Heterogeneous Contributing Factors in MPM Disease Development and Progression: Biological Advances and Clinical Implications

Abstract
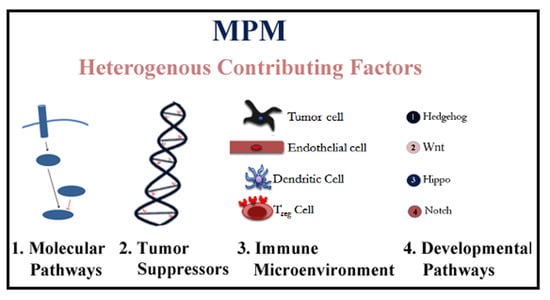
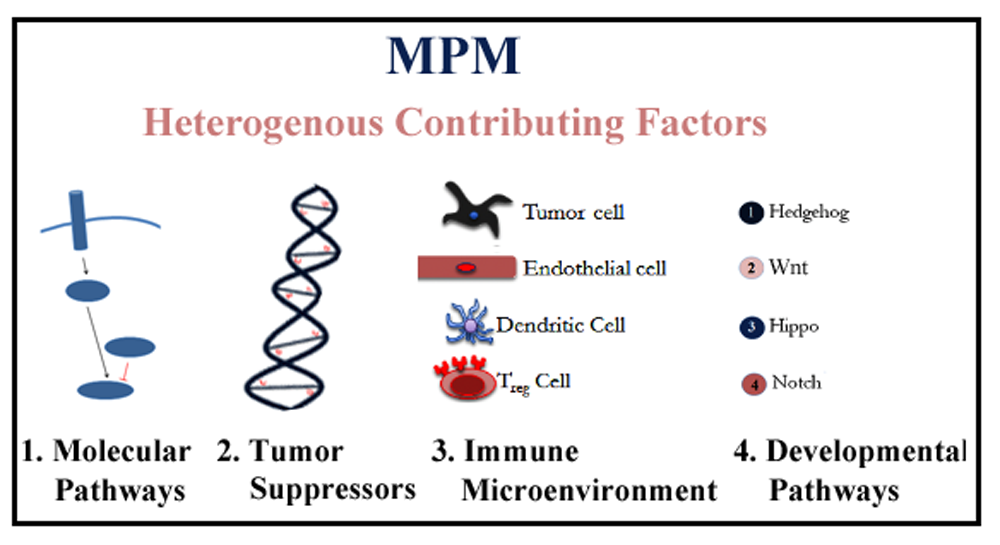
:
1. Introduction
2. Molecular Pathways in Malignant Pleural Mesothelioma
2.1. Angiogenesis
2.2. Apoptosis
2.2.1. FAK (Focal Adhesion Kinase) and Src, Fas Receptor and ROS (Reactive Oxygen Species)
2.2.2. Calcium Ions
2.2.3. TRAIL
2.3. Cell Cycle Effectors
2.4. TERT
2.5. Growth Factors Implicated in Mesothelioma
2.5.1. EGFR
2.5.2. PDGFR
2.5.3. FGF
3. Genomic Landscape Prevalent with Tumor Suppressor Inactivation
3.1. CDKN2A
3.2. NF2
3.3. BAP1
4. Immune Microenvironment of MPM Tumors
4.1. The Stromal Compartment
4.2. The Immune Population
4.3. Tumor Cells
5. Developmental Pathways in Malignant Pleural Mesothelioma
5.1. Hedgehog Pathway
5.2. Wnt/β-Catenin Pathway
5.3. Notch Pathway
5.4. Hippo/YAP Pathway
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tarver, T. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2012. [Google Scholar]
- Mortality and Morbidity Weekly Report; Center for Disease Control: Atlanta, GA, USA, 2009.
- Carbone, M.; Ly, B.H.; Dodson, R.F.; Pagano, I.; Morris, P.T.; Dogan, U.A.; Gazdar, A.F.; Pass, H.I.; Yang, H. Malignant mesothelioma: Facts, myths, and hypotheses. J. Cell. Physiol. 2012, 227, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Zucali, P.A.; Ceresoli, G.L.; Vincenzo, F.D.; Simonelli, M.; Lorenzi, E.; Gianoncelli, L.; Santoro, A. Advances in the biology of malignant pleural mesothelioma. Cancer Treat. Rev. 2011, 37, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Perrot, M. Radio-immunotherapy and chemo-therapy as a novel treatment paradigm in malignant pleural mesothelioma. Transl. Lung Cancer Res. 2017, 6, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.; Wistuba, I.; Roth, J.; Kindler, H. Malignant Pleural Mesothelioma. J. Clin. Oncol. 2009, 27, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.; Marx, A.; Nicholson, A.G. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- National Lung Cancer Audit. National Lung Cancer Audit Report 2014: Mesothelioma Report for the Period 2008–2012; Health and Social Care Information Centre: Leeds, UK, 2014.
