Transcriptome Analysis of Tomato Leaf Spot Pathogen Fusarium proliferatum: De novo Assembly, Expression Profiling, and Identification of Candidate Effectors

Abstract
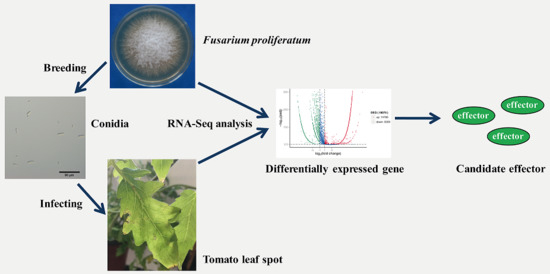
:
1. Introduction
2. Results and Discussion
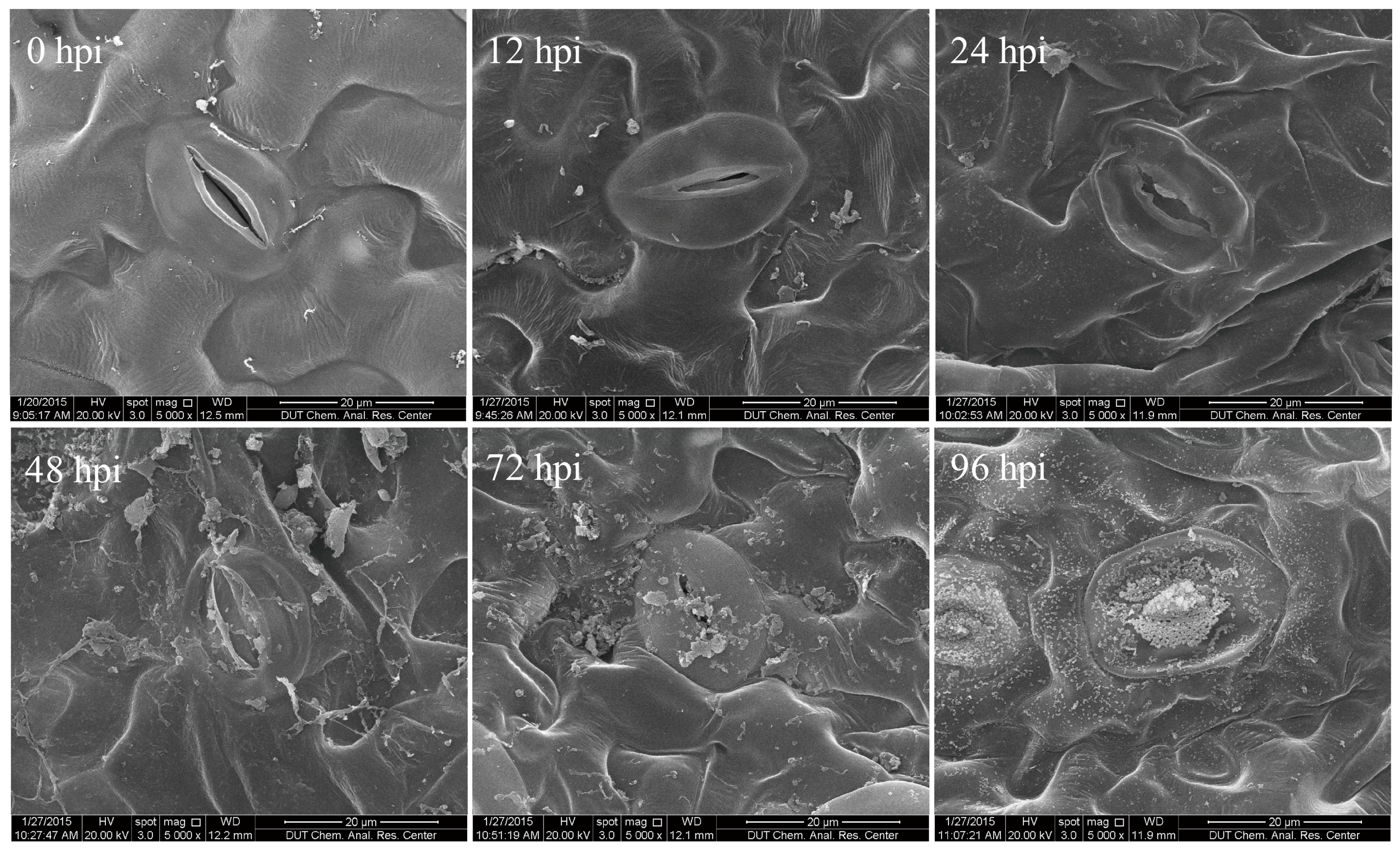
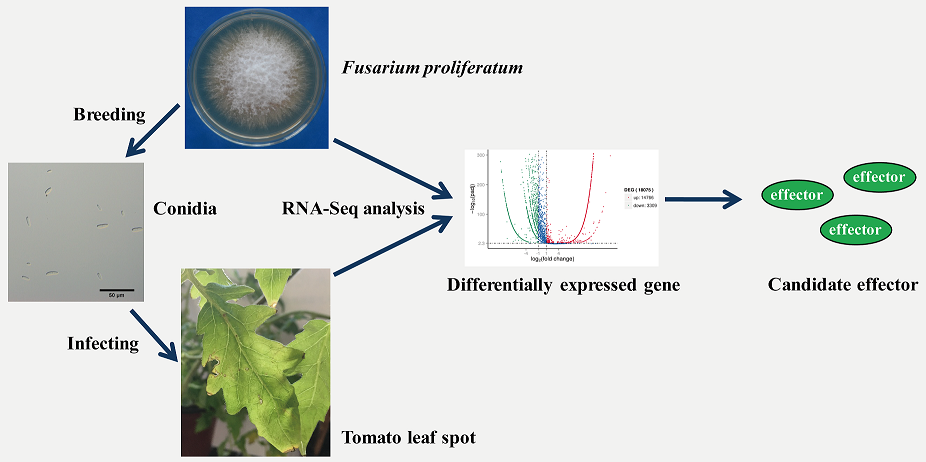
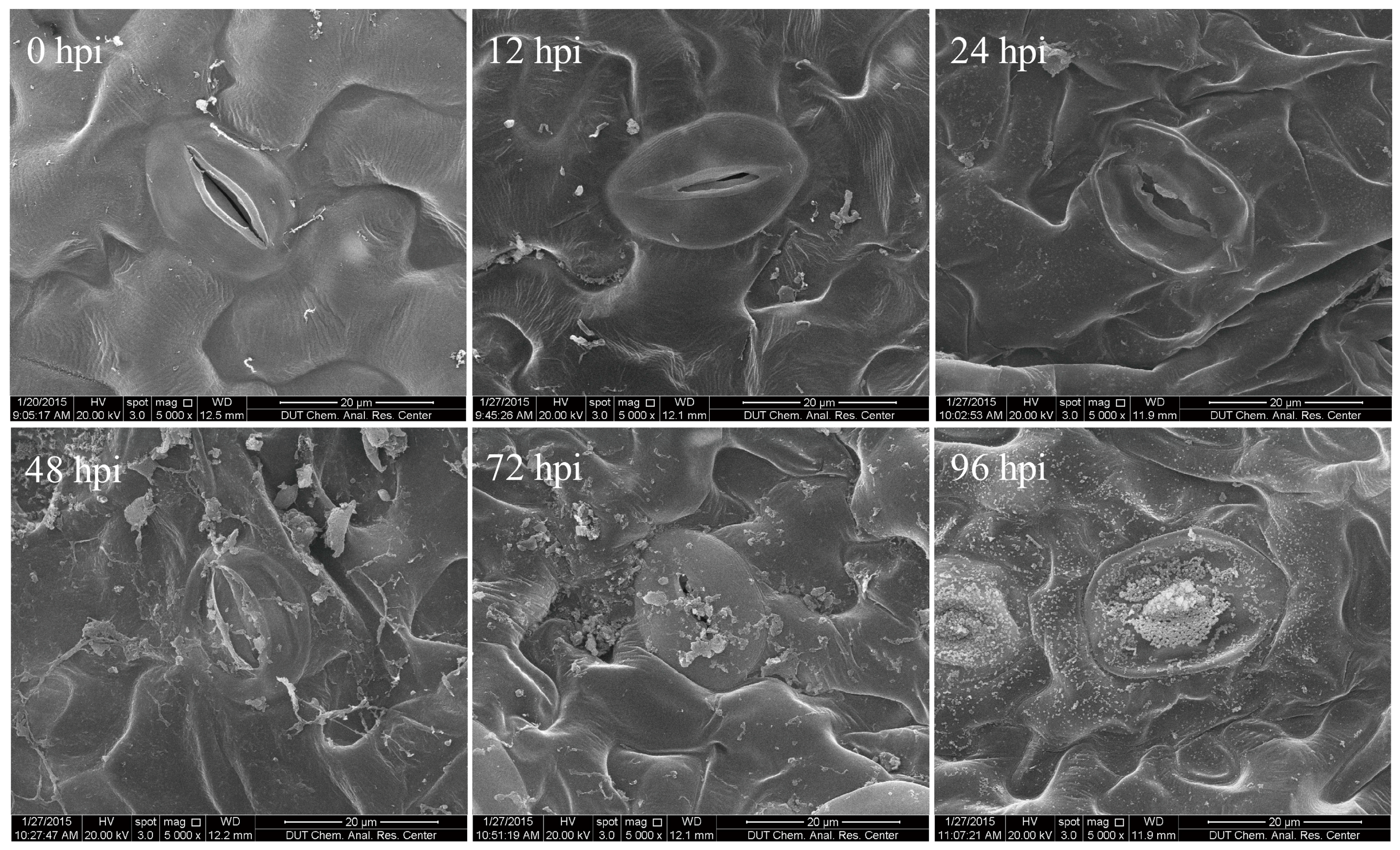
2.1. Characterization of Time Course of F. proliferatum-Infected Tomato Leaves
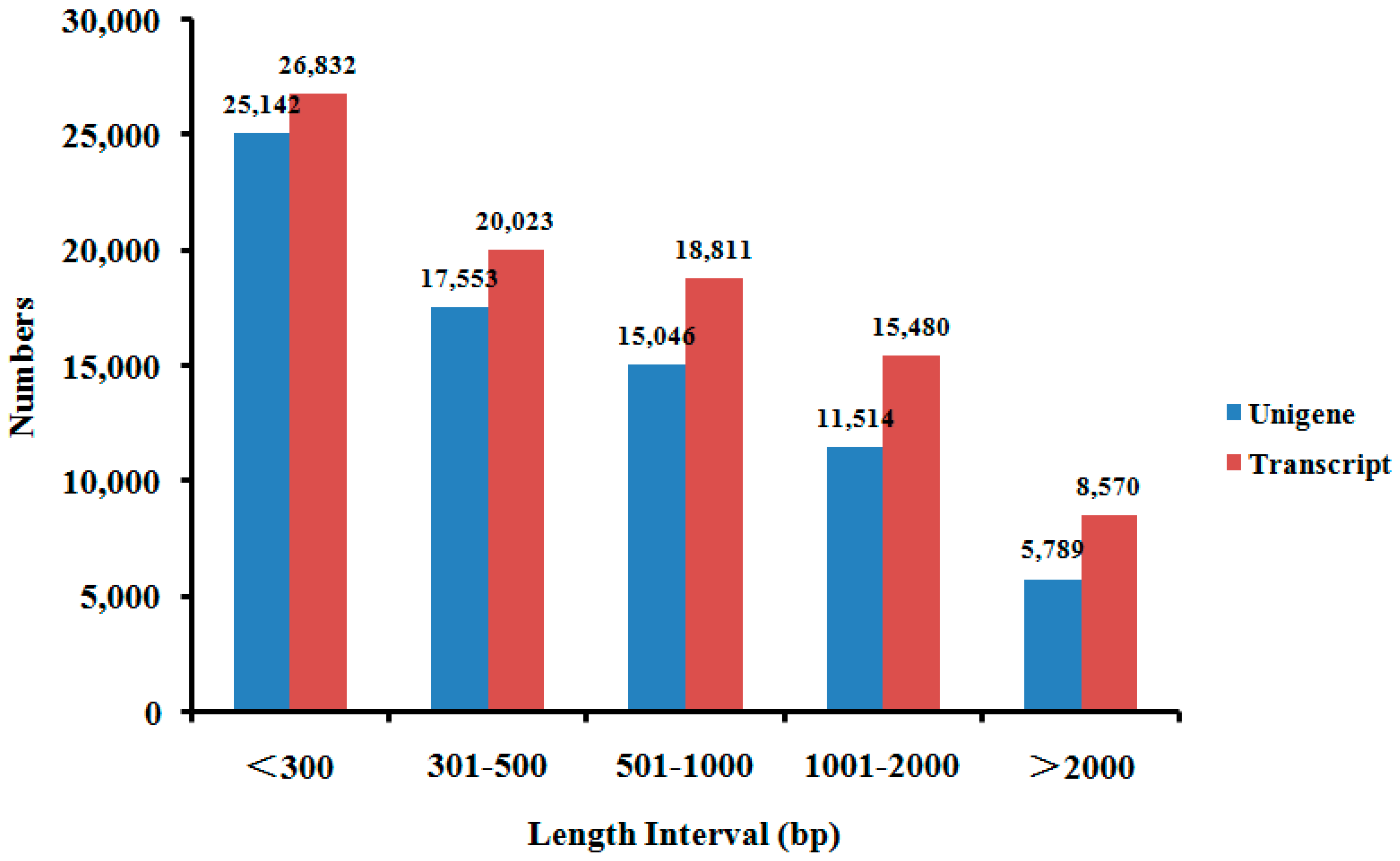
2.2. De novo Assembly of F. proliferatum Transcriptome
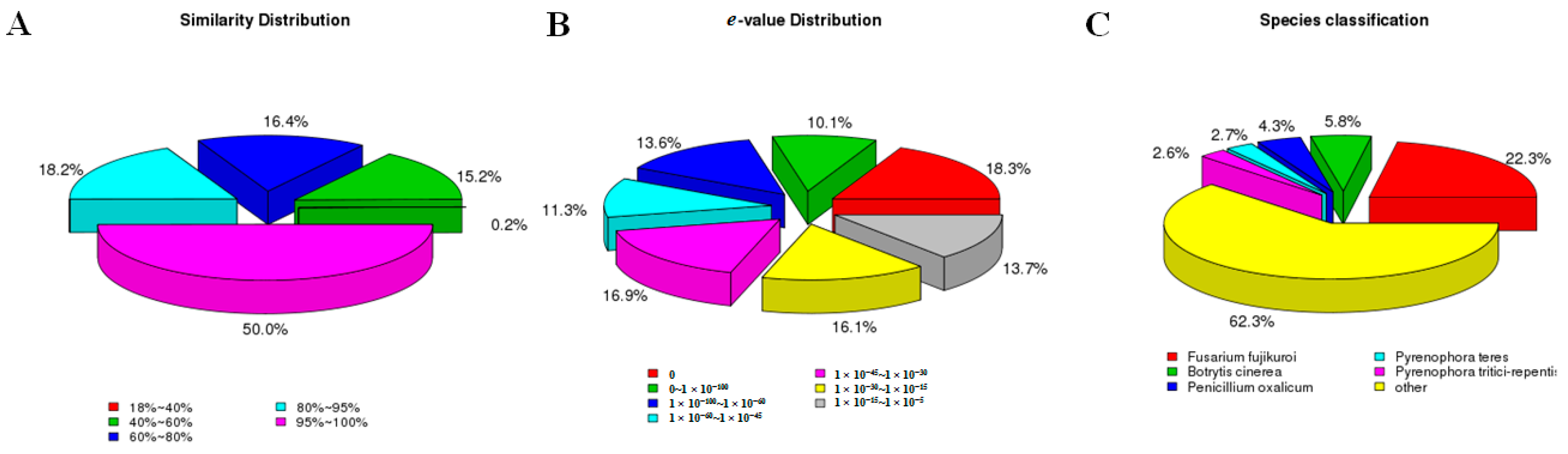
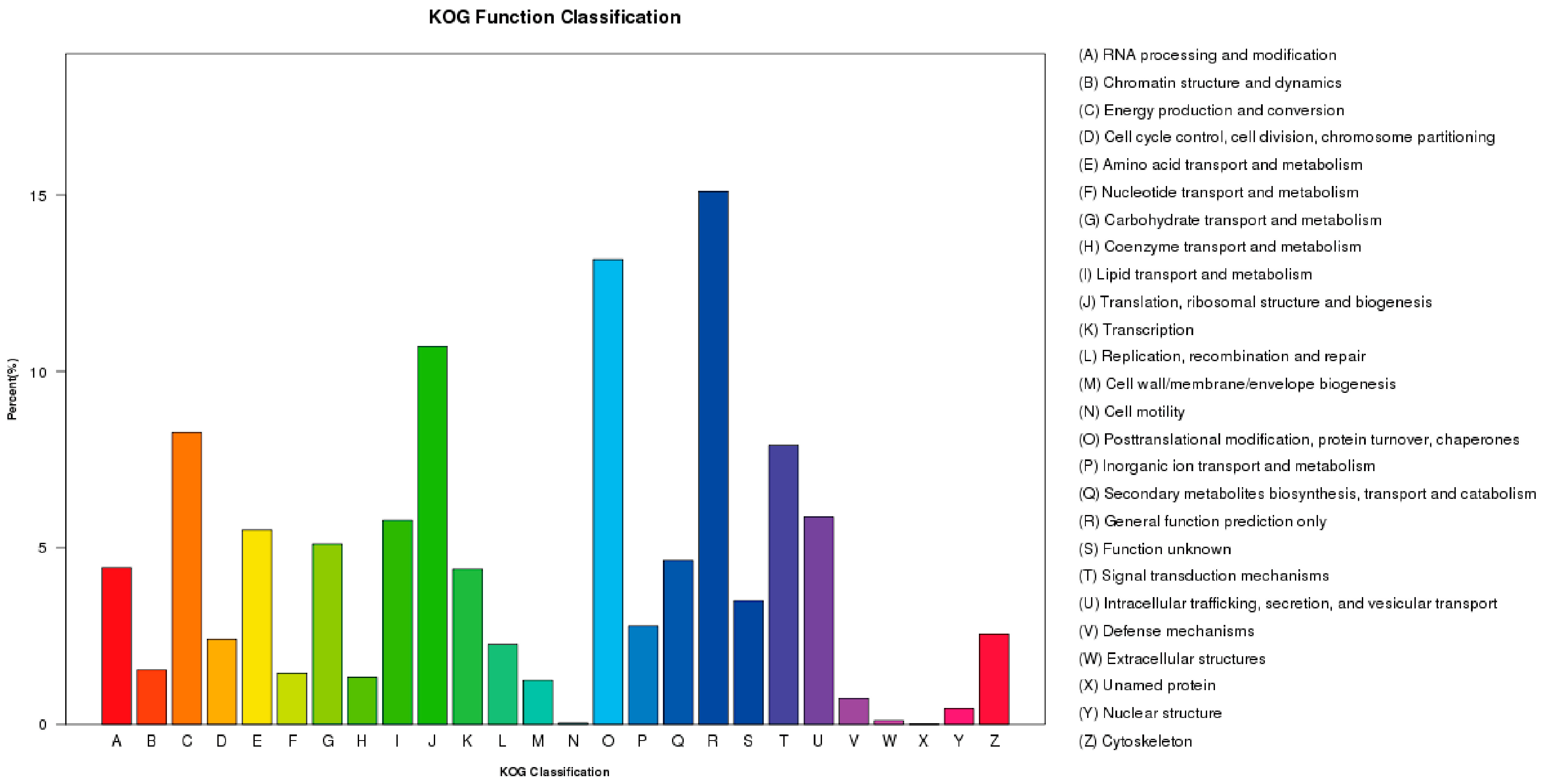
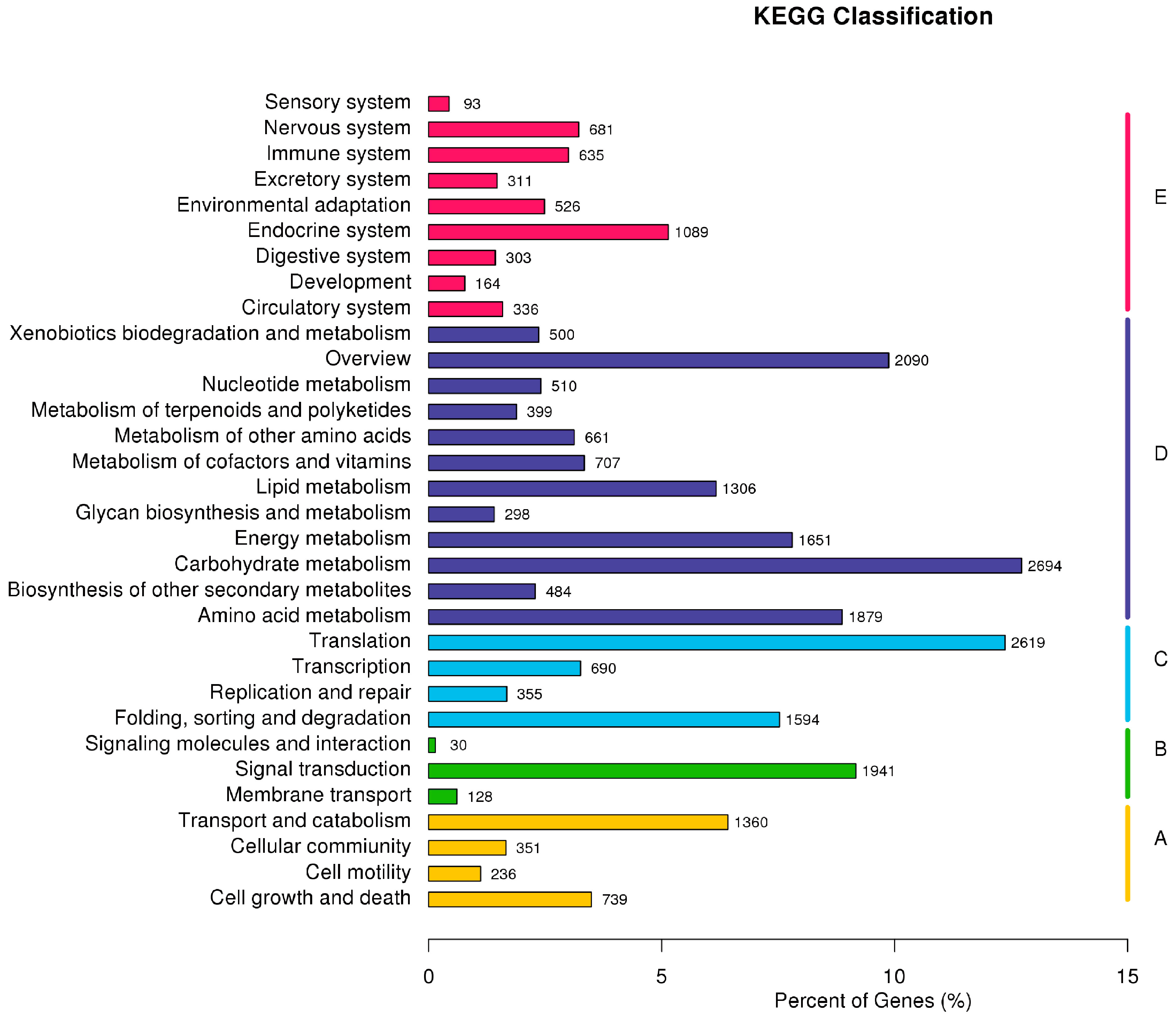
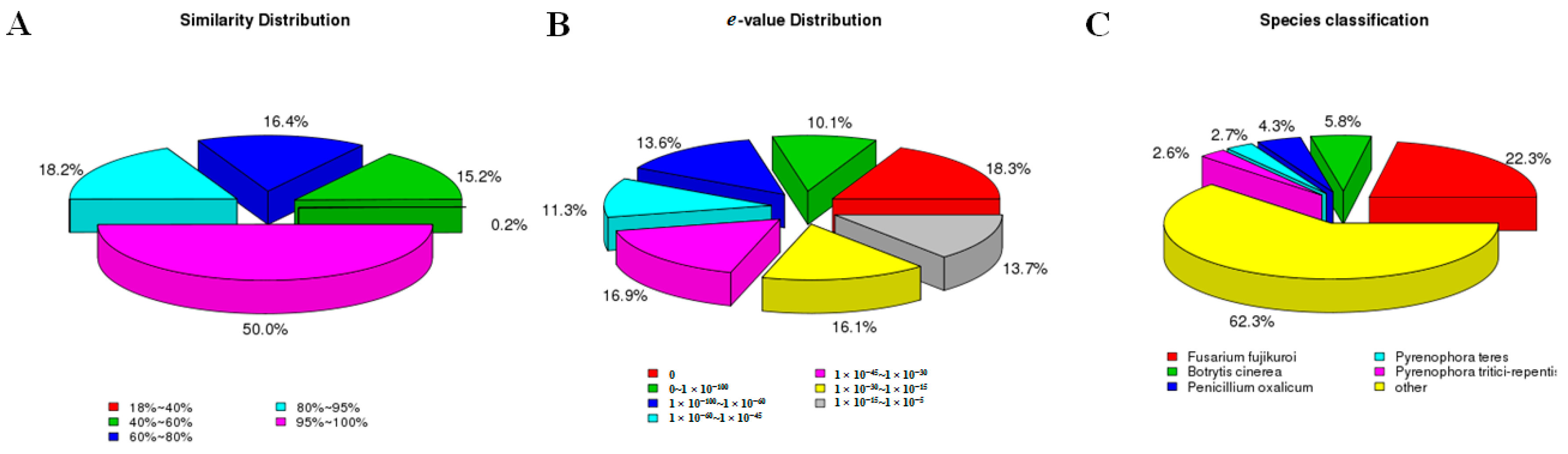
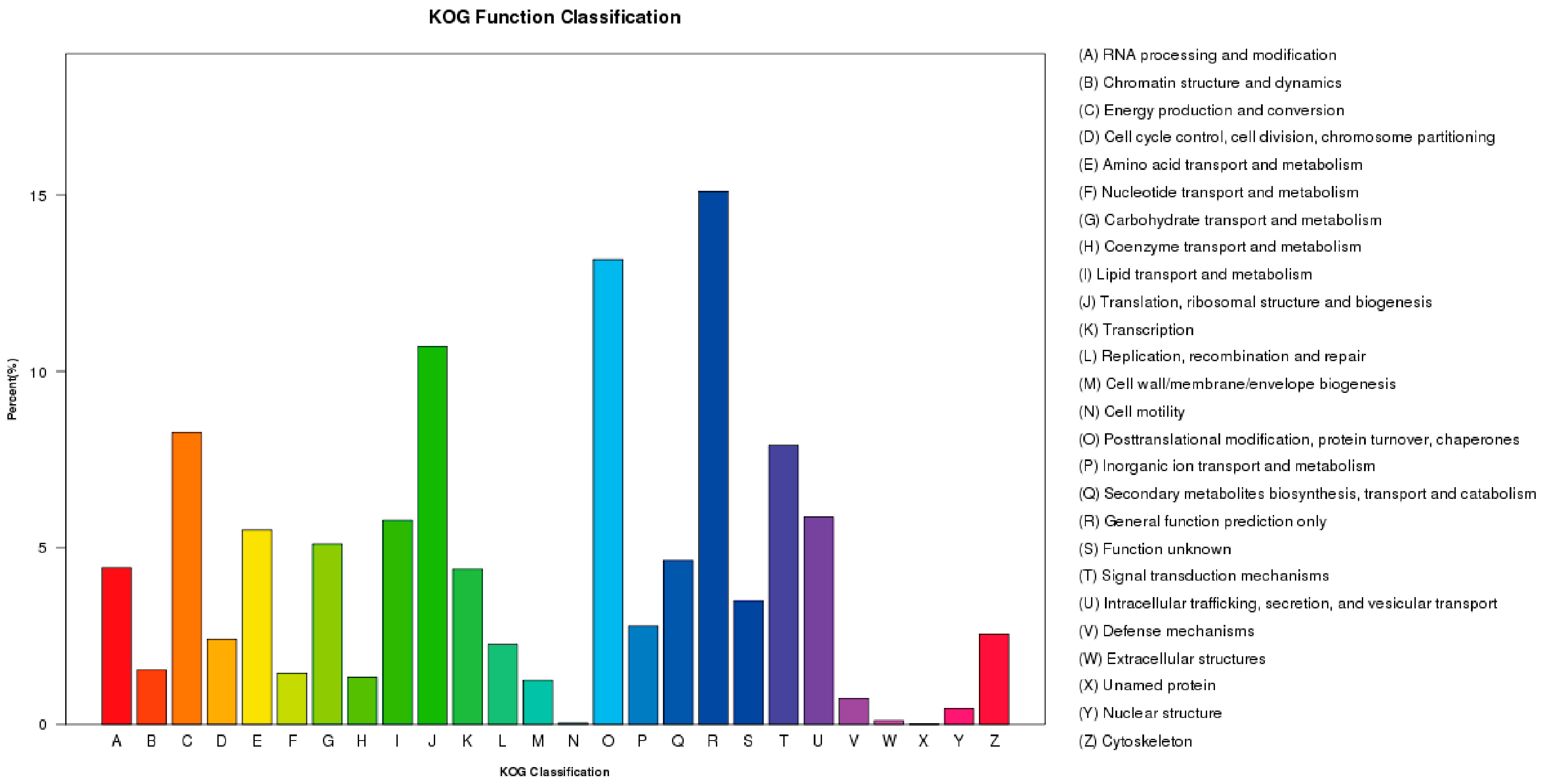
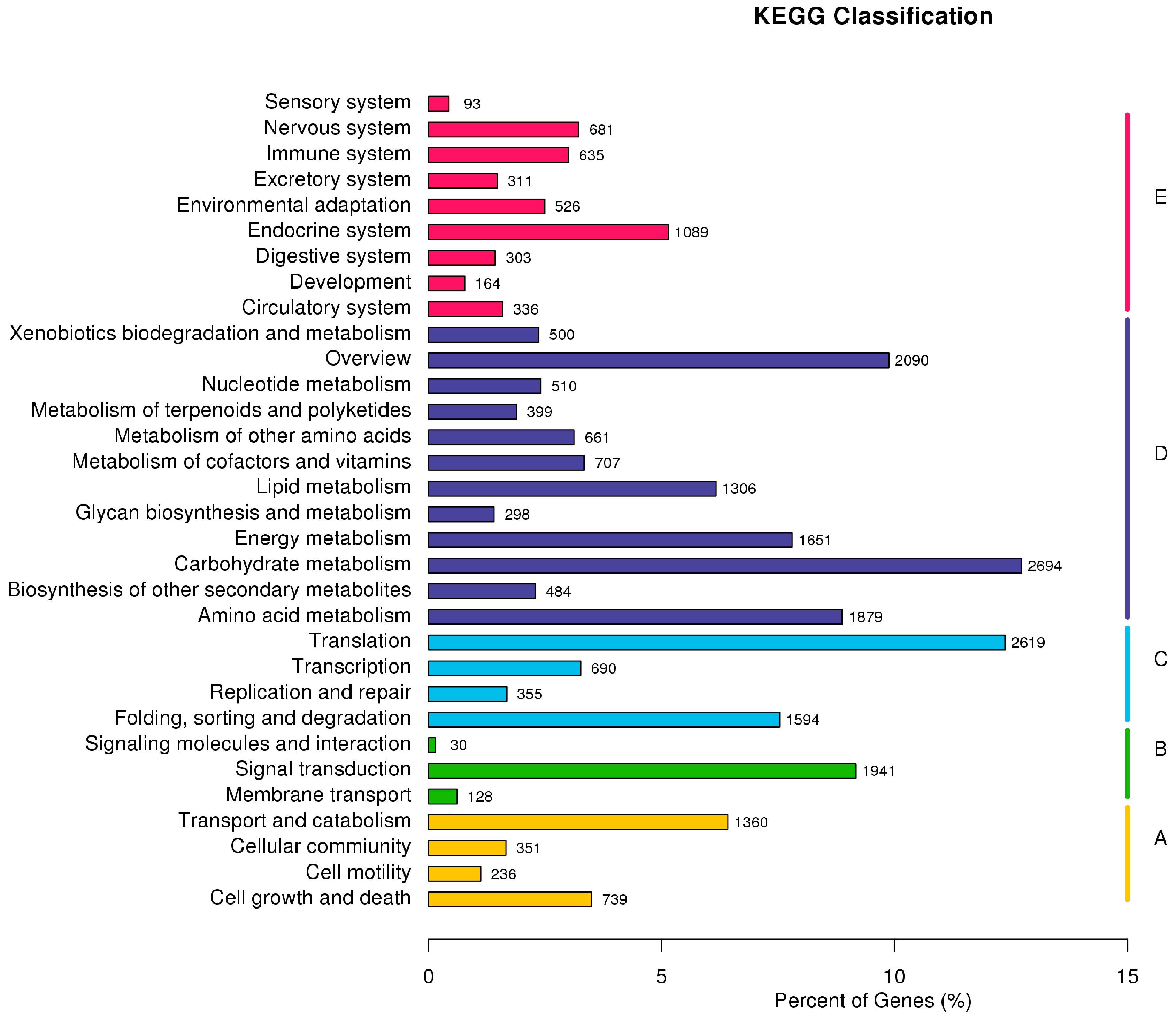
2.3. F. proliferatum Reference Transcriptome Annotation
2.4. F. proliferatum Differental Gene Expression in KS_1
2.5. F. proliferatum Candidate Effectors
3. Materials and Methods
3.1. Biological Material and Inoculation Assays
3.2. Evaluation of Time Course of F. proliferatum Infection
3.3. Transcriptome Profiling
3.4. Transcriptome Annotation and DEG
3.5. Candidate Effector Gene Prediction
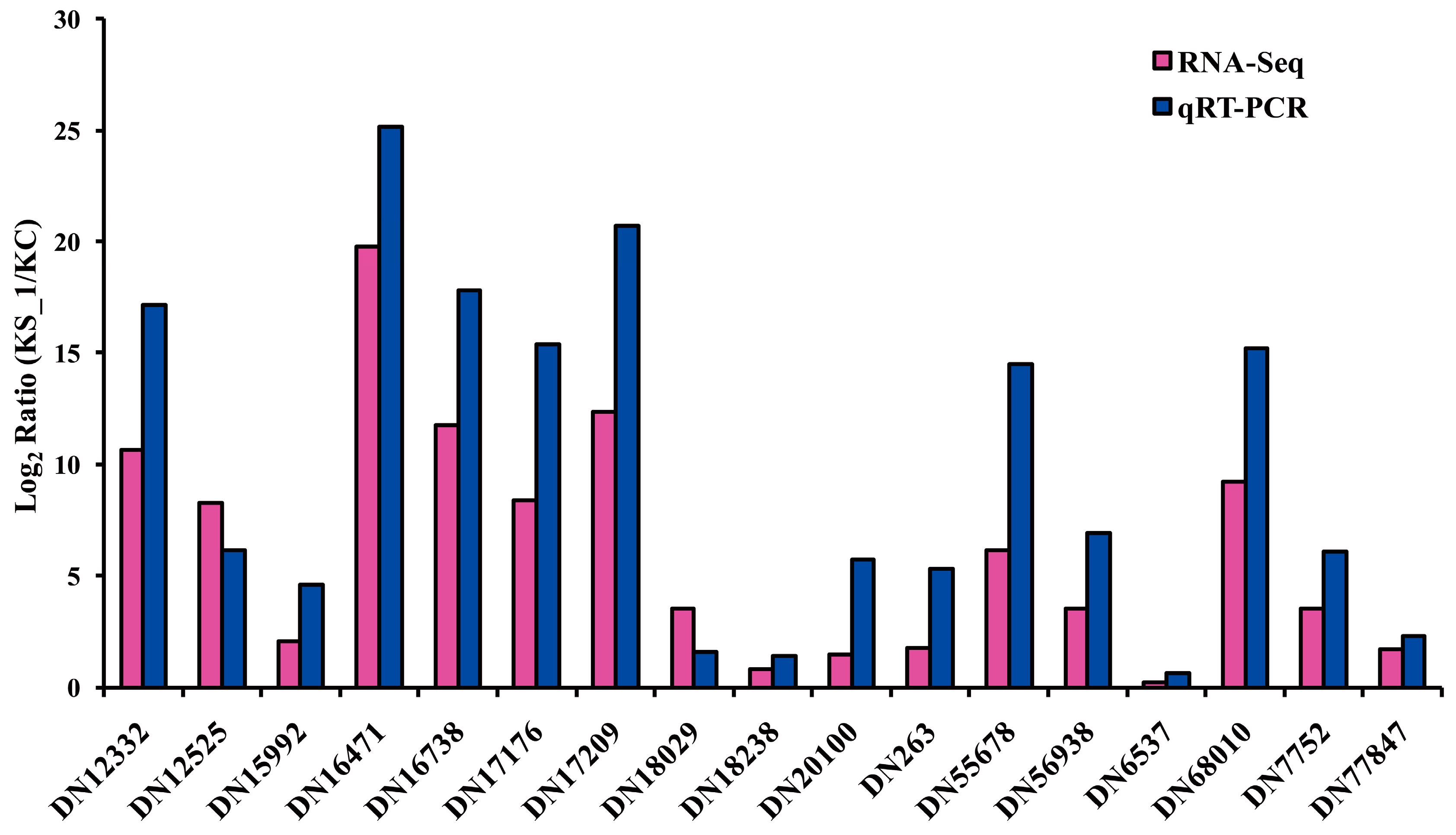
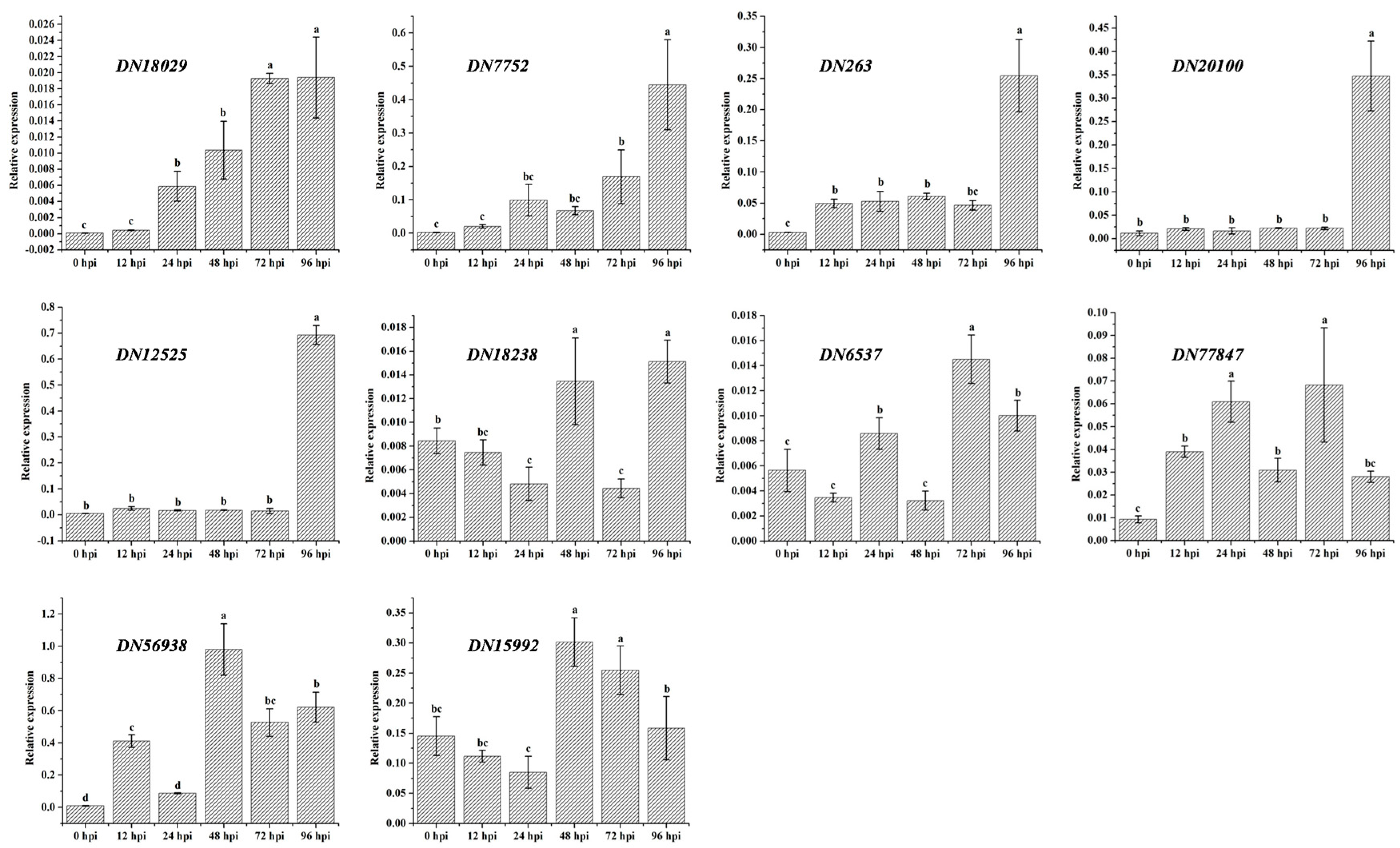
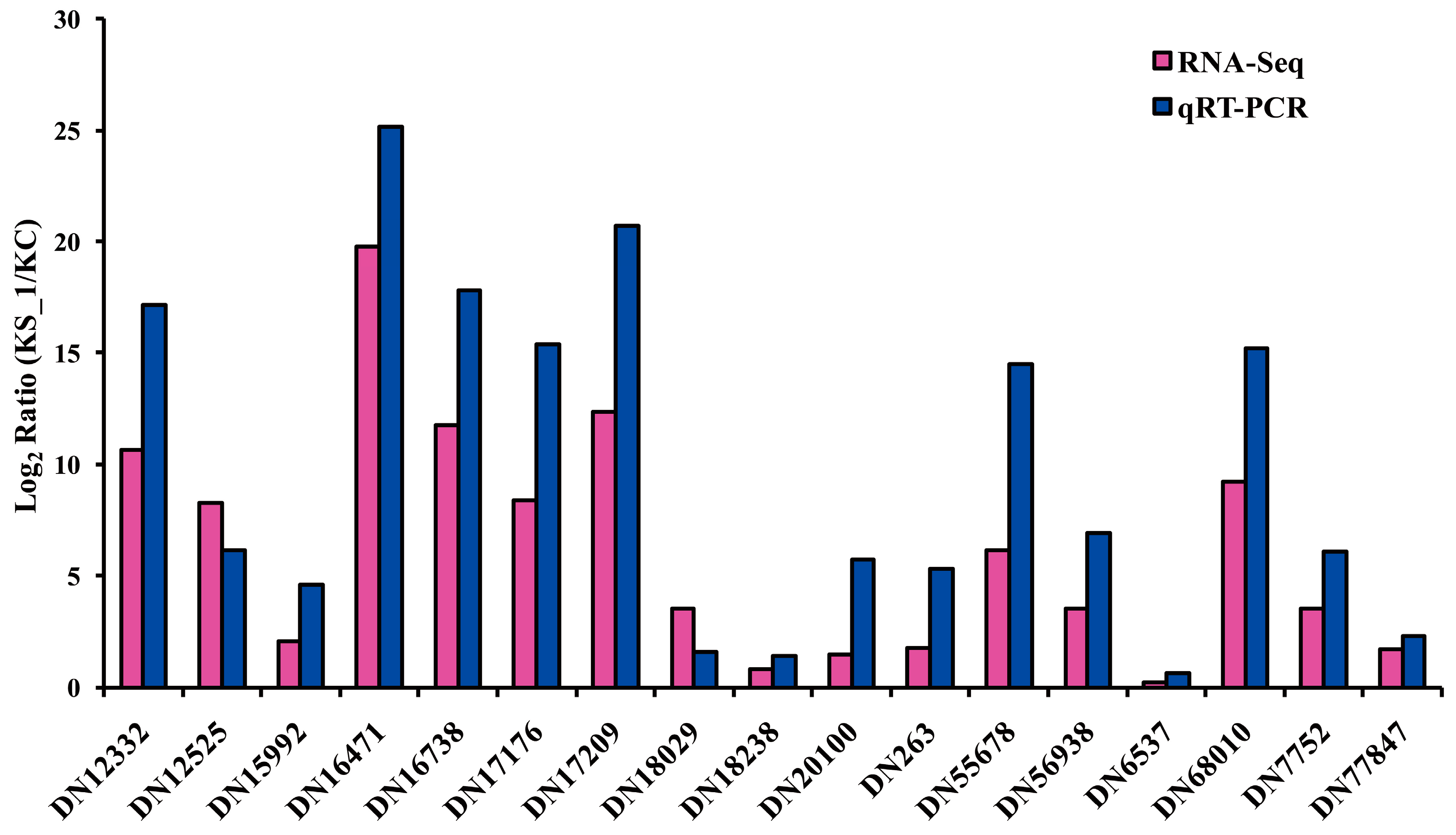
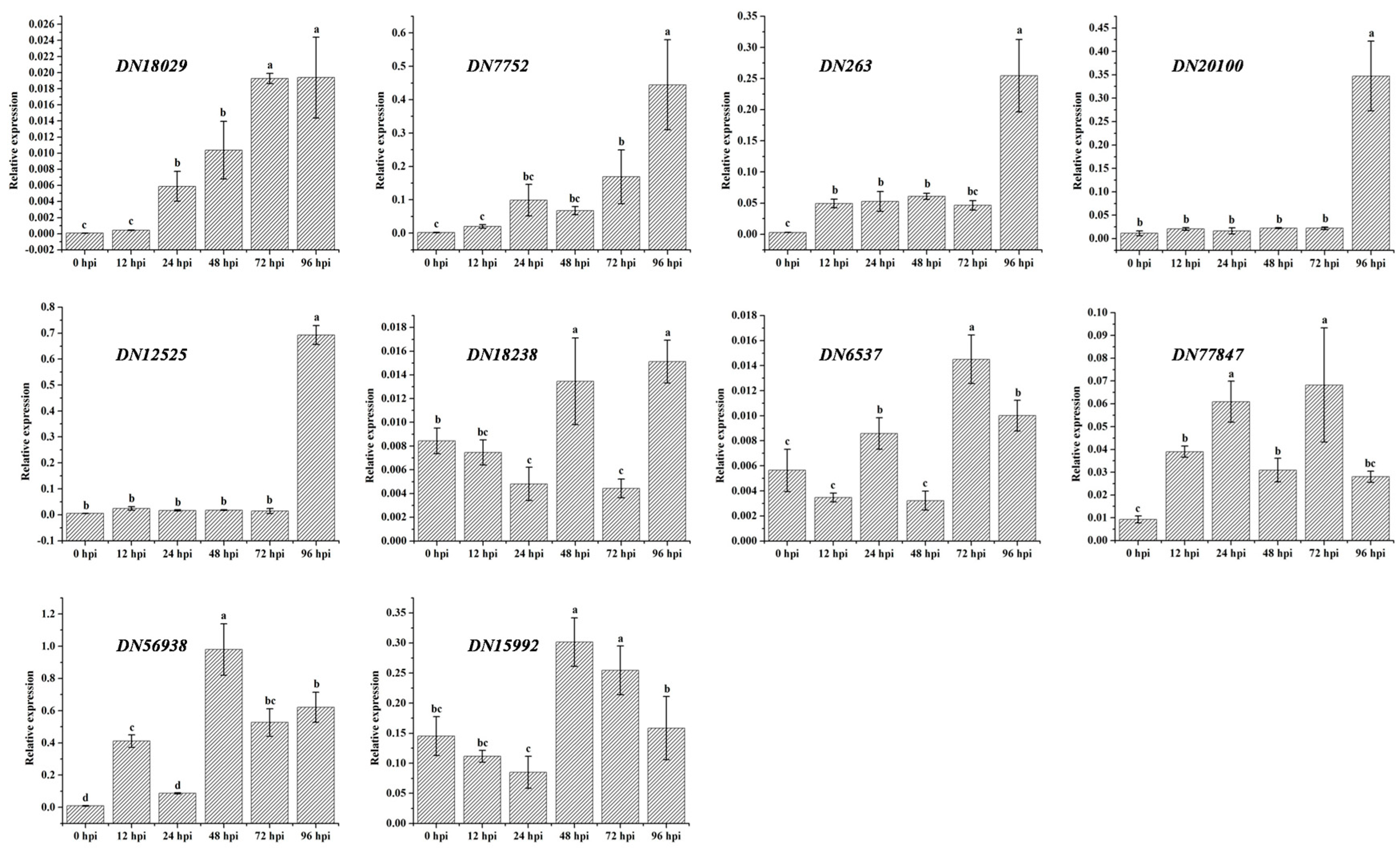
3.6. Quantitative RT-PCR Assay
3.7. Accession Numbers
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes |
| qRT–PCR | Quantitative real-time PCR |
| SIX | Secreted in xylem |
| SGE1 | Secreted gene expression |
| NCBI | National Center of Biotechnology Information |
