beltrame, F.; Raiteri, R.; Scaglione, S. Microenvironment complexity and matrix stiffness regulate breast cancer cell activity in a 3D in vitro model. Sci. Rep. 2016, 6, 35367. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef] [PubMed]
- Majety, M.; Pradel, L.P.; Gies, M.; Ries, C.H. Fibroblasts influence survival and therapeutic response in a 3D co-culture model. PLoS ONE 2015, 10, e0127948. [Google Scholar] [CrossRef] [PubMed]
- Eder, T.; Weber, A.; Neuwirt, H.; Grunbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-associated fibroblasts modify the response of prostate cancer cells to androgen and anti-androgens in three-dimensional spheroid culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, H.; Gage, J.; Leonard, F.; Srinivasan, S.; Souza, G.R.; Dave, B.; Godin, B. Three-dimensional in vitro co-culture model of breast tumor using magnetic levitation. Sci. Rep. 2014, 4, 6468. [Google Scholar] [CrossRef] [PubMed]
- Brancato, V.; Comunanza, V.; Imparato, G.; Cora, D.; Urciuolo, F.; Noghero, A.; Bussolino, F.; Netti, P.A. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 2017, 49, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Santo, V.E.; Estrada, M.F.; Rebelo, S.P.; Abreu, S.; Silva, I.; Pinto, C.; Veloso, S.C.; Serra, A.T.; Boghaert, E.; Alves, P.M.; et al. Adaptable stirred-tank culture strategies for large scale production of multicellular spheroid-based tumor cell models. J. Biotechnol. 2016, 221, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Ahn, J.; Son, K.; Kim, S.; Jeon, N.L. Biomimetic Model of Tumor Microenvironment on Microfluidic Platform. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, A.Y.; Torisawa, Y.S.; Tung, Y.C.; Sud, S.; Taichman, R.S.; Pienta, K.J.; Takayama, S. Microfluidic system for formation of PC-3 prostate cancer co-culture spheroids. Biomaterials 2009, 30, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, M.; Heaysman, J.E. Observations on the social behaviour of cells in tissue culture. I. Speed of movement of chick heart fibroblasts in relation to their mutual contacts. Exp. Cell Res. 1953, 5, 111–131. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E.; Karthauser, H.M. Social behaviour of cells in tissue culture. III. Mutual influence of sarcoma cells and fibroblasts. Exp. Cell Res. 1957, 13, 276–291. [Google Scholar] [CrossRef]
- Parish, R.W.; Schmidhauser, C.; Schmidt, T.; Dudler, R.K. Mechanisms of tumour cell metastasis. J. Cell Sci. Suppl. 1987, 8, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Astin, J.W.; Nobes, C.D. Regulation of contact inhibition of locomotion by Eph-ephrin signalling. J. Microsc. 