- Strizzi, L.; Catalano, A.; Vianale, G.; Orecchia, S.; Casalini, A.; Tassi, G.; Puntoni, R.; Mutti, L.; Procopio, A. Vascular Endothelial Growth Factor is an Autocrine Growth Factor in Human Malignant Mesothelioma. J. Pathol. 2001, 193, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Masood, R.; Kundra, A.; Zhu, S.; Xia, G.; Scalia, P.; Smith, D.L.; Gill, P.S. Malignant Mesothelioma Growth Inhibition by Agents that Target the VEGF and VEGF-C Autocrine Loop. Int. J. Cancer 2003, 104, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Shridhar, V.; Bright, R.K.; Kalemkerian, G.P.; Du, W.; Carbone, M.; Watanabe, Y.; Pass, H.I. VEGF and VEGF type C Play an Important Role in Angiogenesis and Lymphangiogenesis in Human Malignant Mesothelioma Tumors. Br. J. Cancer 1999, 81, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Singh, S.; Weyler, J.; Martin, M.J.; Vermeulen, P.B.; Van MArck, E. Angiogenic Cytokines in Mesothelioma: A Study of VEGF, FGF-1 and -2, and TGF β Expression. J. Pathol. 1999, 189, 72–78. [Google Scholar] [CrossRef]
- Tolnay, E.; Kuhnen, C.; Wiethege, T.; Konig, J.E.; Voss, B.; Muller, K.M. Hepatocyte Growth Factor/Scatter Factor and its Receptor c-Met are Overexpressed and Associated with an Increased Microvessel Density in Malignant Pleural Mesothelioma. J. Cancer Res. Clin. Oncol. 1998, 124, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Karrison, T.G.; Gandara, D.R.; Lu, C.; Krug, L.M.; Stevenson, J.P.; Janne, P.A.; Quinn, D.I.; Koczywas, M.N.; Brahmer, J.R.; et al. Multicenter, Double-blind, Placebo-controlled, Randomized Phase II Trial of Gemcitabine/Cispltain Plus Bevacizumab or Placebo in Patients with Malignant Mesothelioma. J. Clin. Oncol. 2012, 30, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Levin, P.A.; Dowell, J.E. Spotlight on bevacizumab and its potential in the treatment of malignant pleural mesothelioma: The evidence to date. Onco Targets Ther. 2017, 10, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Jahan, T.; Gu, L.; Kratzke, R.; Dudek, A.; Otterson, G.A.; Wang, X.; Green, M.; Vokes, E.E.; Kindler, H.L. Vatalanib in Malignant Mesothelioma: A Phase II Trial by the Cancer and Leukemia Group B (CALGB 30107). Lung Cancer 2012, 76, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, I.; Yano, S.; Trung, V.T.; Hanibuchi, M.; Goto, H.; Li, Q.; Wang, W.; Yamada, T.; Ogino, H.; Kakiuchi, S.; et al. E7080, a Multi-tyrosine Kinase Inhibitor, Suppresses the Progression of Malignant Pleural Mesothelioma with Different Proangiogenic Cytokine Production Profiles. Clin. Cancer Res. 2009, 15, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.; Boogerd, W.; Dalesio, O.; Haringhuizen, A.; Custers, F.; van Zandwijk, N. Thalidomide in patients with malignant pleural mesothelioma. Lung Cancer 2005, 48, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Wang, X.F.; Krug, L.M.; Hodgson, L.; Vokes, E.E.; Kindler, H.L. Sorafenib in malignant pleural mesothelioma (MM): A phase II trial of the Cancer and Leukemia Group B (CALGB 30307). J. Clin. Oncol. 2007, 25, 2007–7707. [Google Scholar]
- Nowak, A.K.; Millward, M.J.; Francis, R.; van der Schaaf, A.; Musk, A.W.; Byrne, M.J. Phase II study of sunitinib as second-line therapy in malignant pleural mesothelioma (MPM). J. Clin. Oncol. 2008. [Google Scholar] [CrossRef]
- Harada, A.; Unchino, J.; Harada, T.; Nakagaki, N.; Hisasue, J.; Fujita, M.; Takayama, K. Vascular Endothelial Growth Factor Promoter-based Conditional Replicative Adenoviruses Effectively Suppress Growth of Malignant Pleural Mesothelioma. Cancer Sci. 2017, 108, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Sakai, K.; Ueki, J.; Xu, Q.; Nakamura, T.; Shimada, H.; Nakamura, T.; Matsumoto, K. Inhibition of Met/HGF Receptor and Angiogenesis by NK4 Leads to Suppression of Tumor Growth and Migration in Malignant Pleural Mesothelioma. Int. J. Cancer 2010, 127, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Identifier NCT00309946; National Library of Medicine (US): Bethesda, MD, USA, 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT00309946 (accessed on 10 September 2017).
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.A.; Rudd, R.M. Defective Core-Apoptosis Signaling in Diffuse Malignant Pleural Mesothelioma: Opportunities for Effective Drug Development. Lancet Oncol. 2004, 5, 354–362. [Google Scholar] [CrossRef]