| RNA-seq | Ribonucleic acid sequencing |
| Hpi | Hourspost-inoculation |
| PDA | Potato dextrose agar |
| Nr | Non-redundant |
| Nt | Nucleotide |
| KEGG | Kyoto encyclopedia of genes and genomes |
| KO | Kyoto encyclopedia of genes and genomes Orthology |
| Pfam | Protein family |
| KOG | euKaryotic Ortholog Groups |
| BLAST | Basic Local Alignment Search Tool |
| GO | Gene ontology |
| FPKM | Fragments per kilobase of exon per million fragments mapped |
| GPI | Glycosylphosphatidylinol |
| CFEM | Cysteine-rich fungal effector motif |
| BYS1 | Blastomyces yeast-phase-specific |
| Poly(A) | Polyadenylated (Poly(A)) |
| ORFs | Open reading frames |
| RSEM | RNA-seq expression estimation by expectationmaximization |
| CT | Threshold cycle |
References
- Consortium, T.T.G. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Suwannarach, N.; Kumla, J.; Nitiyon, S.; Limtong, S.; Lumyong, S. First report of sour rot on tomato caused by Galactomyces reessii in Thailand. J. Gen. Plant Pathol. 2016, 82, 228–231. [Google Scholar] [CrossRef]
- Gao, M.L.; Luan, Y.S.; Yu, H.N.; Bao, Y.M. First report of tomato leaf spot caused by Fusarium proliferatum in China. Can. J. Plant Pathol. 2016, 38, 400–404. [Google Scholar] [CrossRef]
- Peltomaa, R.; Vaghini, S.; Patiño, B.; Benito-Peña, E.; Moreno-Bondi, M.C. Species-specific optical genosensors for the detection of mycotoxigenic Fusarium fungi in food samples. Anal. Chim. Acta 2016, 935, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Seefelder, W.; Gossmann, M.; Humpf, H.U. Analysis of Fumonisin B1 in Fusarium proliferatum-Infected Asparagus Spears and Garlic Bulbs from Germany by Liquid Chromatography−Electrospray Ionization Mass Spectrometry. J. Agric. Food Chem. 2002, 50, 2778–2781. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.F.; Hwang, S.F.; Conner, R.L.; Ahmed, H.U.; Zhou, Q.; Turnbull, G.D.; Strelkov, S.E.; Mclaren, D.L.; Gossen, B.D. First report of Fusarium proliferatum causing root rot in soybean (Glycine max L.) in Canada. Crop Prot. 2015, 67, 52–58. [Google Scholar] [CrossRef]
- Palmero, D.; De Cara, M.; Nosir, W.; Galvez, L.; Cruz, A.; Woodward, S.; Gonzalez-Jaen, M.T.; Tello, J.C. Fusarium proliferatum isolated from garlic in Spain: Identification, toxigenic potential and pathogenicity on related Allium species. Phytopathol. Mediterr. 2012, 51, 207–218. [Google Scholar]
- Rheeder, J.P.; Marasas, W.F.O.; Vismer, H.F. Production of Fumonisin Analogs by Fusarium Species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Gil-Serna, J.; Gálvez, L.; París, M.; Palmero, D. Fusarium proliferatum from rainwater and rooted garlic show genetic and pathogenicity differences. Eur. J. Plant Pathol. 2016, 146, 199–206. [Google Scholar] [CrossRef]
- Isack, Y.; Benichis, M.; Gillet, D.; Gamliel, A. A selective agar medium for isolation, enumeration and morphological identification of Fusarium proliferatum. Phytoparasitica 2014, 42, 541–547. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Dehne, H.W.; Steiner, U. Histopathological assessment of the infection of maize leaves by Fusarium graminearum, F. proliferatum, and F. verticillioides. Fungal Biol. 2016, 120, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Vágány, V.; Jackson, A.C.; Harrison, R.J.; Rainoni, A.; Clarkson, J.P. Identification of pathogenicity-related genes in Fusarium oxysporum f. sp. cepae. Mol. Plant Pathol. 