2013, 251, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Paddock, S.W.; Dunn, G.A. Analysing collisions between fibroblasts and fibrosarcoma cells: Fibrosarcoma cells show an active invasionary response. J. Cell Sci. 1986, 81, 163–187. [Google Scholar] [PubMed]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Abu-Kaoud, N.; Al Thani, H.; Rafii, A. Epithelial to mesenchymal transition in a clinical perspective. J. Oncol. 2015, 2015, 792182. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Theodorou, K.; Donners, M.; Ludwig, A. Fine tuning cell migration by a disintegrin and metalloproteinases. Mediat. Inflamm. 2017, 2017, 9621724. [Google Scholar] [CrossRef] [PubMed]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Danuser, G.; Allard, J.; Mogilner, A. Mathematical modeling of eukaryotic cell migration: Insights beyond experiments. Annu. Rev. Cell Dev. Biol. 2013, 29, 501–528. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Sahai, E.; Weiss, S.; Yamada, K.M. New dimensions in cell migration. Nat. Rev. Mol. Cell Biol. 2012, 13, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Abu-Kaoud, N.; Abdesselem, H.; Madani, A.; Hoarau-Vechot, J.; Thawadi, H.A.; Vidal, F.; Couderc, B.; Favre, G.; Rafii, A. SDF-1α concentration dependent modulation of RhoA and Rac1 modifies breast cancer and stromal cells interaction. BMC Cancer 2015, 15, 569. [Google Scholar] [CrossRef] [PubMed]
- Loessner, D.; Stok, K.S.; Lutolf, M.P.; Hutmacher, D.W.; Clements, J.A.; Rizzi, S.C. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials 2010, 31, 8494–8506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, D.L.; Windus, L.C.; Avery, V.M. Chemokine receptor expression on integrin-mediated stellate projections of prostate cancer cells in 3D culture. Cytokine 2013, 64, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Winters, B.S.; Shepard, S.R.; Foty, R.A. Biophysical measurement of brain tumor cohesion. Int. J. Cancer 2005, 114, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Zhang, Z.; Fouladdel, S.; Deol, Y.; Ingram, P.N.; McDermott, S.P.; Azizi, E.; Wicha, M.S.; Yoon, E. Single cell dual adherent-suspension co-culture micro-environment for studying tumor-stromal interactions with functionally selected cancer stem-like cells. Lab Chip 2016, 16, 2935–2945. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, S.; Ramos Valdes, Y.; Bertrand, M.; McGee, J.; Prefontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. TGFβ signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocr. Relat. Cancer 2016, 23, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Xie, J.; Gurler, H.; Muralidhar, G.G.; Sacks, J.D.; Burdette, J.E.; Barbolina, M.V. Versican regulates metastasis of epithelial ovarian carcinoma cells and spheroids. J. Ovarian Res. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Y.; Wei, X.; Zhou, X.; Gong, C.; Zhang, T.; Jin, P.; Xu, S.; Ma, D.; Gao, Q. Co-targeting EGFR and autophagy impairs ovarian cancer cell survival during detachment from the ECM. Curr. Cancer Drug. Targets 2015, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Peart, T.; Ramos Valdes, Y.; Correa, R.J.; Fazio, E.; Bertrand, M.; McGee, J.; Prefontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget 2015, 6, 22424–22438. [Google Scholar] [CrossRef] [PubMed]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, L.; Maffini, M.V.; Soto, A.; Sonnenschein, C.; Kaplan, D.L. A complex 3D human tissue culture system based on mammary stromal cells and silk scaffolds for modeling breast morphogenesis and function. Biomaterials 2010, 31, 3920–3929. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- El-Gewely, M.R. Biotechnology Annual Review; Elsevier: Amsterdam, The Netherlands, 2008; Volume 14, pp. 1–475. [Google Scholar]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Osuna, J.A., Jr. Mechanisms of anti-cancer agents: Emphasis on oxidative stress and electron transfer. Curr. Pharm. Des. 2000, 6, 277–309. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chow, A.B.; Mattingly, R.R. Three-dimensional overlay culture models of human breast cancer reveal a critical sensitivity to mitogen-activated protein kinase kinase inhibitors. J. Pharmacol. Exp. Ther. 2010, 332, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.V.; Nadanaciva, S.; Oliveira, P.J.; Will, Y. The contribution of oxidative stress to drug-induced organ toxicity and its detection in vitro and in vivo. Expert Opin. Drug Metab. Toxicol. 2012, 8, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Moan, J.; Peng, Q. An outline of the hundred-year history of PDT. Anticancer Res. 2003, 23, 3591–3600. [Google Scholar] [PubMed]
- Chen, Y.C.; Lou, X.; Zhang, Z.; Ingram, P.; Yoon, E. High-throughput cancer cell sphere formation for characterizing the efficacy of photo dynamic therapy in 3D cell cultures. Sci. Rep. 2015, 5, 12175. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zietarska, M.; Maugard, C.M.; Filali-Mouhim, A.; Alam-Fahmy, M.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.M. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Mol. Carcinog. 2007, 46, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cuddihy, M.J.; Kotov, N.A. Three-dimensional cell culture matrices: State of the art. Tissue Eng. Part B Rev. 2008, 14, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.M.; Amann, A.; Koeck, S.; Lorenz, E.; Kelm, J.M.; Obexer, P.; Zwierzina, H.; Gamerith, G. Evaluation of assays for drug efficacy in a three-dimensional model of the lung. J. Cancer Res. Clin. Oncol. 