- De Assis, L.V.; Locatelli, J.; Isoldi, M.C. The Role of Key Genes and Pathways involved in the Tumorigenesis of Malignant Mesothelioma. Biochim. Biophys. Acta 2014, 1845, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Hopkins-Donaldson, S.; Belyanskaya, L.L.; Simoes-Wust, A.P.; Sigris, B.; Kurtz, S.; Zangemeister-Wittke, U.; Stahel, R. p53-induced Apoptosis Occurs in the Absence of P14(ARF) in Malignant Pleural Mesothelioma. Neoplasia 2006, 8, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.J.; Mani, M.; Mukhopadhyay, L.; Dong, L.; Edenfield, H.R.; Glickman, J.N.; Yeap, B.Y.; Sugarbaker, D.J.; Bueno, R. Expression Patterns of Inhibitor of Apoptosis Proteins in Malignant Pleural Mesothelioma. J. Pathol. 2007, 211, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Mohuiddin, I.; Ece, F.; McConkey, D.J.; Smythe, W.R. Histone deacetylase inhibitor downregulation of bcl-xl gene expression leads to apoptotic cell death in mesothelioma. Am. J. Respir. Cell Mol. Biol. 2001, 25, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Ozvaran, M.K.; Cao, X.X.; Miller, S.D.; Monia, B.A.; Hong, W.K.; Smythe, W.R. Antisense oligonucleotides directed at the bcl-xl gene product augment chemotherapy response in mesothelioma. Mol. Cancer Ther. 2004, 3, 545–550. [Google Scholar] [PubMed]
- Cao, X.; Rodarte, C.; Zhang, L.; Morgan, C.D.; Littlejohn, J.; Smythe, W.R. Bcl2/bcl-xL inhibitor engenders apoptosis and increases chemosensitivity in Mesothelioma. Cancer Biol. Ther. 2007, 6, 1–7. [Google Scholar] [CrossRef]
- Ou, W.B.; Lu, M.; Ellers, G.; Li, H.; Ding, J.; Meng, X.; Wu, Y.; He, Q.; Sheng, Q.; Zhou, H.M.; Fletcher, J.A. Co-targeting of FAK and MDM2 Triggers Additive Anti-proliferative Effects in Mesothelioma via a Coordinated Reactivation of P53. Br. J. Cancer 2016, 115, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; He, D.; Saigal, B.; Liu, S.; Lee, J.J.; Bakkannagari, S.; Ordonez, N.G.; Hong, W.K.; Wistuba, I.; Johnson, F.M. Inhibition of c-Src Expression and Activation in Malignant Pleural Mesothelioma Tissues Leads to Apoptosis, Cell Cycle Arrest, and Decreased Migration and Invasion. Mol. Cancer Ther. 2007, 6, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.H.; Tran, T.L.; Levi, N.; Tsai, W.S.; Schrump, D.S.; Nguyen, D.M. The Essential Role of the Mitochondria and Reactive Oxygen Species in Cisplatin-mediated Enhancement of Fas Ligand-induced Apoptosis in Malignant Pleural Mesothelioma. J. Surg. Res. 2007, 141, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Giorgi, C.; Maniero, S.; Missiroli, S.; Maniscalco, P.; Bononi, I.; Martini, F.; Cavallesco, G.; Tognon, M.; Pinton, P. The Endoplasmic Reticulum Mitochondrial Calcium Cross Talk is Downregulated in Malignant Pleural Mesothelioma Cells and Plays a Critical Role in Apoptosis Inhibition. Oncotarget 2015, 6, 23427–23444. [Google Scholar] [CrossRef] [PubMed]
- Lathrop, M.J.; Sage, E.K.; Macura, S.L.; Brooks, E.M.; Cruz, F.; Bonenfant, N.R.; Sokocevic, D.; MacPherson, M.B.; Beuschel, S.L.; Dunaway, C.W.; et al. Antitumor Effects of TRAIL-expressing Mesenchymal Stromal Cells in a Mouse Xenograft Model of Human Mesothelioma. Cancer Gene Ther. 2015, 22, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rios, F.; Chuai, S.; Flores, R.; Shimizu, S.; Ohno, T.; Wakahara, K.; Illei, P.B.; Hussain, S.; Krug, L.; Zakowski, M.F.; et al. Global Gene Expression Profiling of Pleural Mesothelioma: Overexpression of Aurora Kinases and P16/CDKN2A Deletion as Prognostic Factors and Critical Evaluation of Microarray-based Prognostic Prediction. Cancer Res. 2006, 66, 2970–2979. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Godleski, J.J.; Marsit, C.J.; Houseman, E.A.; Lopez-Fagundo, C.Y.; Longacker, J.L.; Bueno, R.; Sugarbaker, D.J.; Nelson, H.H.; Kelsey, K.T. Asbestos Exposure Predicts Cell Cycle Control Gene Promoter Methylation in Pleural Mesothelioma. Carcinogenesis 2008, 29, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, S.; Fasoli, E.; Vaira, V.; Falleni, M.; Pellegrini, C.; Catania, A.; Roncalli, M.; Marchetti, A.; Santambrogio, L.; Coggi, G.; et al. Identification of Potential Therapeutic Targets in Malignant Mesothelioma Using Cell-Cycle Gene Expression Analysis. Am. J. Pathol. 