2016, 17, 1032–1047. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.O.; Laurence, M.H.; Ludowici, V.A.; Puno, V.I.; Lim, C.C.; Tesoriero, L.A.; Summerell, B.A.; Liew, E.C.Y. Putative effector genes detected in Fusarium oxysporum from natural ecosystems of Australia. Plant Pathol. 2016, 65, 914–929. [Google Scholar] [CrossRef]
- Rep, M.; van der Does, H.C.; Meijer, M.; van Wijk, R.; Houterman, P.; Dekker, H.; de Koster, C.; Cornelissen, B. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 2010, 53, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Li, Y.; Zhao, M.; Jing, M.; Liu, X.; Liu, M.; Guo, X.; Zhang, X.; Chen, Y.; Liu, Y. Global Genome and Transcriptome Analyses of Magnaporthe oryzae Epidemic Isolate 98-06 Uncover Novel Effectors and Pathogenicity-Related Genes, Revealing Gene Gain and Lose Dynamics in Genome Evolution. PLoS Pathog. 2015, 11, e1004801. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Gardiner, D.M.; Thatcher, L.F.; Lyons, R.; Singh, K.B.; Manners, J.M.; Taylor, J.M. Genome-Wide Analysis in Three Fusarium Pathogens Identifies Rapidly Evolving Chromosomes and Genes Associated with Pathogenicity. Genome Biol. Evol. 2015, 7, 1613–1627. [Google Scholar] [CrossRef] [PubMed]
- Hogenhout, S.A.; van der Hoorn, R.A.; Terauchi, R.; Kamoun, S. Emerging concepts in effector biology of plant-associated organisms. Mol. Plant Microbe Interact. 