2016, 142, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Soleimani, M.; Atashi, A.; Saglio, G.; Abdollahi, M.; Nikougoftar Zarif, M. Mimicking the acute myeloid leukemia niche for molecular study and drug screening. Tissue Eng. Part C Methods 2017, 23, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.C.; Lim, T.C.; Tai, D.; Xiao, G.; van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramos, C.; Lebourg, M. Three-dimensional constructs using hyaluronan cell carrier as a tool for the study of cancer stem cells. J. Biomed. Mater. Res. B Appl. Biomater. 2015, 103, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Rafii, A. Role of the microenvironment in ovarian cancer stem cell maintenance. Biomed. Res. Int. 2013, 2013, 630782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N., Jr.; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zeng, J.; Liang, B.; Zhao, Z.; Sun, L.; Cao, D.; Yang, J.; Shen, K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp. Mol. Pathol. 2011, 91, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kievit, F.M.; Erickson, A.E.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. Culture on 3D chitosan-hyaluronic acid scaffolds enhances stem cell marker expression and drug resistance in human glioblastoma cancer stem cells. Adv. Healthc. Mater. 2016, 5, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Takayama, S.; Park, J. Formation and manipulation of cell spheroids using a density adjusted PEG/DEX aqueous two phase system. Sci. Rep. 2015, 5, 11891. [Google Scholar] [CrossRef] [PubMed]
- Saadatpour, Z.; Bjorklund, G.; Chirumbolo, S.; Alimohammadi, M.; Ehsani, H.; Ebrahiminejad, H.; Pourghadamyari, H.; Baghaei, B.; Mirzaei, H.R.; Sahebkar, A.; et al. Molecular imaging and cancer gene therapy. Cancer Gene Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Padma, V.V. An overview of targeted cancer therapy. Biomedicine 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Belin, L.; Paoletti, X.; Bieche, I.; Kamal, M. Precision medicine: Lessons learned from the SHIVA trial—Authors’ reply. Lancet Oncol. 2015, 16, e581–e582. [Google Scholar] [CrossRef]
- Garraway, L.A.; Janne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Scotto, D.; Josephs, D.H.; Mele, S.; Crescioli, S.; Bax, H.J.; Pellizzari, G.; Wynne, M.D.; Nakamura, M.; Hoffmann, R.M.; et al. BRAF inhibitors: Resistance and the promise of combination treatments for melanoma. Oncotarget 2017, 8, 78174–78192. [Google Scholar] [CrossRef] [PubMed]
- Pickl, M.; Ries, C.H. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 2009, 28, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.M.; Barbone, D.; Fennell, D.A.; Broaddus, V.C. Bcl-2 family proteins contribute to apoptotic resistance in lung cancer multicellular spheroids. Am. J. Respir. Cell Mol. Biol. 2009, 41, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Roodhart, J.M.; Voest, E.E. Target practice: Lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist 2007, 12, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Guerrouahen, B.S.; Pasquier, J.; Kaoud, N.A.; Maleki, M.; Beauchamp, M.C.; Yasmeen, A.; Ghiabi, P.; Lis, R.; Vidal, F.; Saleh, A.; et al. Akt-activated endothelium constitutes the niche for residual disease and resistance to bevacizumab in ovarian cancer. Mol. Cancer Ther. 2014, 13, 3123–3136. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Pradeep, S.; McGuire, M.; Wu, S.Y.; Ma, S.; Hatakeyama, H.; Lyons, Y.A.; Hisamatsu, T.; Noh, K.; Villar-Prados, A.; et al. Induction of anti-VEGF therapy resistance by upregulated expression of microseminoprotein (MSMP). Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.M.; Brkic, S.; Bovo, E.; Burger, M.; Schaefer, D.J.; Wolff, T.; Gurke, L.; Briquez, P.S.; Larsson, H.M.; Gianni-Barrera, R.; et al. Extracellular matrix and growth factor engineering for controlled angiogenesis in regenerative medicine. Front. Bioeng. Biotechnol. 2015, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Chiew, G.G.Y.; Wei, N.; Sultania, S.; Lim, S.; Luo, K.Q. Bioengineered three-dimensional co-culture of cancer cells and endothelial cells: A model system for dual analysis of tumor growth and angiogenesis. Biotechnol. Bioeng. 2017, 114, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
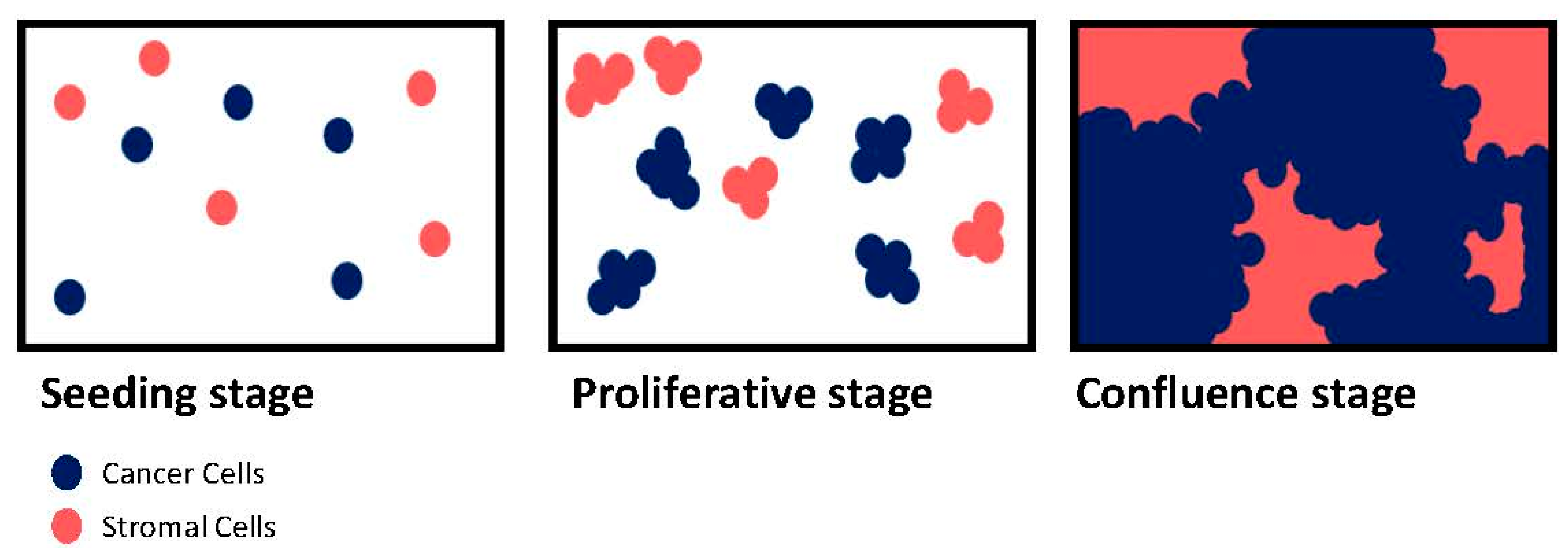{kind=link}
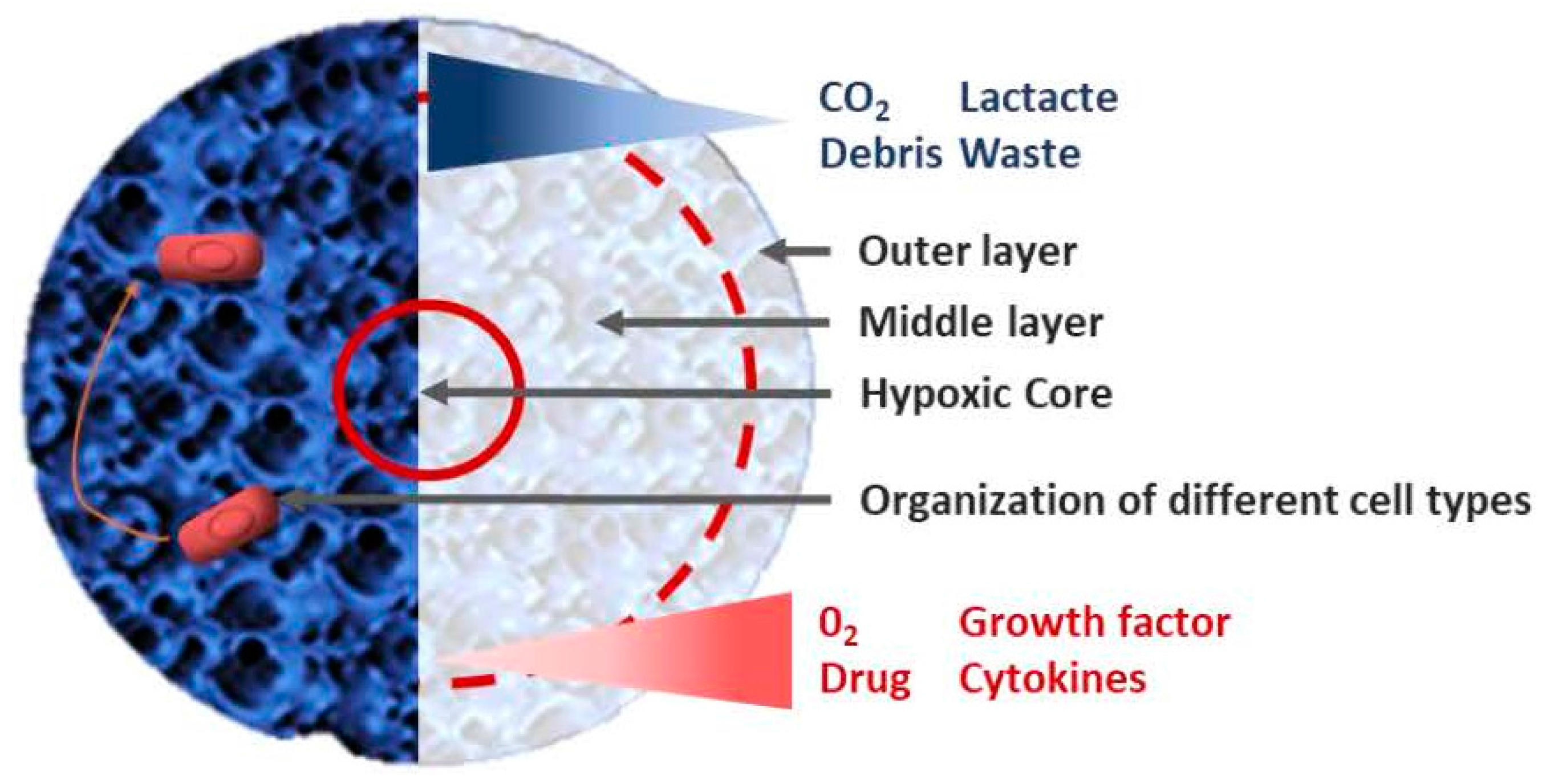{kind=link}
| 2D | 3D | |
| Morphology | Shape changed Polarization lost | Real shape Polarization conserved |
| Genetic profile | Cellular adhesion, proliferation and survival genes are modified compared to in vivo | Better representation of growth factors, pro-angiogenic and adhesion molecule genes |
| Cell differentiation Morphogenesis | Non spontaneous | Could be spontaneous via cellular contact or soluble factors |
| Angiogenesis | Only observational | Can be functional |
| Tumoral heterogeneity | Basic | Better approximation via the proliferation gradient, drug penetration and difference in mobility |
| Mathematical model | Possible | Better geometry, better link between structure and function |
| Reproducibility | Short term only | Controversial |
| Cost | Affordable | More expensive especially with some techniques |
| Multicellular study | Better when studying the immune response | Good in co-culture, but complicated with more than two cell types |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018, 19, 181. https://doi.org/10.3390/ijms19010181
Hoarau-Véchot J, Rafii A, Touboul C, Pasquier J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? International Journal of Molecular Sciences. 2018; 19(1):181. https://doi.org/10.3390/ijms19010181
Chicago/Turabian StyleHoarau-Véchot, Jessica, Arash Rafii, Cyril Touboul, and Jennifer Pasquier. 2018. "Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions?" International Journal of Molecular Sciences 19, no. 1: 181. https://doi.org/10.3390/ijms19010181