2009, 174, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP Induces Malignant Mesothelioma Cell Proliferation by Upregulating Transcription of Cell Cycle-promoting Genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Dhaene, K.; Hubner, R.; Kumar-Singh, S.; Weyn, B.; Van Marck, E. Telomerase activity in human pleural mesothelioma. Thorax 1998, 53, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Whitson, B.A.; Kratzke, R.A. Molecular Pathways in Malignant Pleural Mesothelioma. Cancer Lett. 2006, 239, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Sasaki, H.; Kawano, O.; Yukiue, H.; Yokoyama, T.; Yano, M.; Fujii, Y. Epidermal growth factor receptor gene mutation, amplification and protein expression in malignant pleural mesothelioma. J. Cancer Res. Clin. Oncol. 2008, 134, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Rena, O.; Boldorini, L.R.; Gaudino, E.; Casadio, C. Epidermal growth factor receptor overexpression in malignant pleural mesothelioma: Prognostic correlations. J. Surg. Oncol. 2011, 104, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E., 2nd; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L.; Cancer and Leukemia Group B (CALGB 30101). Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer. Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef] [PubMed]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II study of erlotinib in patients with malignant pleural mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Harun, N.; Fujimoto, J.; Devito, V.; Lee, J.J.; Kuhn, E.; Mehran, R.; Rice, D.; Moran, C.; Hong, W.K.; et al. Elevated PDGFRB gene copy number gain is prognostic for improved survival outcomes in resected malignant pleural mesothelioma. Ann. Diagn. Pathol. 2014, 18, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Catalano, C.; de Santi, C.; Cipollini, M.; Figlioli, G.; Pelle, L.; Barone, E.; Evangelista, M.; Guazzelli, A.; Boldrini, L.; et al. Inhibition of the platelet-derived growth factor receptor β (PDGFRβ) using gene silencing, crenolanib besylate, or imatinib mesylate hampers the malignant phenotype of mesothelioma cell lines. Genes Cancer 2017, 8, 438–452. [Google Scholar] [PubMed]
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Munzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast growth factor receptor inhibition is active against mesothelioma and synergizes with radio- and chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Marek, L.A.; Hinz, T.K.; von Massenhausen, A.; Olszewski, K.A.; Kleczko, E.K.; Boehm, D.; Weiser-Evans, M.C.; Nemenoff, R.A.; Hoffmann, H.; Warth, A.; et al. Nonamplified FGFR1 is a growth driver in malignant pleural mesothelioma. Mol. Cancer Res. 2014, 12, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Strizzi, L.; Vianele, G.; Catalano, A.; Muraro, R.; Mutti, L.; Procopio, A. Basic fibroblast growth factor in mesothelioma pleural effusions: Correlation with patient survival and angiogenesis. Int. J. Oncol. 2001, 18, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Pattarozzi, A.; Carra, E.; Favoni, R.E.; Wurth, R.; Marubbi, D.; Filiberti, R.A.; Mutti, L.; Florio, T.; Barbieri, F.; Daga, A. The inhibition of FGF receptor 1 activity mediates sorafenib antiproliferative effects in human malignant pleural mesothelioma tumor-initiating cells. Stem Cell Res. Ther. 2017, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Houseman, E.A.; Poage, G.M.; Godleski, J.J.; Bueno, R.; Sugarbaker, D.J.; Wiencke, J.K.; Nelson, H.H.; Marsit, C.J.; Kelsey, K.T. Integrated profiling reveals a global correlation between epigenetic and genetic alterations in mesothelioma. Cancer Res. 2010, 70, 5686–5694. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Houseman, E.A.; Godleski, J.J.; Marsit, C.J.; Longacker, J.L.; Roelofs, C.R.; Karagas, M.R.; Wrensch, M.R.; Yeh, R.F.; Nelson, H.H.; et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res. 2009, 69, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Jones, R.E.; Liu, H.; Lizotte, P.H.; Ivanova, M.; Kulkarni, M.; Herter-Sprie, G.S.; Lia, X.; Santos, A.A.; Bittinger, M.A.; et al. Cytotoxic T Cells in PD-L1-Positive Mesothelioma Are Counterbalanced by Distinct Immunosuppressive Factors. Cancer Immunol. Res. 2016, 4, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Menges, C.W.; Xu, J.; Pei, J.; Zhang, L.; Tadevosyan, A.; Neumann-Domer, E.; Liu, Z.; Carbone, M.; Chudoba, I.; et al. Losses of Both Products of the Cdkn2a/Arf Locus Contribute to Asbestos-induced Mesothelioma Development and Cooperate to Accelerate Tumorigenesis. PLoS ONE 2011, 6, e18828. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Zhou, J.; Anderson, D.; Kratzke, R.A. Inactivation of p16INK4a expression in malignant mesothelioma by methylation. Lung Cancer 2002, 38, 131–136. [Google Scholar] [CrossRef]