2009, 22, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Win, J.; Chaparro-Garcia, A.; Belhaj, K.; Saunders, D.G.; Yoshida, K.; Dong, S.; Schornack, S.; Zipfel, C.; Robatzek, S.; Hogenhout, S.A.; et al. Effector biology of plant-associated organisms: Concepts and perspectives. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Edwards, M.C. Genome-Wide Analysis of Small Secreted Cysteine-Rich Proteins Identifies Candidate Effector Proteins Potentially Involved in Fusarium graminearum-Wheat Interactions. Phytopathology 2016, 106, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.W.; Busman, M.; Proctor, R.H. Fusarium verticillioidesSGE1 is required for full virulence and regulates expression of protein effector and secondary metabolite biosynthetic genes. Mol. Plant Microbe Interact. 2014, 27, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.X.; Domier, L.; Radwan, O.; Yendrek, C.; Hudson, M.; Hartman, G.L. Identification of multiple phytotoxins produced by Fusarium virguliforme including a phytotoxic effector (FvNIS1) associated with sudden death syndrome foliar symptoms. Mol. Plant Microbe Interact. 2016, 29, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; Shea, T.; Young, S.; Zeng, Q.; Kistler, H.C. Genome Sequence of Fusarium oxysporum f. sp. melonis Strain NRRL 26406, a Fungus Causing Wilt Disease on Melon. Genome Announc. 2014, 2, e00730-14. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, P.; Sieber, C.M.K.; Bargen, K.W.V.; Studt, L.; Niehaus, E.M.; Espino, J.J.; Huß, K.; Michielse, C.B.; Albermann, S.; Wagner, D. Deciphering the Cryptic Genome: Genome-wide Analyses of the Rice Pathogen Fusarium fujikuroi Reveal Complex Regulation of Secondary Metabolism and Novel Metabolites. PLoS Pathog. 2013, 9, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.M.; Stiller, J.; Kazan, K. Genome Sequence of Fusarium graminearum Isolate CS3005. Genome Announc. 2014, 2, e00227-14. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Yano, K.; Suzuki, A.; Kawamura, S.; Sakurai, N.; Suda, K.; Kurabayashi, A.; Suzuki, T.; Tsugane, T.; Watanabe, M. Large-scale analysis of full-length cDNAs from the tomato (Solanum lycopersicum) cultivar Micro-Tom, a reference system for the Solanaceae genomics. BMC Genom. 2010, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sengupta, A.; Ghosh, R.; Agarwal, G.; Tarafdar, A.; Nagavardhini, A.; Pande, S.; Varshney, R.K. Genome wide transcriptome profiling of Fusarium oxysporum f sp. ciceris conidial germination reveals new insights into infection-related genes. Sci. Rep. 2016, 6, 37353. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. Fungal cell wall chitinases and glucanases. Microbiology 2004, 150, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, S. Groovy times: Filamentous pathogen effectors revealed. Curr. Opin. Plant Biol. 2007, 10, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.G.; Rafiqi, M.; Gan, P.; Chakrabarti, A.; Dodds, P.N. Recent progress in discovery and functional analysis of effector proteins of fungal and oomycete plant pathogens. Curr. Opin. Plant Biol. 2009, 12, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Stergiopoulos, I.; de Wit, P.J. Fungal Effector Proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.D.; Kelkar, H.S.; Dean, R.A. An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem. Sci. 2003, 28, 118–121. [Google Scholar] [CrossRef]