- Jennings, C.J.; Murer, B.; O’Grady, A.; Hearn, L.M.; Harvey, B.J.; Kay, E.W.; Thomas, W. Differential p16/INK4A Cyclin-dependent Kinase Inhibitor Expression Correlates with Chemotherapy Efficacy in a Cohort of 88 Malignant Pleural Mesothelioma Patients. Br. J. Cancer 2015, 113, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Protocol for Study CDKO-125a-005, Version 29 September 2008. Nerviano Medical Sciences, 2008. Document No CDKO-125a-005-P. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=CDKO-125a-005 (accessed on 10 September 2017).
- Cho, J.H.; Lee, S.J.; Oh, A.Y.; Yoon, M.H.; Woo, T.G.; Park, B.J. NF2 blocks Snail-mediated p53 suppression in mesothelioma. Oncotarget 2015, 6, 10073–10085. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Zauderer, M.G.; Krug, L.M.; Ito, T.; McMillan, R.; Bott, M.; Giancotti, F. New strategies in pleural mesothelioma: BAP1 and NF2 as novel targets for therapeutic development and risk assessment. Clin. Cancer Res. 2012, 18, 4485–4490. [Google Scholar] [CrossRef] [PubMed]
- Sekido, Y. Inactivation of Merlin in malignant mesothelioma cells and the Hippo signaling cascade dysregulation. Pathol. Int. 2011, 61, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Thurneysen, C.; Opitz, I.; Kurtz, S.; Weder, W; Stahel, R.A.; Felley-Bosco, E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer 2009, 64, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Masuda, M.; Tsuta, K.; Kawasaki, K.; Nakamura, Y.; Sakuma, T.; Asamura, H.; Gemma, A.; Yamada, T. Hippo pathway gene mutations in malignant mesothelioma: Revealed by RNA and targeted exon sequencing. J. Thorac. Oncol. 2015, 5, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Sato, A.; Tsujimura, T.; Emi, M.; Morinaga, T.; Fukuoka, K.; Yamada, S.; Murakami, A.; Kondo, N.; Matsumoto, S.; et al. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci. 2012, 103, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, W.; Yamada, T.; Matsumoto, K.; Sakai, K.; Bando, Y.; Uehara, H.; Nishioka, Y.; Sone, S.; Iwakiri, S.; et al. Pleural Mesothelioma Instigates Tumor-Associated Fibroblasts to Promote Progression via a Malignant Cytokine Network. Am. J. Pathol. 2011, 179, 1483–1493. [Google Scholar]
- Sekido, Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis 2013, 34, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Hegmans, J.P.; Hemmes, A.; Hammad, H.; Boon, L.; Hoogsteden, H.C.; Lambrecht, B.N. Mesothelioma Environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur. Respir. J. 2006, 27, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Lievense, L.A.; Cornelissen, R.; Bezemer, K.; Kaijen-Lambers, M.E.; Hegmans, J.P.; Aerts, J.G. Pleural Effusion of Patients with Malignant Mesothelioma Induces Macrophage-Mediated T Cell Suppression. J. Thorac. Oncol. 2016, 11, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Anraku, M.; Cunningham, K.S.; Yun, Z.; Tsao, M.S.; Zhang, L.; Keshavjee, S.; Johnston, M.R.; de Perrot, M. Impact of Tumor-infiltrating T cells on Survival in Patients with Malignant Pleural Mesothelioma. J. Thorac. Cardiovasc. Surg. 2008, 135, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Pure, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol. Res. 2016, 4, 541–551. [Google Scholar] [CrossRef] [PubMed]
- DeLong, P.; Tanaka, T.; Kruklitis, R.; Henry, A.C.; Kapoor, V.; Kaiser, L.R.; Sterman, D.H.; Albelda, S.M. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Res. 2003, 63, 7845–7852. [Google Scholar] [PubMed]
- Kiyotani, K.; Park, J.H.; Inoue, H.; Husain, A.; Olugbile, S.; Zewde, M.; Nakamura, Y.; Vigneswaran, W.T. Integrated Analysis of Somatic Mutations and Immune Microenvironment in Malignant Pleural Mesothelioma. Oncoimmunology 2017, 6, e1278330. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Oizumi, S.; Kikuchi, E.; Shinagawa, N.; Konishi-Sakakibara, J.; Ishimine, A.; Aoe, K.; Gemba, K.; Kishimoto, T.; Torigoe, T.; et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol. Immunother. 2010, 59, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Cornwall, S.M.; Wikstrom, M.; Musk, A.W.; Alvarez, J.; Nowak, A.K.; Nelson, D.J. Human Mesothelioma induces Defects in Dendritic Cell Numbers and Antigen-processing Function which predicts Survival Outcomes. Oncoimmunology 2015, 5, e1082028. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, R.; Lievense, L.A.; Maat, A.P.; Hendriks, R.W.; Hoogsteden, H.C.; Bogers, A.J.; Hegmans, J.P.; Aerts, J.G. Ratio of Intratumoral Macrophage Phenotypes is a Prognostic Factor in Epithelioid Malignant Pleural Mesothelioma. PLoS ONE 2014, 9, e106742. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kadota, K.; Sima, C.S.; Sadelain, M.; Rusch, V.W.; Travis, W.D.; Adusumilli, P.S. Chronic Inflammation in Tumor Stroma is an Independent Predictor of Prolonged Survival in Epithelioid Malignant Pleural Mesothelioma Patients. Cancer Immunol. Immunother. 2011, 60, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 6, 1503–1521. [Google Scholar] [CrossRef] [PubMed]
- Beachy, P.A.; Hymowitz, S.G.; Lazarus, R.A.; Leahy, D.J.; Siebold, C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010, 18, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Svard, J.; Heby-Henricson, K.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergstrom, A.; Ericson, J.; Toftgard, R.; Teglund, S. Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Disc. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. BioMed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Noura, U.; Opitz, I.; Soltermann, A.; Rehrauer, H.; Thies, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Role of Hedgehog signaling in malignant pleural mesothelioma. Clin. Cancer Res. 2012, 18, 4646–4656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, J.; Zhang, F.; Li, H.; Yue, D.; Wang, C.; Jablons, D.M.; He, B.; Lui, N. SMO expression level correlates with overall survival in patients with malignant pleural mesothelioma. J. Exp. Clin. Cancer Res. 2013, 32, 7. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Berard, K.; Felley-Bosco, E.; Lauk, O.; Vrugt, B.; Boss, A.; Kenkel, D.; Broggini-Tenzer, A.; Stahel, R.A.; Arni, S.; et al. Antagonizing the Hedgehog Pathway with Vismodegib Impairs Malignant Pleural Mesothelioma Growth In Vivo by Affecting Stroma. Mol. Cancer Ther. 2016, 15, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Felley-Bosco, E.; Opitz, I.; Meerang, M. Hedgehog Signaling in Malignant Pleural Mesothelioma. Genes 2015, 6, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lui, N.; Cheng, T.; Tseng, H.H.; Yue, D.; Giroux-Leprieur, E.; Do, H.T.; Sheng, Q.; Jin, J.Q.; Luh, T.W.; et al. Gli as a novel therapeutic target in malignant pleural mesothelioma. PLoS ONE 2013, 8, e57346. [Google Scholar] [CrossRef] [PubMed]
- Felley-Bosco, E.; Stahel, R. Hippo/YAP pathway for targeted therapy. Transl. Lung Cancer Res. 2014, 3, 75–83. [Google Scholar] [PubMed]
- Kang, Y.; Ding, M.; Tian, G.; Guo, H.; Wan, Y.; Tao, Z.; Li, B.; Lin, D. Overexpression of Numb suppresses tumor cell growth and enhances sensitivity to cisplatin in epithelioid malignant pleural mesothelioma. Oncol. Rep. 2013, 30, 313–319. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Varona-Santos, J.; Singh, S.; Robbins, D.J.; Savaraj, N.; Nguyen, D.M. Targeting of the Hedghog signal transduction pathway suppresses survival of malignant pleural mesothelioma in vitro. J. Thorac. Cardiovasc. Surg. 2014, 147, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Waternam, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/B-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, K.; Kanazawa, S.; You, L.; He, B.; Xu, Z.; Li, K.; Peterlin, B.M.; McCormick, F.; Jablons, D.M. Wnt pathway activation in mesothelioma: Evidence of Dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res. 2003, 63, 4547–4551. [Google Scholar] [PubMed]