- Guzmán-Guzmán, P.; Alemán-Duarte, M.I.; Delaye, L.; Herrera-Estrella, A.; Olmedo-Monfil, V. Identification of effector-like proteins in Trichoderma spp. and role of a hydrophobin in the plant-fungus interaction and mycoparasitism. BMC Genet. 2017, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Burg, E.F., III; Smith, L.H., Jr. Cloning and characterization of bys1, a temperature-dependent cDNA specific to the yeast phase of the pathogenic dimorphic fungus Blastomyces dermatitidis. Infect. Immun. 1994, 62, 2521–2528. [Google Scholar] [PubMed]
- Ismail, I.A.; Able, A.J. Secretome analysis of virulent Pyrenophora teres f. teres isolates. Proteomics 2016, 16, 2625–2636. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D. The Positive False Discovery Rate: A Bayesian Interpretation and the q-Value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Thatcher, L.F.; Gardiner, D.M.; Kazan, K.; Manners, J.M. A highly conserved effector in Fusarium oxysporum is required for full virulence on Arabidopsis. Mol. Plant Microbe Interact. 2011, 25, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; Preter, K.D.; Pattyn, F.; Poppe, B.; Roy, N.V.; Paepe, A.D.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Library | Raw Reads | Clean Reads | Clean Bases (Gb) | Error (%) | Q20 (%) 1 | Q30 (%) 2 | GC (%) |
|---|---|---|---|---|---|---|---|
| KC | 52,509,182 | 37,091,012 | 6.46 | 0.03 | 96.28 | 92.16 | 51.58 |
| KS_1 | 63,479,768 | 61,614,408 | 7.70 | 0.03 | 96.13 | 92.29 | 53.15 |
| Category | Total Number | Min Length (bp) | Mean Length (bp) | Median Length (bp) | Max Length (bp) | N50 |
|---|---|---|---|---|---|---|