- Abutaily, A.S.; Collins, J.E.; Roche, W.R. Cadherins, catenins and PACE in pleural malignant mesothelioma. J. Pathol. 2003, 201, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; You, L.; He, B.; Xu, Z.; Twogood, S.; Lee, A.Y.; Reguart, N.; Batra, S.; Mikami, I.; Jablons, D.M. Wnt2 as a new therapeutic target in malignant pleural mesothelioma. Int. J. Cancer 2005, 117, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Huang, C.L.; Sonobe, M.; Kikuchi, R.; Ishikawa, M.; Kitamura, J.; Miyahara, R.; Menju, T.; Iwakiri, S.; Itoi, K.; et al. Intratumoral Wnt2B expression affects tumor proliferation and survival in malignant pleural mesothelioma patients. Exp. Ther. Med. 2012, 3, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; He, B.; You, L.; Dadfarmay, S.; Xu, Z.; Mazieres, J.; Mikami, I.; McCormick, F.; Jablons, D.M. Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene 2004, 23, 6672–6676. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Zheng, Q.; Chen, Z.; Kinoshita, H.; Okamoto, J.; Kratz, J.; Li, H.; Lui, N.; Do, H.; Cheng, T.; et al. Wnt7a is a putative prognostic and chemosensitivity marker in human malignant pleural mesothelioma. Oncol. Rep. 2015, 33, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Gee, G.V.; Koestler, D.C.; Christensen, B.C.; Sugarbaker, D.J.; Ugolini, D.; Ivaldi, G.P.; Resnick, M.B.; Houseman, E.A.; Kelsey, K.T.; Marsit, C.J. Downregulated microRNAs in the differential diagnosis of malignant pleural mesothelioma. Int. J. Cancer 2010, 127, 2859–2869. [Google Scholar] [CrossRef] [PubMed]
- Ables, J.L.; Breunig, J.J.; Eisch, A.J.; Rakic, P. Not(ch) just development: Notch signaling in the adult brain. Nat. Rev. Neurosci. 2011, 12, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Graziani, I.; Eliasz, S.; De Marco, M.A.; Chen, Y.; Pass, H.I.; De May, R.M.; Strack, P.R.; Miele, L.; Bocchetta, M. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008, 68, 9678–9685. [Google Scholar] [CrossRef] [PubMed]
- Oswald, F.; Tauber, B.; Dobner, T.; Bourteele, S.; Kostezka, U.; Adler, G.; Liptay, S.; Schmid, R.M. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell. Biol. 2001, 21, 7761–7774. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Matsuno, K.; Fortini, M.E. Notch signaling. Science 1995, 268, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Mikolaenko, I.; Elhassan, I.; Ni, X.; Wang, Y.; Ball, D.; Brat, D.J.; Perry, A.; Eberhart, C.G. Notch1 and notch2 have opposite effects on embryonal brain tumor. Cancer Res. 2004, 64, 7787–7793. [Google Scholar] [CrossRef] [PubMed]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Bocchetta, M.; Miele, L.; Pass, H.I.; Carbone, M. Notch-1 induction, a novel activity of S40 required for the growth of SV40-transformed human mesothelial cells. Oncogene 2003, 22, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Zhang, J.; Zhou, C.; Shen, H.; Gagea, M.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Sood, A.K.; Beretta, L. Differentiation therapy for hepatocellular carcinoma: Multifaceted effects of miR-148a on tumor growth and phenotype and liver fibrosis. Hepatology 2016, 63, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high Notch pathway activation predicts response to y secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Long, H.; Yang, Y.L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2a down-regulates Notch1 signaling in lung cancer cells. J. Cell.Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Badouel, C.; McNeill, H. SnapShot: The hippo signaling pathway. Cell 2011, 145, 484. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genome Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, R.; Quetel, L.; Tallet, A.; Meiller, C.; Reiner, A.; de Koning, L.; de Reynies, A.; Le Pimpec-Barthes, F.; Zucman-Rosi, J.; Jaurand, M.C.; et al. Co-occurring Mutations of Tumor Suppressor Genes, LATS2 and NF2, in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2017, 23, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Xu, Q.; Zhou, L.; Pavlovic, M.; Ojeda, V.; Moulick, K.; de Stanchina, E.; Poirier, J.T.; Zauderer, M.; Rudin, C.M.; et al. Combined Inhibition of NEDD8-Activating Enzyme and mTOR Suppresses NF2 Loss-Driven Tumorigensis. Mol. Cancer Ther. 2017, 16, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Berard, K.; Friess, M.; Bitanihirwe, B.K.; Soltermann, A.; Vrugt, B.; Felley-Bosco, E.; Bueno, R.; Richards, W.G.; Seifert, B.; et al. Low Merlin expression and high Survivin labeling index are indicators for poor prognosis in patients with malignant pleural mesothelioma. Mol. Oncol. 2016, 10, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
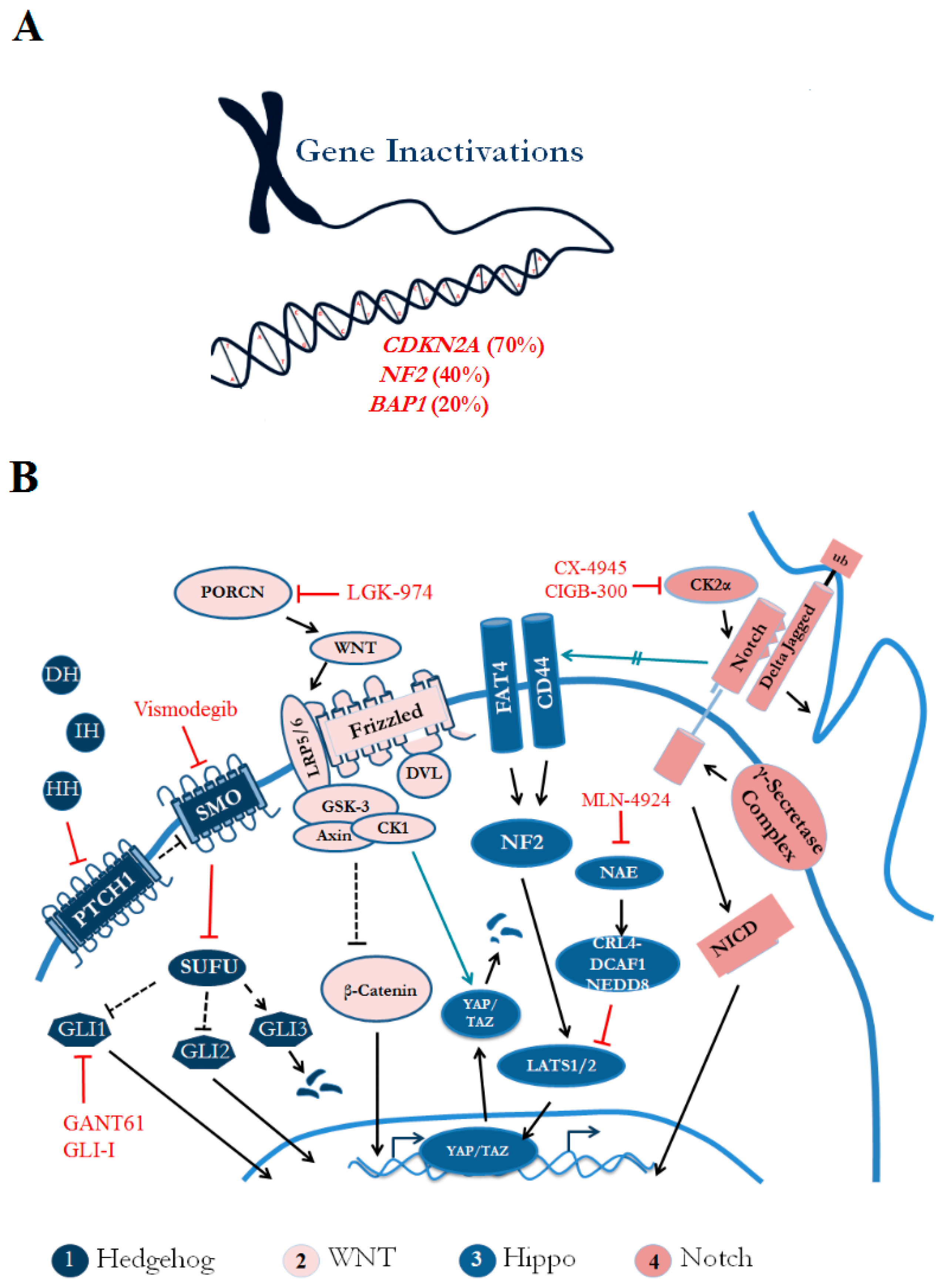

{kind=link}
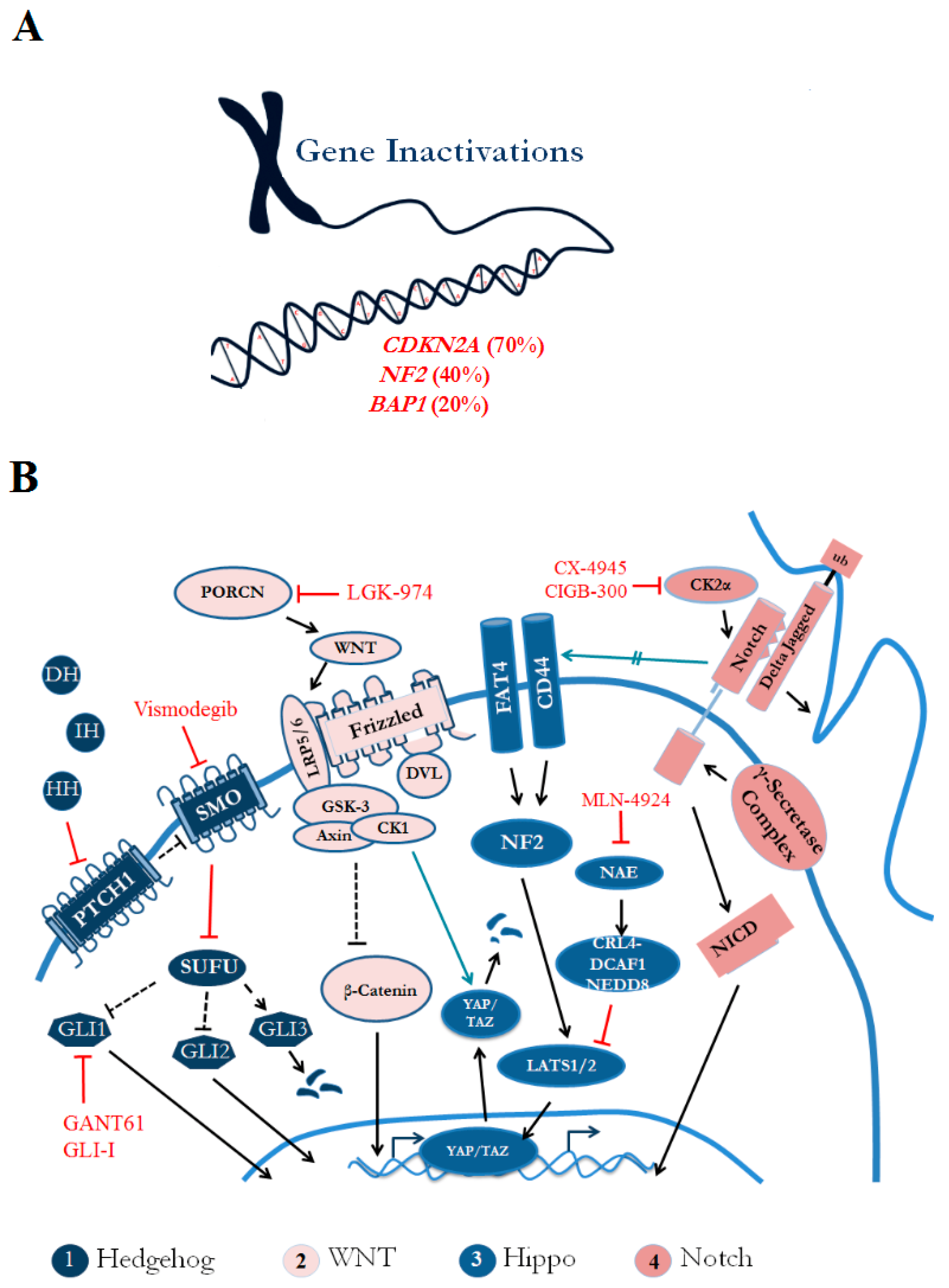{kind=link}
| Pathway | Target(s) | Prevalence of Target in MPM (%) | Therapeutic | Clinical Trial Status |
|---|---|---|---|---|
| Angiogenesis | VEGF | 30% expression | Bevacizumab | Approved for colon cancer |
| VEGF | 30% expression | Bevacizumab + pemetrexed and cisplatin | Clinical trial phase III completed | |
| VEGFR | 20% expression | Dovitinib; nintedanib; cediranib | Under clinical investigation | |
| VEGFR | 20% expression | Vatalanib | Under clinical investigation | |
| VEGF, FGF | 30%, 50% overexpression | Lenvatinib | Approved for thyroid/kidney Cancers | |
| HGF | 85% overexpression | Adenovirus containing an HGF variant, NK-4 | - | |
| ? | ? | Thalidomide | Approved for multiple myeloma | |
| VEGFR, PDGFR | 20%, 30% overexpression | Sorafenib | Approved for thyroid, kidney, and liver cancer | |
| VEGFR, PDGFR | 20%, 30% overexpression | Sunitinib | Approved for kidney and GI cancer | |
| Apoptosis | p53 | 20–25% mutated | - | - |
| Survivin | - | Antisense oligonucleotides + chemotherapy | - | |
| Bcl-xL | - | Small molecule HDAC inhibitors + antisense oligonucleotides | - | |
| Bcl-xL/Bcl-2 | - | 2-Methoxy antimycin A3 + chemotherapy | - | |
| Src | ~50% expression | Dasatinib | Approved for leukemia | |
| Fas Ligand | Selective FasL-positive cells | Fas ligand + cisplatin | - | |
| Calcium Channels | Primary samples showed reduced calcium ion uptake | Exgogenous calcium ions or mitochondrial calcium uniporter | - | |
| TRAIL | - | Administration of MSCs genetically engineered to express TRAIL | - | |
| Cell Cycle | CHEK1 | 50% overexpression | CHEK1 silencing + doxorubicin | - |
| YAP | ~70% expression | YAP silencing | - | |
| Growth Factor | EGFR | ~40% Expression | Gefitinib; Erlotinib | - |
| PDGFR | 20%, 30% overexpression | PDGFR silencing | ||
| FGFR1 | 50% overexpression | FGFR-1 inhibitor PD-166866 | - | |
| FGFR1 | 50% overexpression | Sorafenib | Approved for thyroid, kidney, and liver cancer | |
| FGFR1 | 50% overexpression | Ponatinib | Approved for thyroid, kidney, and liver cancer | |
| DNA Replication | TERT | 90% overexpression | Anti-telomerase drugs + other targeted therapies | - |
| Tumor Suppressor-associated targets | CDK2 | 70% homozygous deletion of CDKN2A known to inhibit CDK2 | Milciclib/PHA-848125AC | Phase II for hepatocellular carcinoma |
| Snail-p53 | 30–45% NF2 deletion known to prevent p53 inhibition | GN25 | - | |
| EZH2 | 60% BAP1 inactivations known to modulate EZH2 | Pinometostat (EPZ5676) and other methyltransferase inhibitors | - | |
| Stromal Compartment | PD1 | (%?) tumor microenvironment is immunosuppressive | Nivolumab | Phase II for MPM |
| Hedgehog | Smoothened | Inhibits Hh signaling (%?) | Vismodegib | Approved for basal cell carcinoma |
| Gli | 90% Gli1/Gli2 active | GANT61 and GLI-I | - | |
| Wnt/β-catenin | PORCN | Inhibits Wnt signaling (5%) | LGK-974 | Phase I for solid tumors |
| Notch | γ secretase | Inhibits Notch signaling (%?) | Semagacestat (LY450139) | Phase III for Alzheimer’s disease |
| CK2α | Down-regulates Notch1 signaling (%?) | Silmitasertib (CX-4945) | Phase I for solid tumors and multiple myeloma | |
| Hippo/YAP | PI3K-AKT-mTOR | LATS2 altered in 11% | PF-04691502 | - |
| Nedd8 activating enzyme (NAE) | Interferes with YAP (70%) activation | Pevonedistat (MLN4924) | Phase I for hematological malagnancies and melanoma | |
| YAP-TEAD | NF2 (40%) and YAP (70%) overactive | Verteporfin | Phase I for prostate cancer |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolani, B.; Acevedo, L.A.; Hoang, N.T.; He, B. Heterogeneous Contributing Factors in MPM Disease Development and Progression: Biological Advances and Clinical Implications. Int. J. Mol. Sci. 2018, 19, 238. https://doi.org/10.3390/ijms19010238
Tolani B, Acevedo LA, Hoang NT, He B. Heterogeneous Contributing Factors in MPM Disease Development and Progression: Biological Advances and Clinical Implications. International Journal of Molecular Sciences. 2018; 19(1):238. https://doi.org/10.3390/ijms19010238
Chicago/Turabian StyleTolani, Bhairavi, Luis A. Acevedo, Ngoc T. Hoang, and Biao He. 2018. "Heterogeneous Contributing Factors in MPM Disease Development and Progression: Biological Advances and Clinical Implications" International Journal of Molecular Sciences 19, no. 1: 238. https://doi.org/10.3390/ijms19010238