| Transcripts | 89,716 | 201 | 853 | 471 | 17,632 | 1443 |
| Unigenes | 75,044 | 201 | 767 | 419 | 17,632 | 1283 |
| Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in Nr | 46,292 | 61.68 |
| Annotated in Nt | 62,514 | 83.3 |
| Annotated in KO | 21,172 | 28.21 |
| Annotated in SwissPort | 39,854 | 53.10 |
| Annotated in Pfam | 37,595 | 50.09 |
| Annotated in GO | 38,947 | 51.89 |
| Annotated in KOG | 24,338 | 32.43 |
| Annotated in all databases | 12,073 | 16.08 |
| Annotated in at least one database | 71,787 | 95.78 |
| Total unigenes | 75,044 | 100 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Yao, S.; Liu, Y.; Yu, H.; Xu, P.; Sun, W.; Pu, Z.; Hou, H.; Bao, Y. Transcriptome Analysis of Tomato Leaf Spot Pathogen Fusarium proliferatum: De novo Assembly, Expression Profiling, and Identification of Candidate Effectors. Int. J. Mol. Sci. 2018, 19, 31. https://doi.org/10.3390/ijms19010031
Gao M, Yao S, Liu Y, Yu H, Xu P, Sun W, Pu Z, Hou H, Bao Y. Transcriptome Analysis of Tomato Leaf Spot Pathogen Fusarium proliferatum: De novo Assembly, Expression Profiling, and Identification of Candidate Effectors. International Journal of Molecular Sciences. 2018; 19(1):31. https://doi.org/10.3390/ijms19010031
Chicago/Turabian StyleGao, Meiling, Siyu Yao, Yang Liu, Haining Yu, Pinsan Xu, Wenhui Sun, Zhongji Pu, Hongman Hou, and Yongming Bao. 2018. "Transcriptome Analysis of Tomato Leaf Spot Pathogen Fusarium proliferatum: De novo Assembly, Expression Profiling, and Identification of Candidate Effectors" International Journal of Molecular Sciences 19, no. 1: 31. https://doi.org/10.3390/ijms19010031